Paeds · gastroenterology-hepatology-and-nutrition
Hirschsprung disease
Also known as Congenital aganglionic megacolon · Aganglionosis · Hirschsprung-associated enterocolitis · Suction rectal biopsy · Soave pull-through · Swenson pull-through · Duhamel pull-through · Total colonic aganglionosis · RET proto-oncogene · Transition zone
Fellowship guide to Hirschsprung disease, built around the rule that a term neonate who has not passed meconium within 48 hours has a functional obstruction until proven otherwise. The page covers the embryology of failed neural crest cell migration that leaves a non-relaxing aganglionic distal bowel, the suction rectal biopsy with calretinin staining that confirms the diagnosis, the levelling biopsy and three pull-through operations (Swenson, Soave, Duhamel), and the recognition and aggressive treatment of Hirschsprung-associated enterocolitis as the leading cause of death.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture a full-term baby boy born after an uncomplicated pregnancy. He feeds well for the first day, but by 48 hours he has not passed any meconium. His abdomen is becoming full and tight. That single delayed milestone, meconium not passed within 48 hours of birth, is the signature that turns a well-looking neonate into a child you must investigate for Hirschsprung disease. [1] [6]
Hirschsprung disease is a congenital disorder in which a variable length of the distal bowel lacks enteric ganglion cells. The affected segment, usually the rectosigmoid colon, cannot relax and cannot push stool onward, so it behaves like a fixed obstruction. Stool and gas pile up behind it, and the proximal bowel dilates. The disease takes its name from Harald Hirschsprung, the Danish physician who described two infants with megacolon in 1887, though the absence of ganglion cells that defines it was not understood until the mid-twentieth century. [1] [12]
The practical consequence is that Hirschsprung disease sits squarely between two common clinical scenarios: the neonate with low intestinal obstruction and delayed meconium, and the older child with chronic constipation that does not respond to standard laxatives. In both settings the diagnosis is easy to miss, and the consequences of missing it are serious. The leading cause of death is not the obstruction itself but Hirschsprung-associated enterocolitis, a fulminant colitis that can strike before or after surgery. [2] [3]
Classification
Hirschsprung disease is classified by how far up the bowel the aganglionosis extends, because the length of the affected segment changes the presentation, the contrast enema appearance and the surgical approach. The most useful clinical split is between short-segment disease, which involves the rectosigmoid and accounts for roughly 80 per cent of cases, and long-segment disease, which extends proximal to the sigmoid colon. A small but important minority have total colonic aganglionosis, where the entire colon and sometimes part of the terminal ileum lack ganglion cells. [1] [12]
The length matters because it predicts the clinical pattern. Short-segment disease typically presents in the neonatal period with delayed meconium and distension, and is amenable to a transanal pull-through. Long-segment and total colonic disease present earlier and more severely, often with bilious vomiting and a microcolon on contrast enema, and require more complex surgery with ileal pull-through. Rare total intestinal aganglionosis, where most of the gut is affected, is incompatible with long-term survival by current surgical standards. [1] [8]
A second classification axis separates isolated or non-syndromic Hirschsprung disease from syndromic forms that travel with other congenital anomalies. Roughly 70 per cent of cases are isolated, while the remaining 30 per cent have an identifiable syndrome or chromosomal abnormality. The most common association is trisomy 21 (Down syndrome), followed by a cluster of neurocristopathies in which the same neural crest migration defect produces pigmentary, auditory and autonomic features. [12]
Short-segment (rectosigmoid)
- Approximately 80 per cent of all cases
- Neonatal delayed meconium and distension
- Transanal Soave pull-through often feasible
- Male predominance of about four to one
Long-segment
- Extends proximal to the sigmoid colon
- Earlier and more severe presentation
- May need laparoscopic or open levelling biopsies
- Near-equal sex ratio
Total colonic aganglionosis
- Approximately 5 to 10 per cent of cases
- Bilious vomiting, microcolon on enema
- Requires ileal pull-through, often staged
- Higher complication and reoperation rates
Epidemiology & Risk Factors
Hirschsprung disease occurs in approximately one in five thousand live births, making it one of the more common congenital gastrointestinal disorders. Boys are affected roughly four times more often than girls for short-segment disease, but the sex ratio approaches one to one for long-segment disease. This shift suggests that longer segments have a stronger genetic contribution that overrides the male predominance seen in milder cases. [1] [12]
The strongest chromosomal association is trisomy 21. Approximately 10 per cent of all children with Hirschsprung disease have Down syndrome, and conversely about 1 per cent of children with Down syndrome have Hirschsprung disease. A child with trisomy 21 who presents with delayed meconium or chronic constipation therefore has a much higher pre-test probability of Hirschsprung disease than the general population, and a low threshold for rectal biopsy is warranted. [12]
Genetically, Hirschsprung disease is heterogeneous but has a clear heritable component. Mutations in the RET proto-oncogene on chromosome 10 are found in roughly 50 per cent of familial cases and 15 to 20 per cent of sporadic cases. Other genes including EDNRB, EDN3, SOX10 and PHOX2B contribute to syndromic forms. Families with one affected child face a recurrence risk of approximately 3 to 5 per cent in siblings, rising to 10 per cent or more for long-segment disease, so genetic counselling is part of routine family discussion. [12]
Pathophysiology
To understand why Hirschsprung disease causes obstruction, you need to understand how the gut gets its nerves in the first place. During the fifth to twelfth week of gestation, neural crest cells migrate from the hindbrain along the developing gut in a wave that moves from mouth to anus. These cells settle in the gut wall to form two networks of ganglion cells: the myenteric plexus (Auerbach plexus) between the longitudinal and circular muscle layers, and the submucosal plexus (Meissner plexus) beneath the mucosa. Together these plexuses coordinate the peristaltic waves that propel gut contents onward. [12]
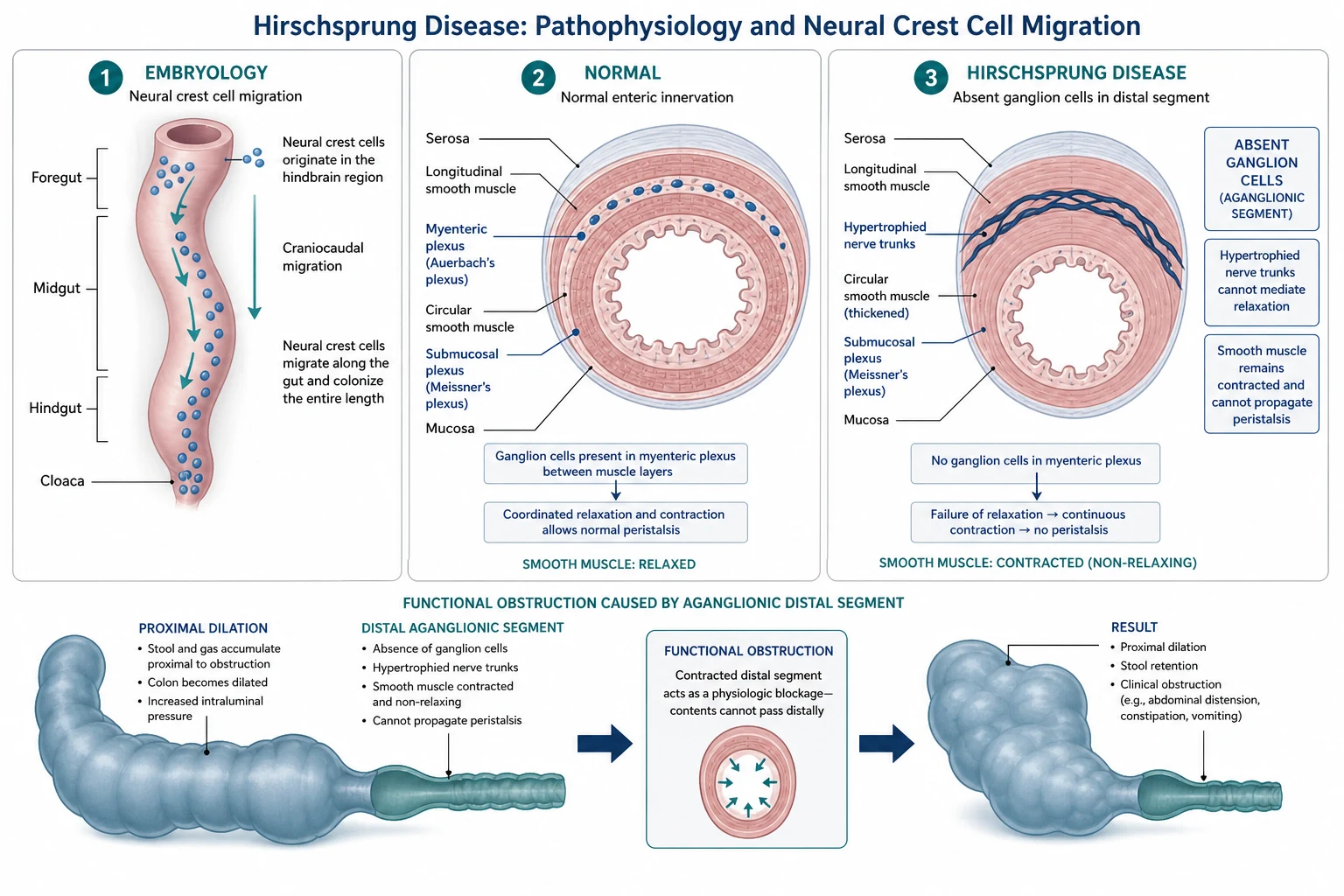
When this migration stalls before it reaches the anal canal, the distal bowel is left without ganglion cells. The aganglionic segment still has extrinsic parasympathetic input from the pelvic nerves, but it lacks the inhibitory interneurons that normally allow the internal anal sphincter and the bowel wall to relax ahead of a peristaltic wave. The result is sustained tonic contraction: the bowel behaves like a tight ring that cannot open. Stool and gas cannot pass, and the proximal ganglionic bowel dilates and hypertrophies as it tries to push against the block. [1] [12]
The junction between the contracted aganglionic segment and the dilated proximal bowel is called the transition zone. This is the area where the calibre of the bowel changes abruptly on contrast enema, and it is the landmark the surgeon uses to decide where to cut. Frozen-section biopsies taken at operation confirm whether ganglion cells are present at the proposed anastomotic level, a step called levelling. The transition zone visible on imaging does not always match the histological level of aganglionosis, which is why levelling biopsies are essential rather than relying on the contrast enema alone. [10]
The pathophysiology of Hirschsprung-associated enterocolitis is less fully understood but involves a dangerous interaction between stasis, bacterial overgrowth and mucosal barrier failure. Stool pooled behind the obstruction becomes a culture medium for bacteria, and the impaired blood supply and mucosal integrity of the obstructed bowel allow translocation of organisms into the bloodstream. The result is a fulminant colitis with explosive diarrhoea, systemic sepsis and, if untreated, progression to toxic megacolon, perforation and death. [4] [3]
Clinical Presentation
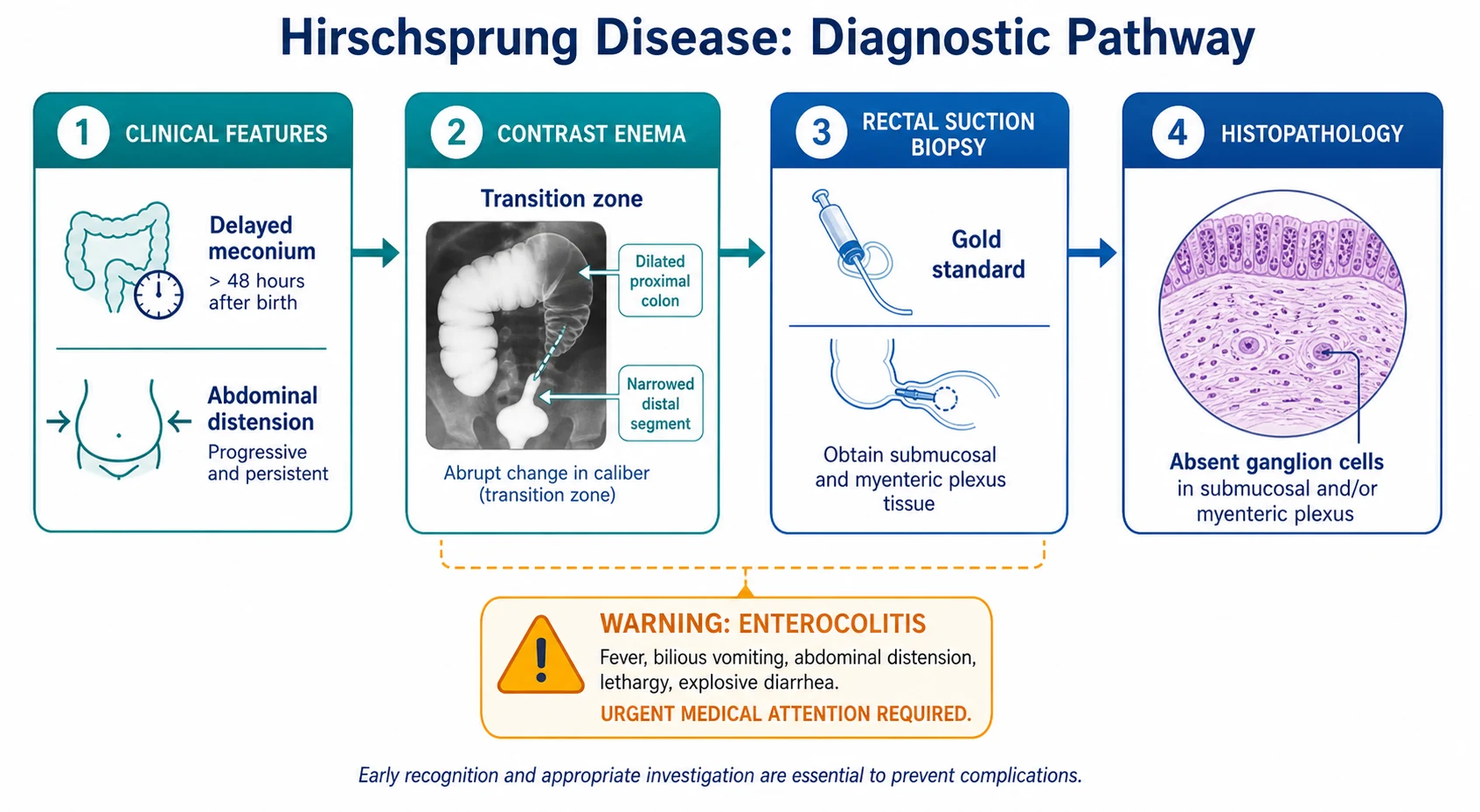
The neonatal presentation is the one every paediatric clinician must recognise. A full-term baby fails to pass meconium within the first 48 hours of life, develops progressive abdominal distension, and may vomit, initially milky then bilious as the obstruction backs up. The baby may appear surprisingly well at first, feeding and alert, which makes the distension easy to dismiss. The clue is the combination of delayed meconium with distension, not either sign alone. Approximately 90 per cent of term neonates with Hirschsprung disease have delayed passage of meconium beyond 48 hours. [1] [6]
The older-infant and child presentation is more insidious and more easily missed. The child has chronic constipation dating from birth, with infrequent large or pellet-like stools, progressive abdominal distension, and failure to thrive from chronic feeding difficulty. Unlike functional constipation, where the rectum is full of palpable stool and the child may soil, the rectum in Hirschsprung disease is characteristically empty on digital examination because the aganglionic segment holds stool proximally. Soiling, a hallmark of functional constipation with faecal impaction, is initially absent. [1] [11]
The third presentation, and the most dangerous, is Hirschsprung-associated enterocolitis. This can occur at any time, including before diagnosis, after stoma formation, and after definitive pull-through surgery. The child develops fever, worsening abdominal distension, and explosive foul-smelling diarrhoea that may be blood-stained. Lethargy, poor feeding and signs of dehydration or septic shock develop rapidly. Enterocolitis is the leading cause of death in Hirschsprung disease, and the index of suspicion must be high in any child with known or suspected disease who deteriorates. [2] [3]
On examination, the abdomen is distended with visible loops and sometimes visible peristalsis. A careful digital rectal examination is diagnostic in the older child: the anal sphincter feels tight, the rectum is empty, and on withdrawal of the finger there may be an explosive release of gas and liquid stool, the so-called blast sign. The perineum must always be inspected to exclude an anorectal malformation, which is a different surgical problem entirely. [1]
Differential Diagnosis
When a neonate presents with delayed meconium and distension, the differential is the full list of low intestinal obstruction causes. The question is whether the obstruction is mechanical, as in ileal atresia or an anorectal malformation, or functional, as in Hirschsprung disease or meconium plug syndrome. The distinction matters because mechanical obstructions need urgent surgery while functional ones may respond to washouts while awaiting definitive diagnosis. [1]
Meconium plug syndrome, also called small left colon syndrome when it occurs in infants of diabetic mothers, is the most common mimic. A plug of thick meconium obstructs the colon, producing distension and delayed passage that resolves with enemas or washouts. Because meconium plug syndrome can be impossible to distinguish from Hirschsprung disease clinically, every infant who presents with this pattern should have a suction rectal biopsy before discharge, even if the obstruction has resolved. Missing Hirschsprung disease because the plug passed and the baby looked well is a classic and dangerous error. [1] [6]
Hirschsprung disease
- Delayed meconium, distension, tight empty rectum
- Absent ganglion cells on biopsy
- Narrowed distal segment with transition zone
- Enterocolitis is the leading cause of death
Meconium plug syndrome
- Transient obstruction from thick meconium
- Resolves with enemas or washouts
- Associated with maternal diabetes
- Still warrants rectal biopsy to exclude Hirschsprung
Functional constipation
- Older child, stool withholding behaviour
- Full rectum on examination, soiling present
- Normal ganglion cells on biopsy
- Responds to laxatives and behavioural therapy
Internal anal sphincter achalasia
- Abnormal relaxation of internal sphincter on manometry
- Normal ganglion cells on biopsy
- Responds to botulinum toxin injection
- A diagnosis of exclusion after Hirschsprung is ruled out
In the older child, the differential is dominated by functional constipation, which is far more common. Functional constipation typically begins after a painful stool, with deliberate withholding behaviour, a palpable faecal mass in a full rectal ampulla, and overflow soiling. Hirschsprung disease in the older child, by contrast, dates from birth, has an empty rectum, lacks soiling, and fails to respond to adequate laxative trials. If the history and examination suggest Hirschsprung disease, a rectal biopsy is indicated regardless of how rare the diagnosis might be in the age group. [1] [11]
Other entities to consider include hypoganglionosis, internal anal sphincter achalasia, and the rare chronic intestinal pseudo-obstruction syndromes. All of these can mimic Hirschsprung disease clinically and may require specialised motility testing and full-thickness biopsy to distinguish. The practical point for the general paediatrician is that any child with chronic constipation from birth that does not fit the functional pattern deserves a suction rectal biopsy. [1] [5]
Clinical & Bedside Assessment
Assessment begins with the pattern recognition that delayed meconium with distension is not normal. In a healthy term infant, 99 per cent pass meconium within 48 hours, so failure to do so is a reliable red flag. The history should establish the exact timing of the first meconium, feeding tolerance, the colour and frequency of any vomitus, and whether there is a family history of Hirschsprung disease, Down syndrome, or any of the associated syndromes. [1] [6]
The examination runs in parallel with resuscitation. Look first at the overall appearance: a lethargic, mottled baby with poor perfusion may already have enterocolitis or perforation. Assess the abdomen for distension, visible peristalsis, tenderness and guarding. A rigid or discoloured abdominal wall suggests perforation or ischaemia and demands immediate surgical review. Inspect the perineum for an anorectal malformation, and check all hernial orifices. [1]
The digital rectal examination is a key discriminator. In Hirschsprung disease the sphincter feels tight, the rectal vault is empty, and withdrawal of the examining finger may trigger an explosive release of gas and stool, the blast sign. In functional constipation, by contrast, the rectum is full of palpable stool. While this finding is useful, it does not replace the biopsy, and you should warn the parents that a rectal examination in a neonate with obstruction can precipitate bacteraemia, so it should be done gently and with aseptic technique. [1]
Record the weight and growth, and check for stigmata of Down syndrome or other syndromic associations: hypotonia, upslanting palpebral fissures, flat facial profile, and the pigmentary, auditory and autonomic features of Waardenburg or Mowat-Wilson syndromes. These associations raise the pre-test probability of Hirschsprung disease and may change the surgical and anaesthetic plan. [12]
Investigations
The suction rectal biopsy is the gold-standard diagnostic test and is the one investigation that definitively confirms or excludes Hirschsprung disease. A suction biopsy device samples the rectal mucosa and submucosa at a point 1 to 3 cm above the dentate line. The sample is examined histologically for ganglion cells in the submucosal plexus: their absence confirms Hirschsprung disease. The biopsy also shows hypertrophied nerve trunks in the aganglionic segment, reflecting the unopposed extrinsic parasympathetic input. Suction biopsy has a sensitivity approaching 100 per cent and a specificity of 100 per cent in experienced centres, with adequate samples obtained in over 95 per cent of neonates. [5] [6]
Modern practice has shifted from acetylcholinesterase staining to calretinin immunohistochemistry. Calretinin is a calcium-binding protein expressed by intrinsic enteric neurons, and its absence in the submucosa of the biopsy sample correlates perfectly with aganglionosis. Calretinin staining is faster, cheaper, more reliable and does not require fresh frozen tissue, making it the preferred method in most modern pathology departments. [5]
A contrast enema is performed as a supportive investigation to delineate the anatomy and help plan surgery. The classical finding is a narrowed distal segment corresponding to the aganglionic bowel, with an abrupt transition to a dilated proximal colon. However, the transition zone seen on contrast enema is only approximately 70 per cent accurate in predicting the actual level of aganglionosis found at operation, and in neonates and breast-fed babies the proximal bowel may not yet be dilated enough to show a clear transition zone. A normal-looking contrast enema therefore does not exclude Hirschsprung disease, and the biopsy always overrides the imaging. [10]
Suction rectal biopsy
Dose
1 to 3 samples
A plain abdominal radiograph may show distended loops of bowel with absent rectal gas, but it is non-specific. Anorectal manometry, which measures the rectoanal inhibitory reflex, can be useful in older children: the reflex is absent in Hirschsprung disease because the internal anal sphincter fails to relax in response to rectal balloon distension. However, manometry is operator-dependent and is mainly used as a screening test in centres where it is available, not as a replacement for biopsy. [5] [1]
Management — Resuscitation
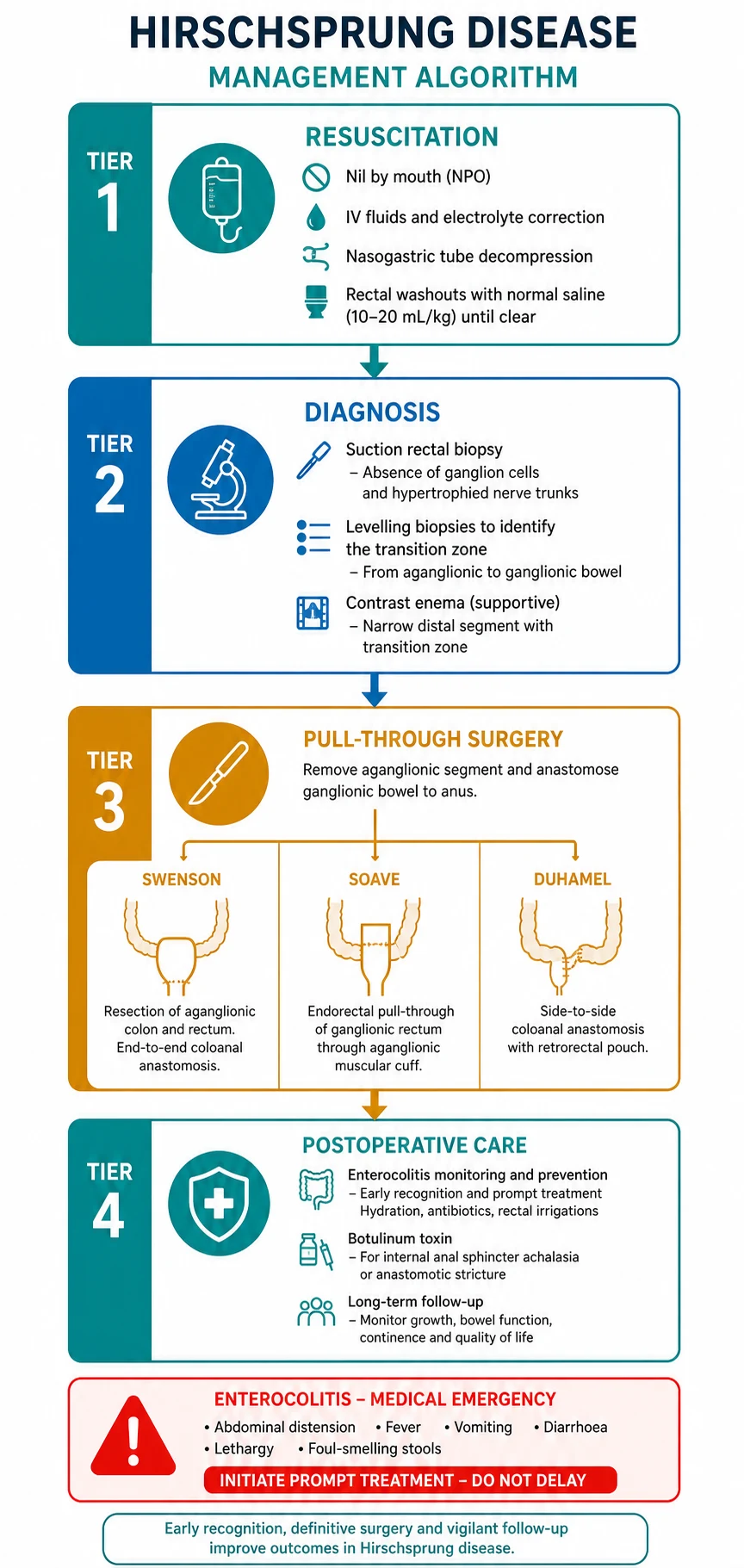
The immediate goal is to decompress the obstructed bowel, correct fluid and electrolyte disturbances, and prevent enterocolitis while awaiting definitive surgery. The child is kept nil by mouth, intravenous access is established, and maintenance fluids with correction of any dehydration or electrolyte derangement are started. A nasogastric tube on free drainage decompresses the stomach and reduces the risk of aspiration. [1]
Regular rectal washouts with warm normal saline are the cornerstone of preoperative decompression. A large-bore catheter is passed into the rectum and warm saline is gently irrigated to wash out the stool and gas pooled behind the obstruction. These washouts are typically performed daily or twice daily until definitive surgery, and they significantly reduce the risk of preoperative enterocolitis by relieving the stasis that drives bacterial overgrowth. [1] [2]
If the child presents with enterocolitis rather than simple obstruction, the resuscitation is more aggressive. Give boluses of isotonic intravenous fluid for shock, start broad-spectrum antibiotics (for example metronidazole plus an aminoglycoside such as gentamicin to target enteric and anaerobic organisms), and perform frequent rectal irrigations to decompress the bowel. A child with severe enterocolitis, septic shock, suspected perforation or toxic megacolon requires urgent paediatric intensive care and may need a diverting stoma as a temporising measure before definitive pull-through. [2] [3]
Enterocolitis treatment bundle
Dose
As per local protocol
Management — Definitive & Stepwise
The principle of definitive surgery is simple: remove or bypass the aganglionic bowel and bring ganglionic bowel down to the anal canal. The aganglionic segment is resected, and the proximal ganglionic colon is pulled through the pelvis and anastomosed to the anal canal just above the dentate line. The exact technique used is less important than ensuring that the anastomosis is made in confirmed ganglionic bowel, verified by frozen-section levelling biopsies taken at operation. [1] [9]
The three classical pull-through operations, all named after their originators, achieve this goal by different routes. The Swenson procedure resects the aganglionic bowel completely and performs an end-to-end anastomosis just above the dentate line. The Soave procedure leaves the outer muscular wall of the aganglionic rectum in place as a sleeve and pulls the ganglionic colon through it, performing the anastomosis at the anal verge. The Duhamel procedure preserves the aganglionic rectum and creates a side-to-side anastomosis between the ganglionic colon pulled down behind it and the native rectum, forming a neorectum. [9] [8]
No single technique has shown clear superiority in long-term outcomes, and meta-analyses comparing Swenson, Soave and Duhamel report broadly equivalent rates of continence, constipation and enterocolitis. Surgeon experience and institutional preference drive the choice more than evidence of one technique's dominance. What matters is that the operation is performed by a surgeon experienced in paediatric colorectal surgery, in a centre with the pathology support to do reliable frozen sections. [9] [8]
The trend over the past two decades has been toward primary pull-through in the neonatal period for uncomplicated short-segment disease, rather than the traditional two-stage approach of initial stoma followed by pull-through months later. Meta-analysis supports the safety and efficacy of primary neonatal Soave pull-through, with shorter hospital stay, no increase in complications, and avoidance of a stoma and a second operation. A staged approach is still used for children with enterocolitis at presentation, long-segment or total colonic disease, or when the surgeon cannot confidently confirm the level of aganglionosis at operation. [7] [8]
The three pull-through operations
Laparoscopic-assisted and transanal approaches have largely replaced open laparotomy for short-segment disease. The transanal Soave pull-through, performed entirely through the perineum without an abdominal incision, avoids the morbidity of laparotomy and can be done in the neonatal period. Laparoscopic-assisted techniques combine abdominal seromuscular biopsies for levelling with a transanal pull-through, and are used when the level of aganglionosis needs to be confirmed from above. Systematic reviews show equivalent or improved outcomes compared with open surgery. [8] [7]
Specific Subtypes & Scenarios
The classic presentation is a term male neonate with delayed meconium and distension. The diagnosis is confirmed by suction rectal biopsy, the child is decompressed with rectal washouts for a few days, and a primary transanal Soave pull-through is performed in the neonatal period or early infancy. This is the scenario most candidates will encounter in exams and in practice. [7] [1]
Total colonic aganglionosis is the severe end of the spectrum, affecting roughly 5 to 10 per cent of patients. The entire colon lacks ganglion cells, and the presentation is earlier and more severe, with bilious vomiting, failure to pass meconium, and a microcolon on contrast enema. Management is more complex, typically requiring an initial ileostomy followed by an ileal pull-through, and long-term outcomes involve higher rates of enterocolitis, frequent stools and perianal excoriation. These children need lifelong specialist follow-up. [1] [8]
The older child or adolescent with previously undiagnosed Hirschsprung disease presents a different challenge. Chronic constipation from birth, an empty rectum, failure to thrive and no response to laxatives should prompt a rectal biopsy regardless of the child's age. The surgical approach is the same, but these children may need preoperative bowel preparation and decompression over a longer period. Postoperatively they are at higher risk of persistent bowel dysfunction because the chronically dilated proximal bowel may not recover normal motility. [11] [1]
Postoperative obstructive symptoms affect a significant minority of children after pull-through. Persistent distension, recurrent constipation, or episodes suggestive of enterocolitis may be due to a retained aganglionic muscular cuff (after Soave), an anastomotic stricture, or internal anal sphincter achalasia. Botulinum toxin injection of the internal anal sphincter is an effective first-line intervention for obstructive symptoms, relieving the functional outlet obstruction by temporarily paralysing the sphincter. The effect lasts several months and can be repeated. [1] [9]
Complications & Pitfalls
Hirschsprung-associated enterocolitis is the single most feared complication and the leading cause of death. It occurs in 20 to 40 per cent of patients before surgery and in up to 25 per cent after pull-through. The risk persists for years, so parents must be taught to recognise the signs and perform emergency rectal washouts at home. Historical mortality was as high as 30 per cent, but early recognition and aggressive treatment have reduced this substantially. The Gosain 2017 consensus guidelines codified the diagnostic criteria and treatment protocol for enterocolitis, and every clinician caring for a child with Hirschsprung disease should be familiar with them. [2] [3]
Postoperative mechanical complications include anastomotic leak and stricture, which may present with distension, vomiting or signs of peritonitis in the early postoperative period. A retained aganglionic segment, if the levelling biopsy was inaccurate or the transition zone was misidentified, leads to persistent obstruction. This requires reoperation with repeat levelling biopsies and revision of the pull-through, a scenario every candidate should mention in a long-case discussion. [9] [1]
Persistent obstructive symptoms after an anatomically correct pull-through are common. The internal anal sphincter may fail to relax normally, producing a functional outlet obstruction. Botulinum toxin injection of the internal anal sphincter, typically performed under general anaesthesia, is the first-line treatment. The dose varies by centre and the child's size; the effect lasts approximately three to six months and can be repeated. For children who do not respond, options include repeat injections, myectomy of the internal sphincter, or reassessment for a mechanical cause. [9]
Other long-term complications include faecal incontinence and soiling, which affect a significant minority of children and persist into adulthood. The mechanism is multifactorial, involving loss of the neorectal reservoir, impaired sensation, and disturbance of the internal anal sphincter mechanism. Chronic constipation also persists in some children, requiring ongoing laxative use and bowel management programmes. These problems underscore the need for long-term follow-up in a dedicated transition clinic. [11] [9]
Prognosis & Disposition
With early diagnosis and skilled surgery, most children with Hirschsprung disease do well. They grow normally, attend school and participate in ordinary activities. However, the picture is not universally positive, and the gap between the optimistic surgical view and the lived experience of patients is one of the most important things a candidate should understand. [11]
Long-term quality-of-life studies paint a more nuanced picture. Adults who had Hirschsprung disease in childhood report lower social functioning and persistent bowel symptoms compared with their peers. Faecal soiling, urinary incontinence and sexual dysfunction are reported by a significant minority. These problems are more common after long-segment disease, total colonic aganglionosis, and reoperation. The message for the clinician is that surgery is not the end of the story, and ongoing bowel management and psychosocial support are essential. [11]
Disposition depends on the clinical stage. A neonate presenting with uncomplicated short-segment disease is admitted for decompression, biopsy and primary pull-through, typically staying in hospital for one to two weeks. A child with enterocolitis requires intensive care and may need a staged surgical approach with a longer hospital stay. After discharge, all children need regular outpatient follow-up, initially every few months and then annually, with a clear plan for transition to adult care in adolescence. [1] [11]
The family must be given a realistic picture. Most children do well, but a significant minority have persistent problems. Parents should be taught to recognise enterocolitis, perform rectal washouts if instructed, and know when to seek urgent medical review. Genetic counselling is offered to the family, with information about recurrence risk and the availability of genetic testing for RET and other genes. [12] [11]
Special Populations
The child with trisomy 21 and Hirschsprung disease deserves particular attention. The association is strong, with roughly 10 per cent of all Hirschsprung patients having Down syndrome. These children have a higher rate of postoperative enterocolitis and may have additional cardiac and gastrointestinal anomalies, such as duodenal atresia or atrioventricular septal defect, that complicate anaesthesia and perioperative care. A low threshold for rectal biopsy in any child with trisomy 21 and constipation is essential. [12] [3]
Syndromic Hirschsprung disease, associated with conditions such as Waardenburg syndrome (pigmentary abnormalities and deafness), Mowat-Wilson syndrome (distinctive facies, intellectual disability and Hirschsprung disease), and congenital central hypoventilation (Ondine curse, requiring ventilatory support), demands a multidisciplinary approach. The neural crest migration defect that causes the aganglionosis also affects melanocytes, auditory neurons and autonomic neurons, so these children need coordinated input from genetics, audiology, neurology and respiratory medicine alongside the surgical team. [12]
Rural and remote families face particular challenges. A neonate born in a regional centre with delayed meconium and distension needs stabilisation and decompression before retrieval to a paediatric surgical centre. Rectal washouts can be started locally after telephone advice from the receiving surgical team, and the child is transferred with a nasogastric tube on free drainage. The family may need to stay near the surgical centre for weeks, with implications for accommodation, work and the care of siblings. [1]
Adolescents with Hirschsprung disease transitioning to adult care are a growing population. These young people may have persistent bowel symptoms, psychosocial issues related to their surgical history, and questions about fertility and genetic risk to their own children. A structured transition clinic, with paediatric and adult gastroenterology and colorectal surgery input, is the ideal model. The goal is to ensure continuity of care, address emerging issues, and empower the young person to manage their own bowel health. [11]
Evidence, Guidelines & Regional Differences
The ERNICA 2020 European guidelines for rectosigmoid Hirschsprung disease (Kyrklund et al) are the most recent comprehensive consensus document and form the backbone of current European and Australasian practice. They recommend suction rectal biopsy for diagnosis, primary transanal pull-through for uncomplicated short-segment disease, and a staged approach for complex cases. They also address enterocolitis prevention through regular rectal washouts and parent education. [1]
The Gosain 2017 consensus guidelines for Hirschsprung-associated enterocolitis codified the diagnostic criteria and treatment protocol for this life-threatening complication. Diagnosis requires a combination of clinical features (fever, distension, diarrhoea) and may be supported by radiographic findings. Treatment involves intravenous fluids, broad-spectrum antibiotics, decompression and rectal irrigations. These guidelines are widely adopted and are the standard reference for enterocolitis management. [2] [4]
The evidence on the optimal pull-through technique remains controversial. Meta-analyses comparing Swenson, Soave and Duhamel show broadly equivalent long-term outcomes, with no single technique demonstrating clear superiority in continence, constipation rates or enterocolitis incidence. Surgeon experience and institutional preference are the main determinants of technique choice. What matters clinically is that the operation is done in a high-volume centre by an experienced paediatric colorectal surgeon. [9] [8]
The trend toward primary neonatal pull-through is supported by meta-analysis evidence showing safety, shorter hospital stay and no increase in complications compared with staged surgery. However, some European and Asian centres still favour a staged approach with initial stoma for selected cases, particularly when the diagnosis is made late or there is uncertainty about the level of aganglionosis. This regional variation is worth knowing for exam answers, as it reflects legitimate differences in surgical philosophy rather than a right or wrong answer. [7] [8]
Exam Pearls
The high-yield facts every candidate must hold: a term neonate who has not passed meconium within 48 hours has Hirschsprung disease until proven otherwise. The gold-standard test is a suction rectal biopsy showing absent ganglion cells, with calretinin immunohistochemistry as the preferred stain. The RET proto-oncogene on chromosome 10 is the major susceptibility gene, and trisomy 21 is the most common chromosomal association, found in roughly 10 per cent of patients. [1] [12]
The three pull-through operations are Swenson, Soave and Duhamel, all aiming to bring ganglionic bowel to the anal canal. No single technique is clearly superior. Primary neonatal pull-through is the modern standard for uncomplicated short-segment disease, avoiding a stoma and a second operation. The levelling biopsy at operation confirms the presence of ganglion cells at the anastomotic level. [9] [7]
Hirschsprung-associated enterocolitis is the leading cause of death. It presents with fever, explosive foul-smelling diarrhoea and abdominal distension, and can occur before or after surgery. Treatment is aggressive: intravenous fluids, broad-spectrum antibiotics including metronidazole, nasogastric decompression and frequent rectal washouts. Parents must be taught the warning signs. [2] [3]
References
- [1]Kyrklund K; Sloots CEJ; de Blaauw I; et al ERNICA guidelines for the management of rectosigmoid Hirschsprung's disease. Orphanet J Rare Dis, 2020.PMID 32586397
- [2]Gosain A; Frykman PK; Cowles RA; et al Guidelines for the diagnosis and management of Hirschsprung-associated enterocolitis. Pediatr Surg Int, 2017.PMID 28154902
- [3]Ziogas IA; Mylonas KS; Tsoucala S; et al Hirschsprung-associated enterocolitis: a comprehensive review. World J Pediatr Surg, 2024.PMID 39410939
- [4]Lewit RA; Svetanoff WJ; Lopez JJ; et al Current understanding of Hirschsprung-associated enterocolitis: Pathogenesis, diagnosis and treatment. Semin Pediatr Surg, 2022.PMID 35690459
- [5]Green N; Cromie W; Griffiths DM; et al Rectal suction biopsy versus incisional rectal biopsy in the diagnosis of Hirschsprung disease. Pediatr Surg Int, 2022.PMID 36171348
- [6]Allen AR; Azmy SE; Munro FD; et al Accuracy of Suction Rectal Biopsy for Diagnosis of Hirschsprung's Disease in Neonates. Eur J Pediatr Surg, 2019.PMID 30068006
- [7]Westfal ML; Hakim J; Chougule A; et al Optimal timing for Soave primary pull-through in short-segment Hirschsprung disease: A meta-analysis. J Pediatr Surg, 2022.PMID 34330420
- [8]Tomuschat C; Zimmer J; Puri P Laparoscopic-assisted pull-through operation for Hirschsprung's disease: a systematic review and meta-analysis. Pediatr Surg Int, 2016.PMID 27369964
- [9]Davidson JR; Quan A; Batey M; et al Comparative cohort study of Duhamel and endorectal pull-through for Hirschsprung's disease. BJS Open, 2022.PMID 35143630
- [10]Haikal Z; Soriano MM; Otero N; et al Accuracy of transition zone in contrast enema to predict intraoperative aganglionosis level in patients with Hirschsprung disease. BMC Res Notes, 2020.PMID 32098631
- [11]Drissi F; Faure C; Cargill G; et al Long-term Outcome of Hirschsprung Disease: Impact on Quality of Life and Social Condition at Adult Age. Dis Colon Rectum, 2019.PMID 30807458
- [12]Amiel J; Sproat-Emison E; Garcia-Barcelo M; et al Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet, 2008.PMID 17965226