Paeds · gastroenterology-hepatology-and-nutrition
Liver transplantation in children
Also known as Paediatric liver transplant · Pediatric liver transplantation · Living donor liver transplantation in children · PELD score and transplant listing · Tacrolimus immunosuppression in children
Fellowship guide to liver transplantation in children: the indications with biliary atresia dominant, graft types including living-donor and split grafts, the Paediatric End-stage Liver Disease score for deceased-donor allocation, tacrolimus-anchored maintenance immunosuppression, acute cellular rejection diagnosed on biopsy and treated with high-dose corticosteroid, and the long-term complications of hepatic artery thrombosis, post-transplant lymphoproliferative disorder and non-adherence.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A child who reaches the point of liver transplantation has travelled one of three roads: chronic decompensated liver disease, most often biliary atresia that has exhausted the Kasai operation; an acute catastrophe such as fulminant hepatic failure; or a metabolic disease in which the native liver, though structurally intact, is biochemically dangerous. The task of the paediatric team is to recognise the destination early, to prioritise the child correctly on the waiting list, and to support a graft and a growing child through decades of immunosuppression. Few areas of paediatrics demand a longer view, and few reward early, structured referral more handsomely. [1]
Liver transplantation is the replacement of a failing native liver with a vascularised graft that restores synthetic, metabolic and excretory function. In children this is rarely a straightforward whole-organ deceased-donor transplant, because the small size of the recipient and the scarcity of size-matched paediatric deceased donors mean that most children receive a segmental graft, whether a reduced-size, split, or living-donor graft. The single most common indication is biliary atresia, which accounts for up to half of all paediatric transplants, followed by metabolic and genetic disease and then acute liver failure. [10]
The discipline rests on three pillars that the candidate must hold together. The first is allocation: the Paediatric End-stage Liver Disease score ranks children on the deceased-donor waiting list and is the numerical backbone of who is transplanted and when. The second is immunosuppression: a tacrolimus-anchored regimen walks the narrow line between rejection and the infections, malignancy and organ toxicity of over-treatment. The third is the long arc of follow-up, in which hepatic artery thrombosis, post-transplant lymphoproliferative disorder, and adolescent non-adherence decide whether a technically perfect transplant succeeds over a lifetime. [8]
Classification
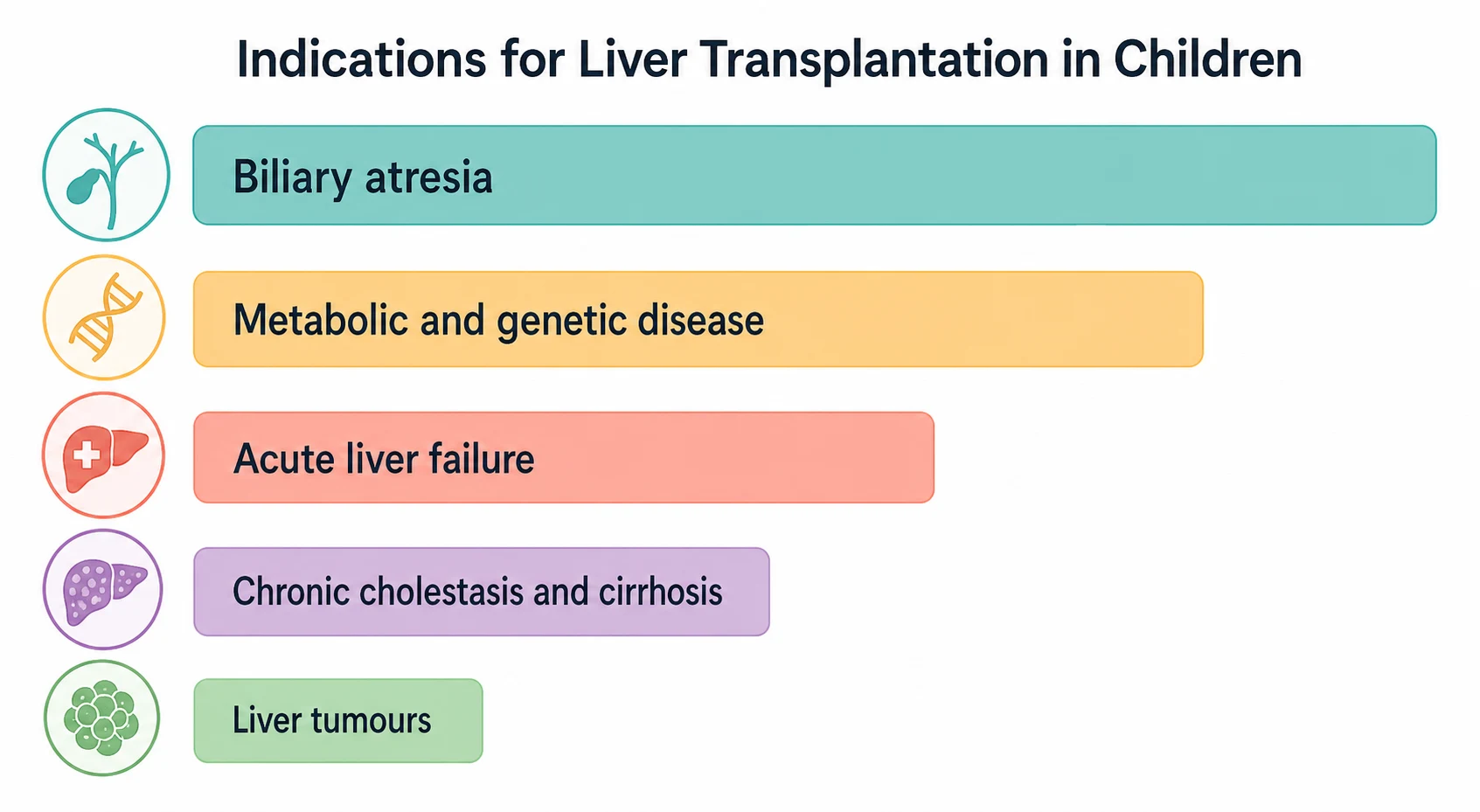
Liver transplantation in children is classified in two ways that the examiner expects the candidate to separate cleanly: by the type of graft used, and by the indication that brought the child to transplant. The graft type matters because it dictates the surgical anatomy, the risk profile, and the ethical calculus, while the indication matters because it predicts the tempo of referral, the urgency of listing, and the long-term behaviour of the graft. [1]
[1]The living-donor and split grafts deserve particular attention because they were developed to solve the central paediatric problem: the smallest infants have the highest waiting-list mortality because size-matched deceased donors are vanishingly rare. A left lateral segment from a living donor, typically a parent, fits an infant and dramatically shortens waiting time, while a split deceased graft can serve a child and an adult from a single organ. The trade-off is donor morbidity in the living-donor case and a higher technical complication rate in the segmental graft, particularly biliary and vascular complications. [2]
The indications divide into five categories. Cholestatic disease leads, overwhelmingly biliary atresia, followed by the progressive familial intrahepatic cholestasis syndromes and Alagille syndrome. Metabolic and genetic disease follows, including alpha-1-antitrypsin deficiency, Wilson disease, tyrosinaemia, glycogen storage disease, Crigler-Najjar syndrome, urea cycle defects and organic acidaemias. Acute liver failure is the time-critical category. Chronic cholestasis and cirrhosis encompasses autoimmune hepatitis, cystic fibrosis liver disease, and sclerosing cholangitis. Liver tumours, principally unresectable hepatoblastoma, form a small but distinct category often requiring transplant under a standardised exception. [10]
Epidemiology & Risk Factors
Biliary atresia is the single disease that defines paediatric liver transplantation. It occurs in roughly one in eight thousand to one in eighteen thousand live births worldwide, and although the Kasai portoenterostomy performed in the first weeks of life can restore bile flow in many infants, a substantial proportion progress to cirrhosis and require transplantation, making biliary atresia responsible for up to half of all paediatric liver transplants. A contemporary United States analysis confirmed it remains the dominant indication, with transplant-free survival after Kasai heavily influenced by the timing and success of the initial operation. [10]
The age distribution is bimodal. A large infant peak reflects biliary atresia and neonatal cholestatic disease, while a second peak in older children and adolescents reflects metabolic disease, autoimmune hepatitis, Wilson disease, and acute liver failure. Waiting-list mortality is highest in the smallest infants, those weighing under five to six kilograms at the time of listing, precisely because size-matched deceased donors are scarce; this is the gap that living-donor and split grafts were designed to close. [2]
The risk factors for a poor post-transplant outcome are important because they shape the conversation with the family and the intensity of follow-up. Low recipient weight, young age under one year, previous upper abdominal surgery including a Kasai, severe portal hypertension, renal dysfunction, and graft-to-recipient weight ratio mismatch all raise the risk of vascular complications, bleeding, and graft loss. Beyond the perioperative window, Epstein-Barr virus seronegativity at transplant marks the child at highest risk of post-transplant lymphoproliferative disorder, and adolescence marks the developmental window of highest non-adherence. [11]
Pathophysiology
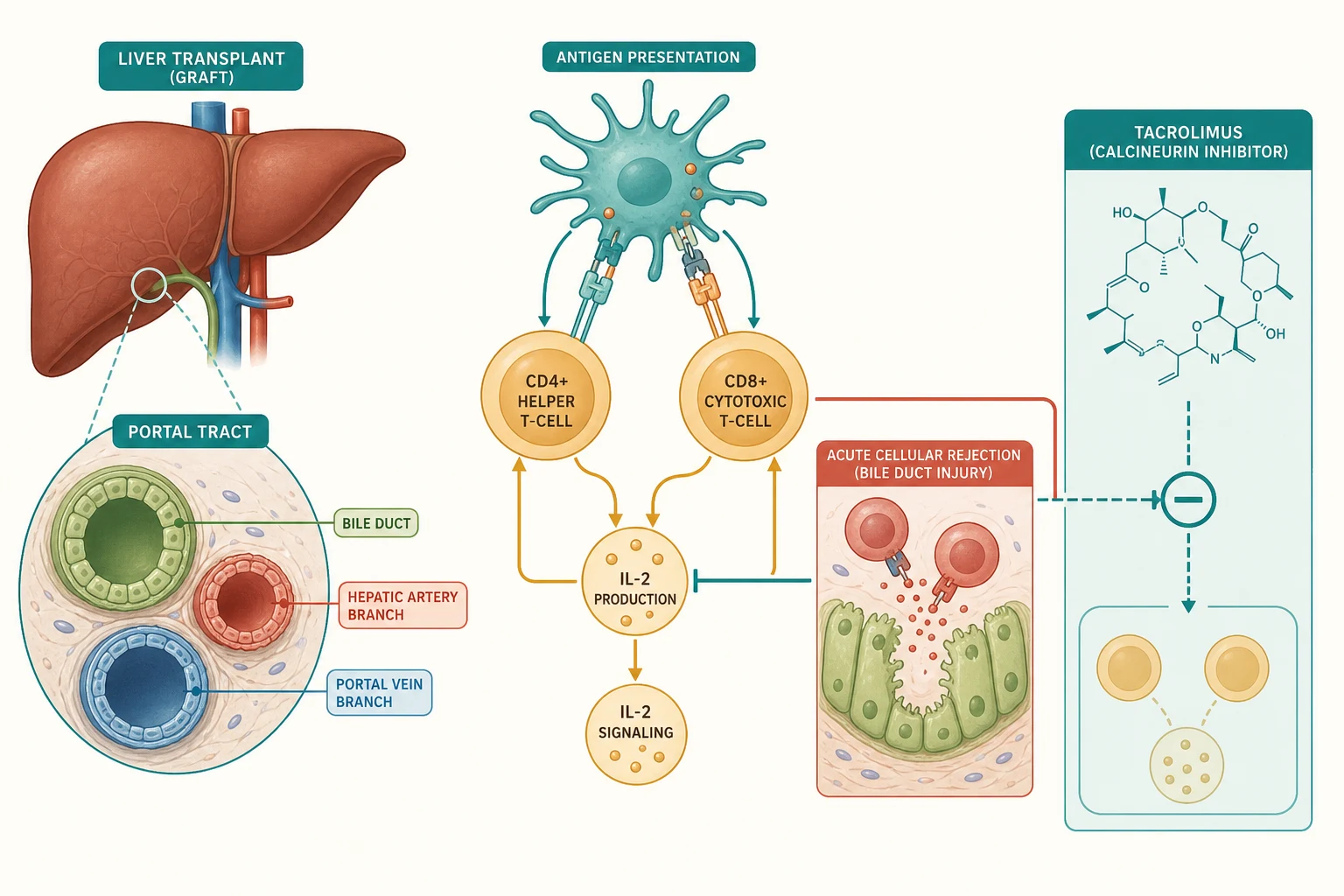
A successful transplant depends on four surgical anastomoses that the candidate should be able to name: the hepatic venous outflow into the inferior vena cava, the portal venous inflow, the hepatic arterial inflow, and the biliary drainage, either as a duct-to-duct anastomosis or a hepaticojejunostomy (a Roux loop). The vulnerability of the graft is written into this anatomy, because the biliary tree is supplied exclusively by the hepatic artery, so any compromise of hepatic arterial flow threatens the bile ducts first and most severely. This single fact explains why hepatic artery thrombosis is the most feared vascular complication. [2]
The biology the trainee must own is the alloimmune response that drives rejection. Donor major histocompatibility complex antigens are picked up by recipient antigen-presenting cells and shown to recipient CD4 helper and CD8 cytotoxic T-cells. The CD8 cells then attack the bile duct epithelium and the vascular endothelium of the graft, which is why the histological signature of acute cellular rejection is a triad of portal inflammation, bile duct damage, and venous endothelialitis. This is a T-cell-mediated process, and it is the direct target of the calcineurin inhibitors. [8]
Tacrolimus works by binding its intracellular receptor, FK-binding protein 12, and the resulting complex inhibits calcineurin, the phosphatase that would otherwise activate nuclear factor of activated T-cells and drive transcription of interleukin-2. Without interleukin-2, T-cell proliferation stalls, and the rejection cascade is held in check. The therapeutic window is narrow: too little and the graft rejects, too much and the child develops nephrotoxicity, neurotoxicity, hypertension, diabetes, and a susceptibility to the opportunistic infections and Epstein-Barr-virus-driven B-cell proliferation that culminate in post-transplant lymphoproliferative disorder. This is why therapeutic drug monitoring of tacrolimus trough concentrations is central to management. [6]
The same suppressed immunity that protects the graft also licences the Epstein-Barr virus. In a seronegative recipient given a seropositive graft or transfusion, primary Epstein-Barr virus infection under the cover of immunosuppression drives B-cell proliferation, which ranges from a reactive polyclonal expansion to a frank monoclonal lymphoma. This is the mechanism behind post-transplant lymphoproliferative disorder, and it is why Epstein-Barr viral load surveillance is so important in the seronegative child. [11]
Clinical Presentation
The pretransplant child presents in one of three ways, and recognising each is the trigger for transplant referral. The chronic presentation is the biliary atresia infant who has had a Kasai but now shows one or more of the accepted indications: synthetic failure with a falling albumin and rising international normalised ratio, intractable pruritus that destroys sleep, growth failure despite nutritional support, recurrent cholangitis, or portal hypertension that is difficult to control. [1]
The acute presentation is the child in acute liver failure who is not recovering. Here the trajectory matters more than any single value: a rising international normalised ratio, deepening encephalopathy, a falling arterial pH, a rising lactate, and emerging multiorgan failure mark the child who needs urgent listing. In regions where deceased donation is scarce, the family is evaluated for live donor transplantation in parallel, because the window between listing and a suitable deceased organ is often too long for a child in fulminant failure. [2]
The metabolic presentation is subtle. The native liver may look structurally intact, but it is biochemically dangerous. A child with Crigler-Najjar syndrome risks kernicterus, a child with a urea cycle defect risks hyperammonaemic crises, and a child with an organic acidaemia risks metabolic decompensation. These children may be candidates for an auxiliary graft, in which part of the native liver is retained to preserve native metabolic capacity and to leave the door open to future gene-directed therapy. [1]
After transplant, the presenting problems change, and the candidate must know the temporal pattern. In the first days, a graft that produces no bile and fails to clear lactate is primary non-function, an indication for urgent retransplantation. In the first weeks, fever with raised liver enzymes is acute cellular rejection until proven otherwise, and a sudden transaminase rise must prompt Doppler ultrasound to exclude hepatic artery thrombosis. After the first month, jaundice and cholangitis point to a biliary complication or stricture. Beyond the first year, the threats are recurrent native disease, chronic rejection, and the insidious appearance of post-transplant lymphoproliferative disorder, while in adolescence non-adherence silently undoes years of careful immunosuppression. [8]
Differential Diagnosis
The post-transplant child with a rising transaminase or new jaundice generates a fixed differential that the candidate should be able to recite in order of frequency and gravity. Acute cellular rejection is the most common cause in the first month. Vascular thrombosis, particularly hepatic artery thrombosis, is graft-threatening and must never be missed. Biliary complications, including leak and stricture, present with jaundice, cholangitis, or a bilious collection. Infection spans bacterial cholangitis, cytomegalovirus, and Epstein-Barr virus. Recurrent native disease can affect autoimmune hepatitis, primary sclerosing cholangitis, and some metabolic conditions. Tacrolimus toxicity and, later, post-transplant lymphoproliferative disorder complete the list. [8]
[11]The temporal context is the single most powerful discriminator, and a strong candidate uses it as the first question. In the first days suspect primary non-function or vascular thrombosis. In the first month suspect acute cellular rejection. After the first month suspect biliary complications, infection, and recurrence. Beyond a year suspect recurrence, post-transplant lymphoproliferative disorder, and chronic rejection. Because hepatic artery thrombosis is the one cause that cannot wait for a biopsy, the first investigation in any early post-transplant deterioration is a Doppler ultrasound of the hepatic artery. [2]
Clinical & Bedside Assessment
Pretransplant assessment has three jobs: to document the severity of the liver disease, to map the vascular and biliary anatomy, and to exclude contraindications. The clinician plots growth on WHO or disease-specific charts, because growth failure is both a marker of severity and, uniquely in children, a component of the allocation score. The abdomen is examined for splenomegaly, ascites, and the stigmata of chronic liver disease, and the chest is examined for the clubbing and cyanosis that suggest hepatopulmonary syndrome. The metabolic diseases demand a search for their extrahepatic features, because these influence anaesthetic and surgical planning. [1]
The active search for contraindications is mandatory because some are absolute and change the plan entirely. Active uncontrolled infection, uncontrolled extrahepatic malignancy, severe irreversible cardiopulmonary disease, and irreversible brain injury are contraindications. Portopulmonary hypertension is a specific and dangerous contraindication that must be excluded by echocardiography, because transplanting a child with uncontrolled portopulmonary hypertension carries a high perioperative mortality. Severe hepatopulmonary syndrome, by contrast, may actually be cured by transplantation and is therefore an indication rather than a barrier. [2]
Post-transplant bedside assessment reads the graft and the immunosuppression at the same time. The candidate looks for graft function in the jaundice, encephalopathy, and synthetic markers; for rejection in the fever and right upper quadrant tenderness; for infection in the fever, line sites, and chest; and for surgical complications in the wound and the abdominal distension of a bile leak or bleed. Tacrolimus toxicity shows itself as a tremor, hypertension, headache, and a rising creatinine. Growth is tracked at every visit, because catch-up growth is one of the most reliable markers that a graft is working and the immunosuppression is not over-suppressing the child. [8]
Investigations
The transplant workup confirms the diagnosis, gauges severity, defines the anatomy, and screens for contraindications, and it runs in parallel with nutritional optimisation and control of portal hypertension. Liver biochemistry and synthetic function (albumin, international normalised ratio), full blood count, and renal function define severity. Abdominal ultrasound with Doppler maps the portal vein, hepatic artery, hepatic veins, and inferior vena cava and assesses parenchyma and splenomegaly. Cross-sectional imaging with computed tomography or magnetic resonance defines the vascular and biliary anatomy in detail before surgery. Echocardiography excludes cardiac disease and screens for portopulmonary hypertension and hepatopulmonary syndrome. [2]
The infectious screen is decisive for post-transplant management. Cytomegalovirus and Epstein-Barr virus serology of both donor and recipient determines the risk stratification and the prophylaxis and surveillance plan. An Epstein-Barr-virus-seronegative recipient is the highest-risk group for post-transplant lymphoproliferative disorder and enters an active viral-load surveillance programme. The standard paediatric immunisations should be completed before transplant wherever possible, because live vaccines are generally avoided once the child is on immunosuppression. [11]
The Paediatric End-stage Liver Disease score is the numerical backbone of paediatric deceased-donor allocation and the calculation every candidate should be able to describe. Introduced in the United States in 2002 to replace a less granular status-based system, it is a continuous score derived from five variables chosen because they predict three-month waiting-list mortality in children. [3]
The accuracy of the PELD score in predicting pretransplant mortality has been validated, and the deliberate weighting of growth failure reflects the paediatric insight that malnutrition is a uniquely informative marker of severity in children that is absent from adult scores. The growth-failure thresholds specifically were shown to influence mortality among candidates, confirming the rationale for their inclusion. [4]
After transplant, a rising transaminase triggers a fixed sequence. The first investigation is a Doppler ultrasound to confirm hepatic artery patency, because hepatic artery thrombosis cannot wait for a biopsy. Once vascular patency is assured, the gold standard for acute cellular rejection is liver biopsy, read against the Rejection Activity Index, which grades portal inflammation, bile duct damage, and venous endothelialitis. Epstein-Barr virus polymerase chain reaction quantification is the surveillance test for post-transplant lymphoproliferative disorder in seronegative recipients. [8]
Management — Resuscitation
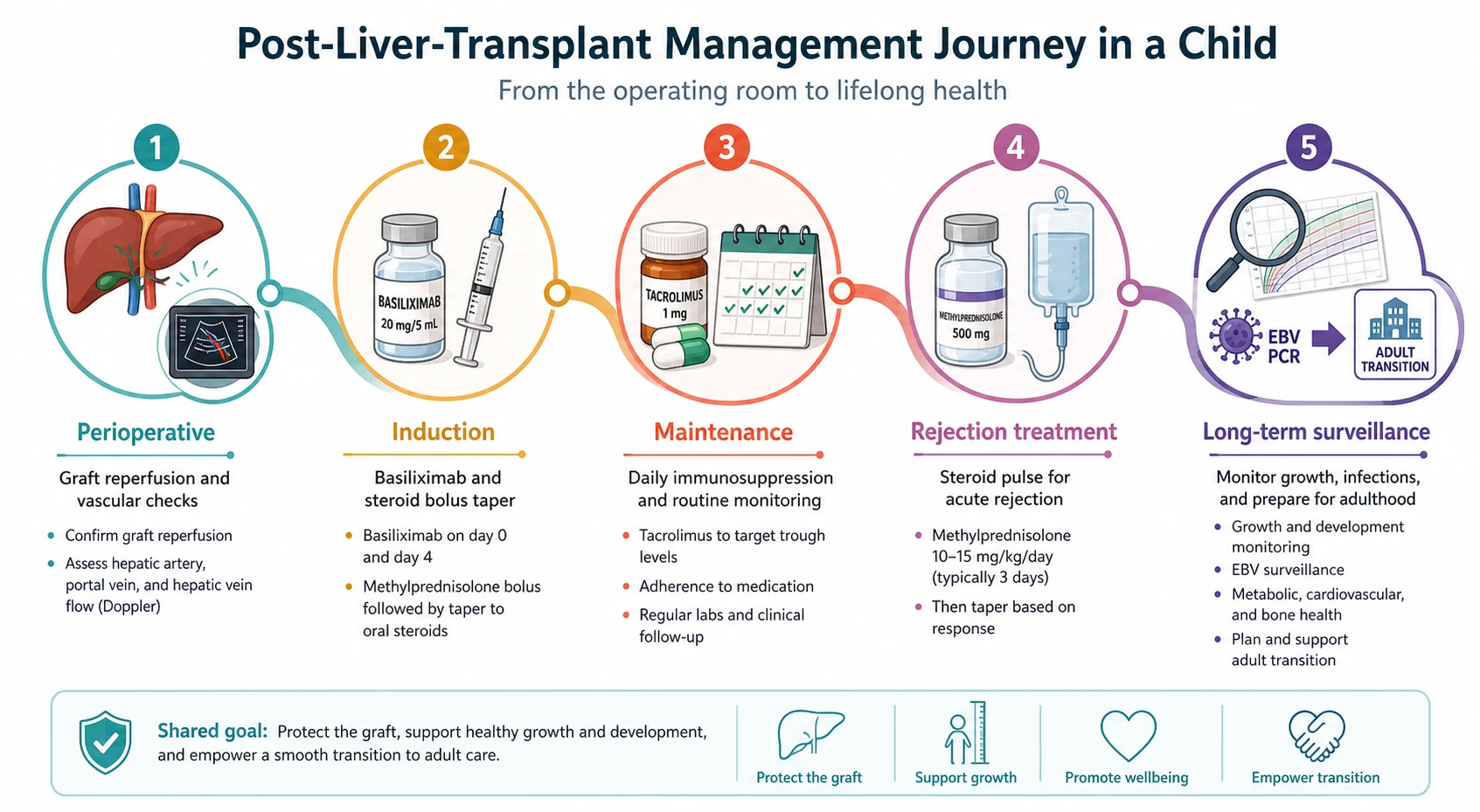
Immediate post-transplant care is surgical and intensive care, and it begins in the operating theatre. The graft is reperfused and its function is judged in real time: bile production, haemodynamic stability, and the clearance of lactate. The four anastomoses are confirmed patent by intraoperative and early Doppler ultrasound. Haemostasis is secured and the abdomen is closed, and the child is observed in paediatric intensive care for graft function, bleeding, and vascular patency over the first days. [1]
Primary non-function is the catastrophe of the first hours: a graft that is reperfused but never works, producing no bile and failing to clear lactate, with a rising international normalised ratio. It is an indication for urgent retransplantation, and recognising it early is essential because the child deteriorates rapidly. The intensive care priorities in parallel are haemodynamic stability, the avoidance of hepatic artery spasm and thrombosis by maintaining adequate perfusion and often antiplatelet aspirin, infection surveillance, and renal protection, because calcineurin-inhibitor nephrotoxicity compounds the haemodynamic and septic insults of the perioperative period. [2]
Induction immunosuppression is begun in theatre or immediately postoperatively. The traditional regimen is a corticosteroid taper with an interleukin-2 receptor antagonist, and contemporary practice has moved increasingly toward steroid-sparing and steroid-free protocols. A steroid-free tacrolimus-basiliximab regimen, in which basiliximab is given on day zero and day four alongside tacrolimus, has been shown to be feasible and to reduce the metabolic and growth penalties of corticosteroids while maintaining low rejection rates, with predictors for steroid requirement identified to guide selection. [7]
Management — Definitive & Stepwise
Maintenance immunosuppression is the keystone of long-term graft survival, and it is anchored on a calcineurin inhibitor, almost always tacrolimus. The dose is individualised by therapeutic drug monitoring of the whole-blood trough concentration, with a typical target around 8 to 12 nanograms per millilitre in the early months, weaning toward 4 to 6 nanograms per millilitre long term as the graft stabilises. Most regimens pair tacrolimus with a steroid taper and frequently add mycophenolate mofetil, with azathioprine as a historical alternative. The contemporary trend is toward minimisation: the lowest effective tacrolimus dose that prevents rejection, to limit nephrotoxicity, neurotoxicity, hypertension, diabetes, and the long-term infective and malignant consequences of over-immunosuppression. [6]
Tacrolimus
Dose
Weight-based, dosed to whole-blood trough
Acute cellular rejection is treated with a high-dose corticosteroid pulse. A typical regimen is intravenous methylprednisolone at 10 milligrams per kilogram per day, in divided doses, for three days, followed by an oral steroid taper. The majority of episodes respond to a single pulse. Steroid-resistant rejection, failure to improve on biopsy and biochemistry after the pulse, escalates to a lymphocyte-depleting agent such as anti-thymocyte globulin. Biopsy confirmation before treatment is the standard, both to confirm rejection and to grade its severity on the Rejection Activity Index, and to exclude the mimics that would be harmed by more immunosuppression. [8]
Methylprednisolone
Dose
10 mg/kg/day in divided doses for 3 days
Over the longer term, management is a deliberate, slow wean toward the lowest effective immunosuppression, integrated with infection prophylaxis and surveillance, growth and nutrition, vaccination, adherence support, and transition. Cotrimoxazole prophylaxis protects against Pneumocystis jirovecii. Antiviral prophylaxis and preemptive Epstein-Barr viral load surveillance manage cytomegalovirus and Epstein-Barr virus. Inactivated vaccines are safe and should be completed before transplant wherever possible; live vaccines are generally avoided on immunosuppression. [11]
A selected minority of stable children may achieve operational tolerance, defined as stable graft function without immunosuppression, under specialist protocolised withdrawal. This is more achievable in paediatric liver recipients than in kidney or heart recipients, but a 10-year prospective study has shown that operational tolerance is not always permanent, with late breakthrough rejection described, so withdrawal remains a specialist decision confined to research protocols and experienced centres rather than routine practice. [9]
Specific Subtypes & Scenarios
Biliary atresia is the paradigmatic indication and the scenario most likely to appear in an examination. The child has had a Kasai in infancy and then develops one of the accepted indications for transplant: synthetic failure, intractable pruritus, growth failure, recurrent cholangitis, or portal hypertension that is difficult to control. The PELD score sets the urgency on the deceased-donor waiting list, and where deceased donation is scarce a parent is evaluated as a living donor for a left lateral segment graft. Outcomes are excellent in the elective setting, with graft and patient survival among the best of any indication. [10]
Acute liver failure is the time-critical indication. The decision to list is dynamic, driven by trends in the international normalised ratio, the depth and progression of encephalopathy, the arterial pH and lactate, and the development of multiorgan failure, rather than by any single adult score. Live donor transplantation is often the only feasible option in regions with deceased-donor scarcity, and the donor evaluation runs in parallel with maximal medical support in intensive care. The principle is early referral to a transplant centre before multiorgan failure is established. [2]
Metabolic disease is the special category in which the graft may be auxiliary. The native liver is partly retained to preserve native metabolic capacity and the possibility of future gene-directed therapy, and the indication is biochemical danger rather than structural failure. Crigler-Najjar syndrome, urea cycle defects, and the organic acidaemias are the classic examples. The graft corrects the biochemical defect while the retained native liver retains synthetic reserve, and in selected cases immunosuppression may eventually be withdrawn if native function recovers or a definitive therapy emerges. [1]
Post-transplant lymphoproliferative disorder is the feared late complication driven by Epstein-Barr virus, and the scenario the candidate must be able to walk through. It presents with fever, lymphadenopathy, and an expanding mass, often with a rising Epstein-Barr viral load, and it ranges from a reactive polyclonal proliferation to a frank monoclonal lymphoma. The first step in management is reduction of immunosuppression to restore T-cell immune surveillance against the Epstein-Barr-virus-infected B-cells, which alone controls many early cases. Refractory or high-grade disease is treated with the anti-CD20 monoclonal antibody rituximab, with chemotherapy reserved for aggressive monoclonal lymphoma. The risk is concentrated in Epstein-Barr-virus-seronegative recipients, who require active viral-load surveillance. [11]
Complications & Pitfalls
Complications divide into surgical, immunological, infectious, and long-term, and the candidate should be able to categorise each presenting problem into one of these streams. The surgical complications are vascular, biliary, and bleeding. Hepatic artery thrombosis is the most feared because it threatens the graft through bile duct ischaemia; portal vein thrombosis and venous outflow obstruction are less common; biliary leaks and strictures present with jaundice, cholangitis, or a collection; and postoperative bleeding demands re-exploration. [2]
The immunological complications are acute cellular rejection, which is the most common and usually steroid-responsive, chronic rejection, which is the ductopenic vanishing-bile-duct pattern that may culminate in graft loss and retransplantation, and recurrent native disease, which affects autoimmune hepatitis, primary sclerosing cholangitis, and some metabolic conditions. Infectious complications follow a recognisable timeline: the first month brings nosocomial bacteria, the one-to-six-month window brings the opportunistic viruses (cytomegalovirus, Epstein-Barr virus) and opportunists such as Pneumocystis, and beyond six months community-acquired infection dominates. [8]
[8]The classic pitfall is non-adherence in adolescence, a leading preventable cause of graft loss. Developmental factors, the shift toward self-management, and the disruption of transition combine to make the adolescent the highest-risk period of the entire transplant journey, and adherence assessment and structured transition planning are therefore central, not optional. Other long-term complications include renal dysfunction from calcineurin-inhibitor nephrotoxicity, hypertension, post-transplant diabetes, de novo malignancy (especially skin cancer), and the metabolic and cardiovascular consequences of chronic corticosteroid exposure. [8]
Prognosis & Disposition
Outcomes have improved dramatically over three decades and are now excellent. The 30-year European study reports patient survival of around 90 percent at one year and roughly 80 percent at ten years, with graft survival somewhat lower because some children are retransplanted. Survival is best in elective biliary atresia transplants and lower in acute liver failure, in the smallest infants, and in retransplantation, reflecting both the technical challenge and the severity of the recipient. [12]
Long-term survival is now limited less by graft loss than by the cumulative consequences of immunosuppression. Renal impairment from calcineurin-inhibitor nephrotoxicity, cardiovascular risk, de novo malignancy, and the psychosocial burden of lifelong treatment and clinic attendance increasingly determine the long-term outlook. The single most important principle in disposition is lifelong specialist follow-up at a transplant centre, with a structured transition from paediatric to adult services that includes formal readiness assessment and a documented transfer of care, because poorly managed transition is a recognised contributor to late graft loss. [8]
Special Populations
The smallest infants, those weighing under five to six kilograms at transplant, face the highest waiting-list mortality and the greatest technical challenge. They benefit most from living-donor and split grafts, but they also carry the highest vascular and biliary complication rates, and they demand meticulous surgical and intensive care. The waiting-list mortality in this group is precisely the gap that living-donor transplantation was developed to close. [2]
Adolescents are the highest-risk group for non-adherence and graft loss. The developmental drive toward autonomy, the wish to be normal, and the disruption of the transition from paediatric to adult care conspire against the relentless daily discipline of immunosuppression. Structured transition programmes, with formal readiness assessment, skills training, and a planned and documented transfer to an adult transplant service, measurably improve outcomes and are now a standard of care. [8]
Epstein-Barr-virus-seronegative recipients are the highest-risk group for post-transplant lymphoproliferative disorder. They require active viral-load surveillance and preemptive immunosuppression reduction if the viral load rises. In regions with scarce deceased donation, the living-donor programme carries the ethical weight of donor risk, and the donor hepatectomy, while safe in experienced hands, carries a small but real morbidity and a rare mortality that must be disclosed and weighed. [11]
Evidence, Guidelines & Regional Differences
The Paediatric End-stage Liver Disease score, introduced for deceased-donor allocation in the United States from 2002, was a landmark because it replaced a coarse status-based system with a continuous, severity-weighted score. It prioritises children by five variables, deliberately weighting growth failure because malnutrition reflects disease severity in children in a way that no adult variable captures. Its accuracy in estimating pretransplant mortality has been validated, and the growth-failure thresholds specifically were shown to influence mortality among candidates. Standardised exception scores exist for conditions whose severity the raw score underestimates, such as metabolic disease and unresectable hepatoblastoma, and their accuracy in reflecting true urgency continues to be studied and refined. [4]
In Australia and New Zealand, deceased donation is supplemented by established living-donor programmes at the specialist paediatric transplant centres, and allocation follows a severity-weighted framework consistent with the PELD principle. Live donor liver transplantation is an accepted and important pathway, particularly for the smallest infants and for acute liver failure, with donor evaluation conducted to the same safety standards as in deceased-donor pathways. [1]
Immunosuppression has moved steadily toward steroid-sparing and steroid-free regimens. The steroid-free tacrolimus-basiliximab protocol has been shown to be feasible and to spare children the growth and metabolic penalties of corticosteroids while maintaining low rejection rates. Operational tolerance is achievable in a selected minority of paediatric liver recipients, more so than in kidney or heart recipients, but the 10-year prospective experience shows it is not always durable, so immunosuppression withdrawal remains a specialist, protocolised decision rather than routine practice. [7]
The controversies centre on the long-term balance of immunosuppression, the role and durability of tolerance protocols, the equity of access to transplantation across regions and health systems, and the safety and timing of transition to adult care. Living-donor transplantation carries donor risk and ethical weight but delivers outcomes comparable to deceased-donor grafts and is indispensable where deceased donation is scarce. [9]
Exam Pearls
BALEM
Biliary atresia is the single most common indication, accounting for up to half of cases. The PELD score is calculated from bilirubin, international normalised ratio, albumin, growth failure, and age at listing, and uniquely weights growth failure because malnutrition reflects severity in children. Tacrolimus is the backbone calcineurin inhibitor, monitored by whole-blood trough, targeting around 8 to 12 nanograms per millilitre early and 4 to 6 long term. [3]
Acute cellular rejection is the most common form of rejection, usually occurs within the first month, is diagnosed by biopsy showing portal inflammation with bile duct damage and endothelialitis graded by the Rejection Activity Index, and is treated with intravenous methylprednisolone 10 milligrams per kilogram per day for three days. Hepatic artery thrombosis is the most feared vascular complication because the bile ducts are exclusively hepatic-artery supplied, and it demands immediate Doppler ultrasound. Post-transplant lymphoproliferative disorder is Epstein-Barr-virus-driven, is most common in seronegative recipients, and is managed first by reduction of immunosuppression. Non-adherence in adolescence is a leading preventable cause of graft loss. Live vaccines are generally avoided on immunosuppression; inactivated vaccines are safe and should be given before transplant wherever possible. [8]
References
- [1]Smith SK, Miloh T Pediatric Liver Transplantation. Clin Liver Dis, 2022.PMID 35868688
- [2]Pham YH, Miloh T Liver Transplantation in Children. Clin Liver Dis, 2018.PMID 30266163
- [3]McDiarmid SV, Merion RM, Dykstra DM, et al Use of a pediatric end-stage liver disease score for deceased donor allocation: the United States experience. Indian J Pediatr, 2007.PMID 17476086
- [4]Chang CH, Bryce CL, Shneider BL, et al Accuracy of the Pediatric End-stage Liver Disease Score in Estimating Pretransplant Mortality Among Pediatric Liver Transplant Candidates. JAMA Pediatr, 2018.PMID 30242345
- [5]Swenson SM, Roberts JP, Rhee S, et al Impact of the Pediatric End-Stage Liver Disease (PELD) growth failure thresholds on mortality among pediatric liver transplant candidates. Am J Transplant, 2019.PMID 31370108
- [6]Reding R Tacrolimus in pediatric liver transplantation. Pediatr Transplant, 2002.PMID 12453195
- [7]Trezeguet Renatti G, Riva N, Minetto J, et al Feasibility of steroid-free tacrolimus-basiliximab immunosuppression in pediatric liver transplantation and predictors for steroid requirement. Liver Transpl, 2024.PMID 37439661
- [8]Antala S, DiNorcia J, Bucuvalas J Balancing immunosuppression in pediatric liver transplantation: Playing the long game. Pediatr Transplant, 2023.PMID 37439035
- [9]Wozniak LJ, Venick RS, Naini BV, et al Operational tolerance is not always permanent: A 10-year prospective study in pediatric liver transplantation recipients. Liver Transpl, 2022.PMID 35395132
- [10]Anouti A, Patel MS, VanWagner LB, et al Biliary atresia and liver transplantation in the United States: A contemporary analysis. Liver Int, 2023.PMID 37548078
- [11]Okamoto T, Okajima H, Uebayashi EY, et al Management of Epstein-Barr Virus Infection and Post-Transplant Lymphoproliferative Disorder in Pediatric Liver Transplantation. J Clin Med, 2022.PMID 35456259
- [12]Baumann U, Karam V, Adam R, et al Prognosis of Children Undergoing Liver Transplantation: A 30-Year European Study. Pediatrics, 2022.PMID 36111446