Paeds · gastroenterology-hepatology-and-nutrition
Pancreatitis and pancreatic disorders
Also known as Pancreatitis · Acute pancreatitis · Chronic pancreatitis · Acute recurrent pancreatitis · Hereditary pancreatitis · Pancreatic exocrine insufficiency · Pancreatitis in children
Fellowship guide to pancreatitis and pancreatic disorders in children: the NASPGHAN classification and two-of-three diagnostic criteria for acute pancreatitis, the premature intracellular trypsin activation pathophysiology, the three paediatric risk-factor domains of toxic-metabolic, genetic and obstructive causes, the INSPPIRE definitions separating acute from acute recurrent and chronic pancreatitis, early aggressive hydration and the shift to early enteral feeding, the management of pain and complications, pancreatic exocrine insufficiency diagnosed by faecal elastase and treated with pancreatic enzyme replacement, hereditary PRSS1 pancreatitis and its cancer risk, autoimmune pancreatitis, traumatic pancreatitis, and total pancreatectomy with islet autotransplantation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
The pancreas is a single gland that does two very different jobs, and pancreatitis in a child is the failure of one of them. The exocrine pancreas releases digestive enzymes into the gut, and the endocrine pancreas releases insulin into the blood. When the gland inflames, enzymes activate inside it instead of in the bowel lumen, the pancreas digests itself, and the child arrives in the emergency department with severe epigastric pain that drives everything that follows. [1]
Acute pancreatitis in a child is defined by the NASPGHAN criteria as the presence of at least two of three features: characteristic epigastric pain that may radiate to the back, a serum lipase or amylase at or above three times the upper limit of normal, and imaging findings consistent with pancreatic inflammation. The definition is deliberately practical, because the child does not always have all three at the first presentation, and the clinician who waits for the complete triad will miss the early window for treatment. [1]
The paediatric disease differs from the adult disease in almost every way that matters. The causes are different — children do not typically have gallstones or alcohol, and the dominant paediatric causes are medications, trauma and genetic variants. The severity tends to be lower, the recovery faster, and the stakes are in the long tail: a child who has one episode may have a genetic predisposition that drives recurrence and the eventual slide into chronic disease with exocrine and endocrine failure. The modern approach is therefore not just to treat the acute attack but to hunt the underlying cause, because finding a PRSS1 mutation or stopping valproate changes the child's whole trajectory. [5] [1]
Classification
The most useful clinical framework holds pancreatitis as a temporal spectrum, because the child who has one episode is not the same patient as the child who has five. The NASPGHAN and INSPPIRE definitions separate three stages along this spectrum, and the distinction matters because each stage carries a different prognosis and demands a different investigation depth. [1] [5]
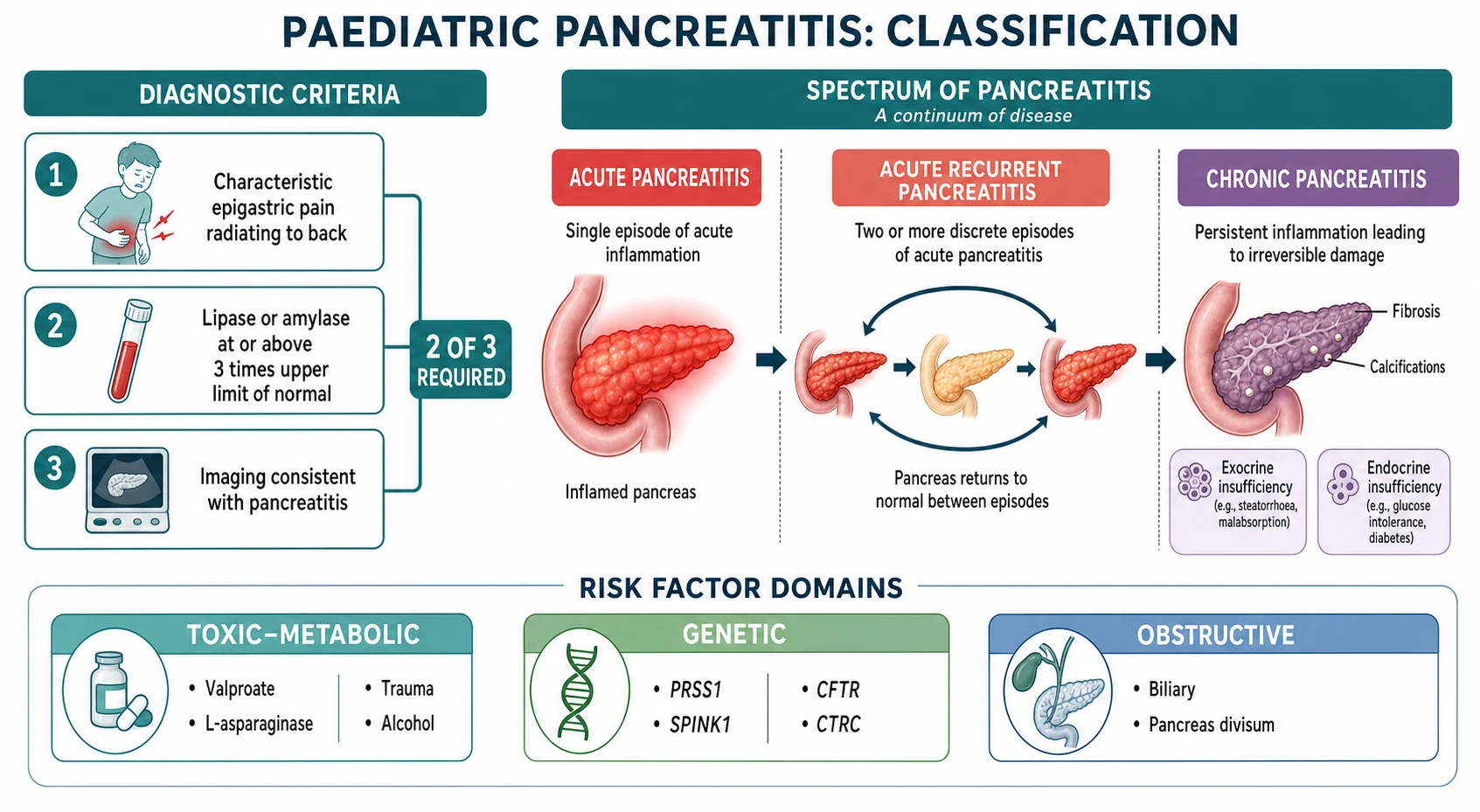
Acute pancreatitis is a single reversible episode. It is further graded as mild, moderately severe, or severe based on the absence or presence of organ dysfunction and local or systemic complications, though the adult-derived Revised Atlanta Classification does not translate cleanly to children, where severe disease and mortality are substantially rarer. The clinical question in acute disease is not just how severe it is now, but what caused it and whether it will recur. [1]
Acute recurrent pancreatitis is defined as two or more discrete episodes of acute pancreatitis with complete recovery, including normalisation of enzymes and resolution of symptoms, between episodes. This is the bridge between a single attack and chronic disease, and it is the point at which a thorough search for genetic, obstructive and metabolic risk factors becomes essential, because roughly a quarter to a third of children with acute recurrent pancreatitis carry a pathogenic genetic variant. [5] [6]
Chronic pancreatitis is defined by irreversible morphological change in the pancreas — ductal distortion, calcification, atrophy or fibrosis on imaging — accompanied by endocrine or exocrine insufficiency. The progression from acute recurrent to chronic disease is the central clinical narrative of paediatric pancreatitis, and it is driven most powerfully by the genetic background. A child with a PRSS1 mutation who presents with recurrent acute attacks is on a trajectory toward chronic disease, exocrine failure, pancreatic diabetes and an elevated lifetime risk of pancreatic cancer, and the clinician who identifies this early can intervene, counsel and survey. [5] [6]
[1] [5]A second classification axis is the cause, and the paediatric risk-factor framework is built around three domains that are easier to hold than the long adult lists. The toxic-metabolic domain includes medications — valproate, L-asparaginase, mesalamine, azathioprine — alcohol in the adolescent, and hypertriglyceridaemia and hypercalcaemia. The genetic domain includes the gain-of-function PRSS1 mutations that cause hereditary pancreatitis, the loss-of-function SPINK1 and CTRC variants that reduce trypsin defence, and CFTR variants that impair ductal clearance. The obstructive domain includes biliary microlithiasis, pancreas divisum and congenital ductal anomalies. In the INSPPIRE cohort, a substantial proportion of children with acute recurrent or chronic pancreatitis had at least one identifiable risk factor, and genetic causes predominated in chronic disease. [5] [6] [9]
Epidemiology & Risk Factors
Paediatric pancreatitis is more common than it was a generation ago, and the rise is real rather than merely artefactual. The estimated incidence of acute pancreatitis in children is between three and thirteen per one hundred thousand children per year, with figures that have climbed steadily over two decades, driven by greater awareness among clinicians, wider availability of serum lipase testing, and improved survival of children with complex chronic conditions that predispose to pancreatic disease. [1] [2]
The risk factors, organised into the three domains, are the facts the examiner rewards for knowing by name. In the toxic-metabolic domain, medications are the single commonest identifiable cause of paediatric acute pancreatitis, and the high-yield drugs are valproate, L-asparaginase used in acute lymphoblastic leukaemia, mesalamine, azathioprine and thiopurines, and certain antiretrovirals. Alcohol is a factor in the adolescent. Blunt abdominal trauma — classically the bicycle handlebar — is a distinctive paediatric cause that may injure the pancreatic duct directly. Hypertriglyceridaemia and hypercalcaemia are metabolic precipitants. [9]
In the genetic domain, the genes are the molecular guardians of the trypsin balance, and their disturbance is the engine of recurrence and chronicity. PRSS1 encodes cationic trypsinogen, and gain-of-function mutations cause hereditary pancreatitis, an autosomal dominant disorder in which recurrent attacks begin in childhood and carry a high lifetime risk of progression to chronic disease and pancreatic cancer. SPINK1 encodes the primary trypsin inhibitor, and loss-of-function variants are recessive modifiers that lower the threshold for pancreatitis in the presence of other triggers. CFTR variants impair ductal bicarbonate and fluid secretion, allowing protein plugs to obstruct the duct, and the link is strong enough that CFTR carriage and atypical cystic fibrosis are well-recognised causes of idiopathic pancreatitis. CTRC encodes chymotrypsin C, which normally degrades prematurely activated trypsin, and loss-of-function variants remove this protective degradation. [5] [6]
In the obstructive domain, biliary disease is far less common in children than in adults, but biliary microlithiasis, sludge and choledochal cysts still cause acute pancreatitis, especially in the adolescent. Pancreas divisum, the commonest congenital pancreatic anomaly, is found in a substantial proportion of children with idiopathic acute recurrent pancreatitis, though its causal role is debated because it is also present in a sizeable fraction of the healthy population. The clinical point is that a child with recurrent pancreatitis needs a thorough search across all three domains, because finding the cause is what prevents recurrence and progression. [5] [9]
Pathophysiology
The pancreas is protected from its own enzymes by a single principle: trypsin must never activate inside the acinar cell. Pancreatitis begins when that principle fails. Trypsinogen, the inactive precursor, is prematurely converted to active trypsin inside the pancreatic acinar cell rather than in the duodenal lumen, and once trypsin is active it converts the other proenzymes — prophospholipase, proelastase, kallikreinogen — to their active forms. The result is autodigestion of the gland from within, and the entire clinical picture flows from this single catastrophic event. [1] [4]
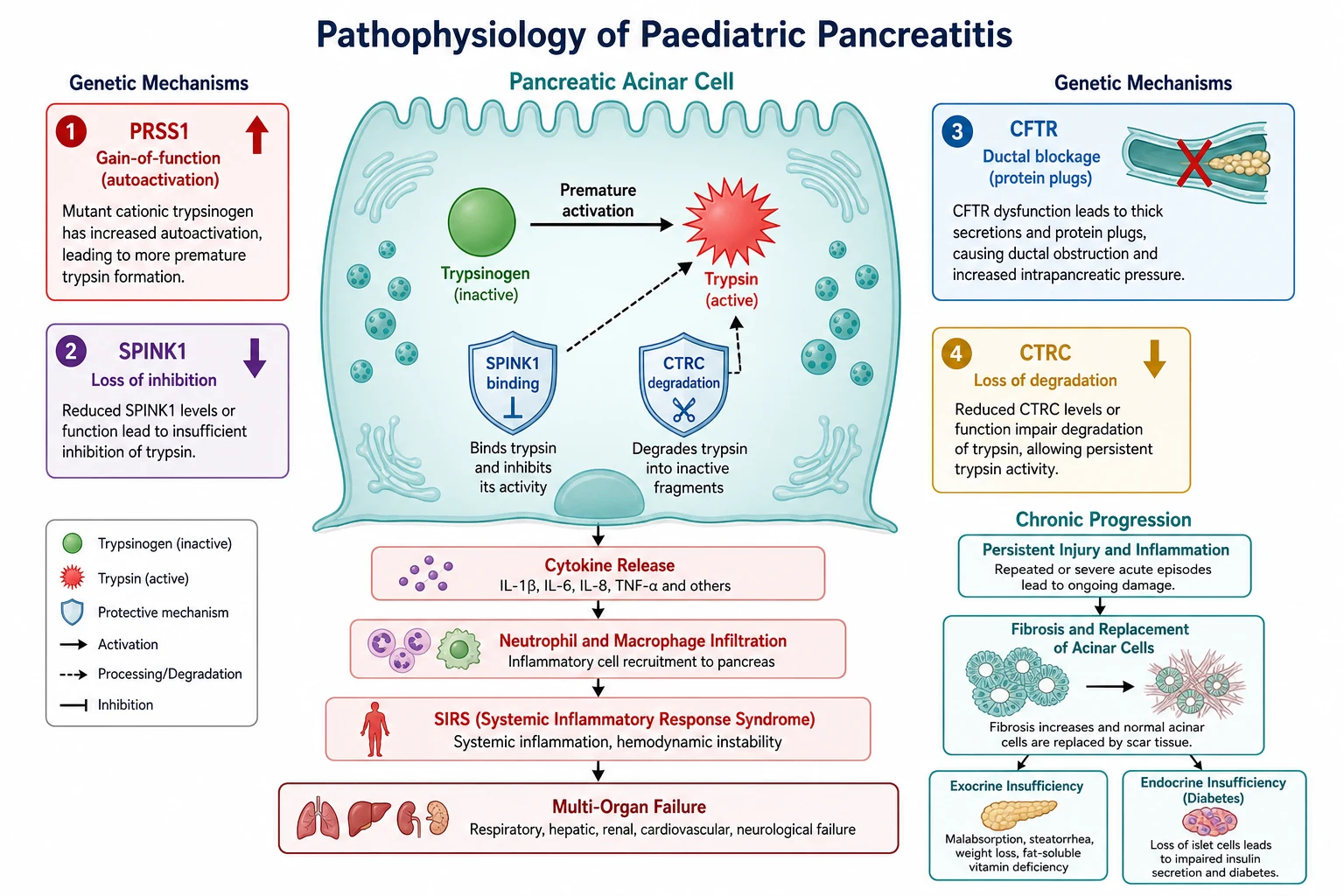
The cell has two protective mechanisms against this event, and the genetic causes of pancreatitis each attack one of them. The first is serine protease inhibitor Kazal type 1, encoded by SPINK1, which binds and inhibits trypsin as it begins to activate. The second is chymotrypsin C, encoded by CTRC, which degrades prematurely activated trypsin, clipping it into inert fragments. When either defence is compromised by a loss-of-function variant, the threshold for autodigestion drops. PRSS1 gain-of-function mutations make the opposite error, increasing the autoactivation of trypsinogen so that it activates more readily than the defences can contain. CFTR variants do not act on trypsin at all but on the duct: they impair the secretion of bicarbonate-rich fluid that flushes the ductal system, allowing viscous protein-rich secretions to plug the ducts, build pressure, and injure the acinar cells upstream. [5] [4]
Once autodigestion begins, the injury amplifies through an inflammatory cascade. The damaged acinar cell releases cytokines, damage-associated molecular patterns and trypsin itself into the interstitium, recruiting neutrophils and macrophages that release further proteases and reactive oxygen species. In mild disease this inflammation stays local. In severe disease it spills into the systemic circulation, producing the systemic inflammatory response syndrome, capillary leak, hypovolaemia, and in the worst cases, acute respiratory distress syndrome and multi-organ failure. The severity of the systemic illness is not proportional to the visible pancreatic damage on imaging, which is why a child can be systemically very unwell with a relatively normal-looking scan. [3] [2]
The progression to chronic disease is the long arc, and it follows the same mechanism repeated. Each acute attack injures acinar and ductal cells, and the healing is imperfect, with fibrosis replacing functional tissue. Ducts distort and narrow from stricture and fibrosis, creating a cycle of obstruction, pressure and further injury. Over months to years, the acinar mass is lost and exocrine insufficiency develops, with steatorrhoea and fat-soluble vitamin deficiency; the islets are eventually destroyed and endocrine insufficiency appears as pancreatic diabetes. The speed of this progression is set by the genetic background and the frequency of attacks, which is why the child with a PRSS1 mutation is on a faster and more dangerous trajectory than the child with a single medication-triggered episode. [5] [6]
Clinical Presentation
The child with acute pancreatitis usually has one dominant symptom: severe, persistent epigastric abdominal pain that may radiate straight through to the back. The pain comes on over hours, is constant rather than colicky, and is made worse by eating. Nausea and vomiting accompany it, and the child often finds that leaning forward eases the pain — a posture the examiner should ask about, because it points to a retroperitoneal inflammatory process. [1] [2]
The presentation is less clear in infants and young children, and this is where the diagnosis is most often missed. A toddler may not localise the pain to the epigastrium and may present instead with irritability, abdominal distension, feeding refusal and non-specific distress that is attributed to gastroenteritis. The index of suspicion must be deliberately raised for the young child with abdominal pain and vomiting who does not settle, because the lipase is the test that separates pancreatitis from the common self-limiting causes and it is not always sent early. [2]
The atypical presentations are the ones the examiner probes deliberately, because they test whether the candidate carries the diagnosis as an active possibility rather than waiting for the textbook picture. The child on valproate for epilepsy who develops abdominal pain is the classic drug-triggered scenario, because valproate is one of the commonest medication causes and the diagnosis is often delayed by attributing the pain to a viral illness. The child on L-asparaginase for leukaemia is another, because the drug is directly toxic to the acinar cell. The child with diabetic ketoacidosis may have concurrent pancreatitis that is masked by the metabolic derangement, and both hyperamylasaemia and genuine pancreatitis occur in DKA. The child who has suffered blunt abdominal trauma, classically from a bicycle handlebar strike to the epigastrium, may have a pancreatic contusion or duct injury whose severity only becomes apparent over hours. [9] [2]
The presentation of chronic pancreatitis is different in character. The pain becomes recurrent or persistent rather than a single discrete attack, and the child may describe episodes over months or years that have been labelled as functional or stress-related. Between the pain episodes, the insidious consequences of glandular destruction appear: steatorrhoea with greasy, foul-smelling stools that are difficult to flush, poor weight gain and growth failure from malabsorption, and eventually glucose intolerance or overt diabetes. The child with chronic pancreatitis often arrives in clinic with these long-term problems rather than with an acute attack, and the growth chart and the stool history are the clues that demand investigation. [5] [4]
[1] [5]Differential Diagnosis
The differential for acute epigastric pain with vomiting in a child is the list of things that can produce the same picture, and pancreatitis is the diagnosis that sits on it quietly until the lipase is sent. The competitors are biliary colic and cholecystitis, which share the epigastric or right upper quadrant pain and vomiting but tend to produce colicky pain that follows fatty food and may have right upper quadrant tenderness and a positive Murphy sign. Perforated peptic ulcer is the must-not-miss surgical diagnosis, producing sudden severe epigastric pain with peritonism and free gas on imaging. Mesenteric adenitis, gastroenteritis and intestinal obstruction all cause abdominal pain and vomiting, but they lack the characteristic epigastric localisation, the radiation to the back, and the three-fold lipase elevation. [2]
Diabetic ketoacidosis deserves special attention because it both mimics and causes pancreatitis. The child with DKA has abdominal pain, vomiting and a metabolic acidosis that may include an elevated amylase, and the question is whether the amylase reflects genuine pancreatitis or a non-pancreatic source. Lipase is more specific than amylase, and a lipase at or above three times the upper limit of normal in a child with DKA warrants imaging to confirm or exclude concurrent pancreatitis, because the management of the two conditions must proceed in parallel. [9] [2]
The differential for chronic or recurrent abdominal pain that might be pancreatitis is wider and more treacherous. Functional abdominal pain, irritable bowel syndrome, peptic ulcer disease, biliary dyskinesia, inflammatory bowel disease and constipation all produce recurrent abdominal pain in children, and the child with chronic pancreatitis is at risk of being labelled functional before the diagnosis is made. The growth chart, the faecal elastase and cross-sectional imaging are the tests that separate organic pancreatic disease from functional pain, and the family history of recurrent abdominal attacks or pancreatic cancer should lower the threshold to investigate. [5] [11]
The paediatric risk-factor domains — TGO
Clinical & Bedside Assessment
Bedside assessment begins, as always in paediatrics, with the growth chart and the vital signs, because both contain information that changes the immediate management. Plot the weight and height and look for the faltering growth that signals chronic disease with exocrine insufficiency, because a child who has been losing weight or falling across centiles between episodes has a different problem from the previously well child with a first acute attack. The vital signs grade the severity of the acute episode: tachycardia, hypotension, fever and tachypnoea suggest systemic inflammatory response syndrome and the need for early aggressive fluid resuscitation. [2] [3]
The abdominal examination seeks the signs of pancreatic inflammation. Epigastric tenderness is the expected finding, and guarding or rigidity suggests peritonism from severe disease or, importantly, an alternative surgical diagnosis such as a perforated viscus. Abdominal distension and reduced or absent bowel sounds indicate an ileus from the retroperitoneal inflammation. The rare signs of Grey Turner — flank bruising — and Cullen — periumbilical bruising — indicate haemorrhagic necrotising pancreatitis and are seldom seen in children, but they carry the same gravity when present. [2]
The history is where the cause is found, and it must be deliberately structured around the three risk-factor domains. Ask about every medication the child takes, with specific attention to valproate, L-asparaginase and other chemotherapy, mesalamine, azathioprine and antiretrovirals, because stopping an offending drug is sometimes the only intervention needed. Ask about recent blunt abdominal trauma, especially a bicycle handlebar or a sports injury to the epigastrium. Ask about alcohol use in the adolescent, using a non-judgemental, developmentally appropriate approach. Ask about a family history of pancreatitis, recurrent abdominal pain, pancreatic cancer or cystic fibrosis, because a positive family history changes the investigation to a genetic workup. Ask about the onset of the pain in relation to meals, the character and radiation, and whether leaning forward eases it. [9] [2]
Investigations
Investigation is built on a single enzyme test and a single first-line image. Serum lipase is the preferred enzyme for paediatric acute pancreatitis, because it is more sensitive and more specific than amylase, it stays elevated for longer after the onset of symptoms, and it is not spuriously elevated by the non-pancreatic sources that inflate amylase. The diagnostic threshold is a lipase at or above three times the upper limit of normal, and this must be paired with the clinical picture rather than read in isolation, because a mildly elevated lipase can occur in gastroenteritis, DKA and renal failure. Amylase is less useful because it normalises faster and is less specific, but it is still checked in many centres as a second line. [1] [2]
Abdominal ultrasound is the first-line imaging modality in children. It avoids radiation, it visualises the pancreas and the biliary tree, and it can identify gallstones, biliary dilatation, peripancreatic fluid and pseudocysts. When the ultrasound is technically limited by bowel gas, when the diagnosis is uncertain, or when severe disease or complications such as necrosis or ductal disruption are suspected, cross-sectional imaging is needed. Contrast-enhanced computed tomography assesses necrosis and local complications but carries a radiation dose, so it is reserved for the situations where the diagnostic yield justifies it. Magnetic resonance cholangiopancreatography is the preferred modality for ductal anatomy and for evaluating pancreas divisum, ductal strictures and biliary anomalies, and it avoids radiation, making it ideal for the recurrent or chronic disease workup. [1] [4]
The investigation of recurrent or chronic pancreatitis goes beyond the acute workup and into the search for the underlying cause. Faecal pancreatic elastase is the preferred non-invasive test for exocrine insufficiency, with a level below two hundred micrograms per gram indicating insufficiency, and it is stable, reproducible and unaffected by pancreatic enzyme replacement. A genetic panel is indicated for any child with acute recurrent or chronic pancreatitis, and the first-line test for suspected hereditary pancreatitis is a PRSS1 gene analysis, followed by a broader panel including SPINK1, CFTR, CTRC and CASR if PRSS1 is negative. An elevated serum triglyceride and calcium screen for metabolic precipitants, and an immunoglobulin G4 level screens for autoimmune pancreatitis. [5] [4]
Management — Resuscitation
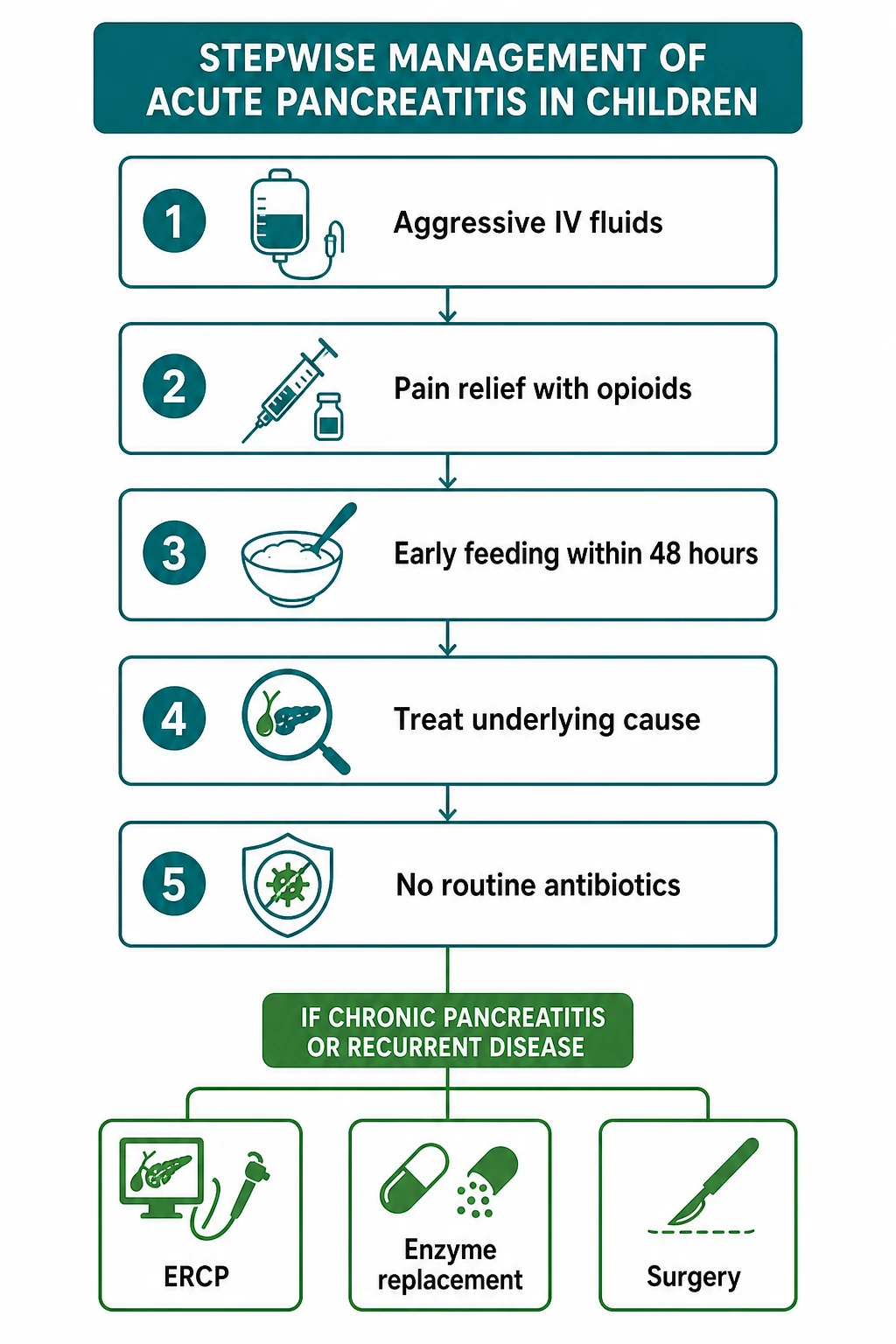
The resuscitation of acute pancreatitis rests on a single principle delivered early: aggressive intravenous hydration. The inflamed pancreas leaks fluid into the retroperitoneum and third space, and the child who is under-resuscitated develops the systemic inflammatory response, hypovolaemia and organ dysfunction that define severe disease. The recommended approach is isotonic crystalloid — normal saline or lactated Ringer solution — at one and a half to two times the maintenance rate in the first twenty-four to forty-eight hours, with regular reassessment of hydration status, urine output and clinical response. Timely fluid resuscitation reduces the severity of the attack and is the single most impactful early intervention. [2] [3]
Early aggressive hydration for acute paediatric pancreatitis
Loading dose
Isotonic crystalloid (normal saline or lactated Ringer solution) at 1.5 to 2 times the maintenance rate for the first 24 to 48 hours, titrated to urine output, hydration status and clinical response
Maintenance dose
Reduce to maintenance rate once the child is clinically euvolaemic and pain is settling; reassess hydration at least every 6 to 8 hours during the aggressive phase
Analgesia is the second pillar, and the modern approach is to provide adequate pain relief rather than to withhold opioids. Acute pancreatitis is severely painful, and the historic concern that opioids would obscure a surgical abdomen does not apply once the diagnosis is established. Age-appropriate opioids — morphine or a similar agent — are used for acute pain, titrated to comfort, alongside simple analgesics such as paracetamol. The child who is still in severe pain despite adequate analgesia needs reassessment for a complication. [2] [3]
The third pillar, and the one that has changed most in the past decade, is nutrition. The old practice of prolonged fasting to rest the pancreas — keeping the child nil by mouth for days — has been overturned by evidence that early enteral feeding within forty-eight to seventy-two hours reduces complications, infection and length of stay compared with bowel rest. The practical approach is to allow oral intake as tolerated once the pain is settling and the child is hungry, starting with clear fluids and advancing to a low-fat diet. When oral intake is not possible, nasogastric or nasojejunal feeding is used. Total parenteral nutrition is reserved for the child who cannot tolerate enteral feeding for a prolonged period. [7] [2]
The first 48 hours of acute paediatric pancreatitis
Assess airway, breathing, circulation and severity; check lipase, full blood count, electrolytes, glucose, calcium and a venous gas
Start early aggressive isotonic crystalloid hydration at 1.5 to 2 times maintenance for the first 24 to 48 hours
Provide adequate analgesia with age-appropriate opioids and simple analgesics
Plan early enteral feeding within 48 to 72 hours, advancing as tolerated
Identify and treat the underlying cause: stop offending drugs, investigate biliary disease, screen for metabolic triggers
Escalate to paediatric intensive care for organ dysfunction, systemic inflammatory response syndrome, or severe local complications
Prophylactic antibiotics are not recommended in acute pancreatitis, including in severe disease with necrosis, unless there is documented or strongly suspected infected necrosis. This is a deliberate departure from older practice and a high-yield exam fact: routine antibiotics do not prevent infection, they select resistant organisms, and they do not change the outcome. Antibiotics are reserved for the child with proven infected necrosis, cholangitis or another documented infection. [2] [3]
Management — Definitive & Stepwise
Definitive management of the acute episode builds on resuscitation by addressing the cause and watching for complications. Stopping the offending drug — valproate, L-asparaginase, mesalamine — is the single most important intervention for medication-induced pancreatitis, and the epilepsy or oncology team should be engaged early to find an alternative agent. Biliary pancreatitis from gallstones requires cholecystectomy after the acute inflammation settles, and an impacted stone at the ampulla may require endoscopic removal. Traumatic pancreatitis with a duct injury may need ERCP stenting or, in severe disruption, surgical repair. [2] [9]
The management of acute recurrent and chronic pancreatitis is a long-term, multidisciplinary project, and its goal is to preserve glandular function, control pain and prevent progression. Lifestyle modification includes a low-fat diet to reduce the secretory demand on the gland and alcohol avoidance in the adolescent. Pain management uses a multi-modal approach that begins with non-opioid analgesics and advances cautiously, because chronic pancreatitis pain is a leading cause of opioid dependence in children with the disease. Antioxidant supplementation has been studied with mixed results, and some centres use it as an adjunct. Pancreatic enzyme replacement is given both for exocrine insufficiency and, in some children with chronic pancreatitis, for a feedback effect that may reduce pain by suppressing exocrine secretion. [11] [7]
Ductal obstruction from strictures, stones or pancreas divisum is treated with therapeutic ERCP, which in the INSPPIRE data was performed safely and effectively in children with acute recurrent and chronic pancreatitis, providing sphincterotomy, stricture dilation, stone removal and stent placement. The aim is to relieve the obstruction, drain the duct and break the cycle of pressure and injury. When endoscopic therapy fails or the disease is refractory, surgical options include drainage procedures and, for carefully selected children with intractable disease, total pancreatectomy with islet autotransplantation, which removes the source of pain while infusing the child's own islet cells into the liver to preserve some beta-cell function. [10] [12]
Pancreatic enzyme replacement therapy for exocrine insufficiency
Loading dose
Pancreatic enzyme capsules (lipase, protease, amylase) at approximately 500 to 1000 lipase units per kg per meal for children, with half that dose per snack, taken with meals and snacks
Maintenance dose
Titrate to symptom control (reduced steatorrhoea, weight gain) and a daily maximum of 10000 lipase units per kg per day to avoid fibrosing colonopathy
Pancreatic enzyme replacement therapy is the cornerstone of managing exocrine insufficiency, whether from chronic pancreatitis, cystic fibrosis or Shwachman-Diamond syndrome. The enzyme capsules contain lipase, protease and amylase, and they are dosed by lipase content. A practical starting dose for children is approximately five hundred to one thousand lipase units per kilogram per meal, with about half that per snack, titrated to symptom control. The daily maximum of ten thousand lipase units per kilogram per day must not be exceeded, because high-dose enzyme replacement is associated with fibrosing colonopathy. The enzymes must be taken with every meal and snack containing fat, and in infants and young children the microspheres can be mixed with acidic soft food. [4]
Total pancreatectomy with islet autotransplantation — when and why
For the child with intractable, debilitating chronic pancreatitis whose pain is refractory to medical and endoscopic management, total pancreatectomy with islet autotransplantation is a last-resort surgical option. The pancreas is removed to eliminate the source of pain, and the islets of Langerhans are isolated and infused into the liver via the portal vein to preserve some endocrine function. The INSPPIRE data and outcome studies show that the procedure reduces opioid use and improves quality of life in carefully selected children, though it causes insulin-dependent diabetes in most. The decision is made by a multidisciplinary team including paediatric gastroenterology, surgery, endocrinology, psychology and pain services, and it is reserved for children in whom all other options have failed. [12] [11]
Specific Subtypes & Scenarios
Hereditary pancreatitis is the subtype that defines the genetic domain, and it is the one the examiner rewards for understanding in depth. It is caused by autosomal dominant gain-of-function mutations in the PRSS1 gene encoding cationic trypsinogen, which make the trypsinogen molecule activate prematurely inside the acinar cell. The affected child typically presents with recurrent episodes of acute pancreatitis beginning in childhood — often before the age of ten — and the disease progresses to chronic pancreatitis in a high proportion of carriers. The lifetime risk of pancreatic cancer is substantially elevated, and the risk rises with the duration of disease, which is why hereditary pancreatitis carries a recommendation for surveillance in adulthood. Genetic counselling of the family is essential, and first-degree relatives should be offered testing. [5] [6]
Medication-induced pancreatitis is the commonest toxic-metabolic cause in children, and the two drugs to know are valproate and L-asparaginase. Valproate causes pancreatitis through a direct toxic or idiosyncratic mechanism, and the pancreatitis can occur at any time during treatment, not just at initiation. L-asparaginase, used in acute lymphoblastic leukaemia, causes pancreatitis in a proportion of treated children and may be severe. The management is to stop the offending drug and provide supportive care, and the oncology or neurology team should be engaged early to find an alternative treatment for the underlying condition. Other drugs to recognise include mesalamine, azathioprine and thiopurines, didanosine, and certain diuretics. [9]
Traumatic pancreatitis has a distinctive paediatric signature. The classic mechanism is a bicycle handlebar striking the epigastrium, compressing the pancreas against the spine and causing a contusion, laceration or duct injury. The child may present hours to days after the injury with worsening abdominal pain, and a pancreatic duct injury is the concern that demands cross-sectional imaging. A duct disruption may be managed with ERCP and stenting, with endoscopic or surgical drainage of collections, or in severe cases with surgical resection. The diagnosis is often delayed because the initial injury seems minor and the symptoms evolve gradually. [2]
Autoimmune pancreatitis in childhood is rare but important because it is steroid-responsive. The INSPPIRE consensus defines it by a combination of elevated serum immunoglobulin G4, characteristic imaging with a diffusely enlarged sausage-shaped pancreas, a dense lymphoplasmacytic infiltrate on histology, and a dramatic response to corticosteroids. The distinction from other forms of chronic pancreatitis matters because the treatment is fundamentally different: a course of corticosteroids can induce remission in autoimmune pancreatitis, whereas steroids have no role in the routine management of other forms. [8]
Complications & Pitfalls
The local complications of acute pancreatitis are the things that go wrong inside the abdomen. A peripancreatic fluid collection forms in the first four weeks from the inflammatory exudate. When this collection organises a wall of fibrous or granulation tissue after four to six weeks, it becomes a pancreatic pseudocyst, which may cause pain, gastric outlet obstruction or become infected. Pancreatic necrosis — the death of pancreatic and peripancreatic tissue — may be sterile or infected, and infected necrosis is the complication that justifies antibiotics and drainage. A ductal disruption can produce a pancreatic fistula to the peritoneum, pleura or skin. [2] [3]
The systemic complications are the things that can kill. Systemic inflammatory response syndrome can progress to sepsis, acute respiratory distress syndrome and multi-organ failure. Hypocalcaemia occurs from fat necrosis saponifying calcium, and hyperglycaemia occurs from impaired insulin secretion. These complications are uncommon in children but they define severe disease, and the child who develops organ dysfunction needs paediatric intensive care. [2] [3]
The long-term complications of chronic pancreatitis are the ones that shape the child's life. Exocrine insufficiency produces steatorrhoea, malabsorption of fat-soluble vitamins A, D, E and K, and faltering growth. Endocrine insufficiency produces pancreatic diabetes, which is typically brittle and difficult to manage because the loss of both insulin and glucagon makes glycaemic control unstable. Chronic pain is the most disabling symptom and is the leading cause of opioid dependence and school absence. And in hereditary pancreatitis, the lifetime risk of pancreatic cancer is substantially elevated, which is the rationale for genetic counselling and, in adulthood, surveillance. [5] [6]
Prognosis & Disposition
Most children with acute pancreatitis recover fully and quickly. The pain and the lipase elevation settle within days to one to two weeks, the mortality is under two per cent in modern paediatric series, and the majority return to normal activity without residual damage. The child who has had a single episode triggered by a clearly reversible cause — a stopped medication, a resolved metabolic disturbance — has a good prognosis and may not recur. [1] [2]
The prognosis is more guarded for the child with acute recurrent pancreatitis and especially for those with a genetic predisposition. In the INSPPIRE-2 cohort, a substantial fraction of children with acute recurrent or chronic pancreatitis carried identifiable genetic risk factors, and these children bore a significant burden of pain, healthcare use, school absence and hospitalisation. The progression from acute recurrent to chronic disease occurs in a meaningful proportion of children, driven most powerfully by the PRSS1 background, and it is this progression that the early genetic workup and cause-directed management aim to interrupt. [6] [11]
Disposition follows the severity and the stage. Mild acute pancreatitis is managed on the general paediatric ward with surgical or gastroenterology input. Severe disease with organ dysfunction requires paediatric intensive care and a multidisciplinary team. Acute recurrent and chronic pancreatitis need long-term follow-up by paediatric gastroenterology, with dietetics for enzyme replacement and nutritional support, endocrinology for glucose surveillance, psychology and pain services for chronic pain, and genetic counselling for the family. Total pancreatectomy with islet autotransplantation is performed in specialist centres for carefully selected children. Transition to adult care is planned in adolescence, with particular attention to the genetic and cancer surveillance needs. [11] [12]
Special Populations
The infant or young child with pancreatitis is a special population because the presentation is non-specific and the underlying cause is more likely to be anatomic, metabolic or genetic. An infant with irritability, abdominal distension and feeding refusal needs a lipase sent early, because the diagnosis is frequently missed as gastroenteritis, and the yield of a thorough metabolic and genetic workup is higher in this age group. Structural causes such as a choledochal cyst or pancreas divisum should be sought on imaging. [2] [9]
The child with cystic fibrosis sits in a paradoxical position. Most children with classic cystic fibrosis have pancreatic exocrine insufficiency from birth, because thick secretions destroy the acinar tissue in utero and in early infancy. It is the child with a milder CFTR genotype and preserved pancreatic sufficiency who is at risk of acute pancreatitis, because the gland still has enough functional acinar tissue to become inflamed. Pancreatitis in a child with cystic fibrosis therefore suggests a pancreatic-sufficient phenotype, and faecal elastase helps to characterise the exocrine reserve. [4]
The adolescent with pancreatitis needs developmentally appropriate assessment and counselling. Alcohol use is a factor in the adolescent age group and should be explored non-judgementally. Medication adherence and interactions become relevant as the adolescent manages their own health. The transition to adult care must address the genetic counselling and cancer surveillance needs of hereditary pancreatitis, the reproductive implications of chronic disease, and the psychological burden of chronic pain and its management. [11]
The Indigenous, remote and migrant child faces barriers that extend the time to diagnosis and the adequacy of treatment. Pancreatitis may present late in remote settings, cross-sectional imaging may be hundreds of kilometres away, and the cost and supply of pancreatic enzyme replacement can limit adherence. Culturally safe shared-care pathways, telehealth gastroenterology support for the local team, and attention to the practical logistics of enzyme supply and dietitian access are what make the management achievable for families far from a specialist centre. [11]
Across Australia, New Zealand, the United Kingdom, the United States and Europe, the diagnostic criteria and the management principles are broadly aligned. The NASPGHAN clinical reports from North America and the ESPGHAN and EPC/HPSG European guidelines converge on early aggressive hydration, adequate analgesia, early enteral feeding, the avoidance of prophylactic antibiotics, and the identification and treatment of the underlying cause. Regional differences are mainly in the access to specialist paediatric gastroenterology, paediatric ERCP and total pancreatectomy with islet autotransplantation, which are concentrated in tertiary centres. The INSPPIRE consortium has built a shared evidence base that underpins practice internationally, and the genetic panels and faecal elastase testing that drive the chronic disease workup are increasingly standard across all high-income settings. [3] [11]
Evidence, Guidelines & Regional Differences
The evidence base for paediatric pancreatitis has matured rapidly, driven largely by the INSPPIRE consortium. The NASPGHAN 2017 classification clinical report defined the diagnostic criteria for paediatric acute pancreatitis using the two-of-three approach — characteristic pain, enzyme elevation at or above three times the upper limit of normal, and imaging — adapting the adult framework to children and establishing the common language for the paediatric disease. [1]
The NASPGHAN 2018 management clinical report transformed practice by shifting away from prolonged fasting towards early enteral feeding, formalising early aggressive hydration, and recommending against prophylactic antibiotics. This was the pivotal evidence-based change in the acute management, and it reduced complications and length of stay. The EPC/HPSG European guidelines, published the same year, converged on the same principles, giving an internationally consistent approach to the acute disease. [2] [3]
The INSPPIRE studies built the evidence base for acute recurrent and chronic disease. The 2016 risk factors study, published in JAMA Pediatrics, established the three-domain framework and showed that at least one risk factor was identifiable in a substantial majority of children with acute recurrent or chronic pancreatitis, with genetic causes predominating in chronic disease. The INSPPIRE-2 analysis expanded the cohort and quantified the disease burden in terms of pain, healthcare use and quality of life. The nutritional position paper set out the principles of early feeding, and the autoimmune pancreatitis consensus defined the steroid-responsive subtype. [5] [6] [7] [8]
The controversies that remain are the ones examiners probe for breadth. The optimal fluid rate and type — normal saline versus lactated Ringer solution, and the exact multiplier on maintenance — is still debated. The threshold for and route of enteral feeding — oral versus nasogastric versus nasojejunal — is individualised rather than protocolised. The role of ERCP versus surgery for ductal disease is centre-dependent. And the timing and selection criteria for total pancreatectomy with islet autotransplantation, which has shown promising results in reducing opioid use and improving quality of life, are still being refined as more centres develop the capability. [10] [12]
Exam Pearls
The diagnostic criteria — Two of Three
References
- [1]Abu-El-Haija M; Kumar S; Szabo F; Jażdżewska M; Ranganathan S; Werlin SL Classification of Acute Pancreatitis in the Pediatric Population: Clinical Report From the NASPGHAN Pancreas Committee. J Pediatr Gastroenterol Nutr, 2017.PMID 28333771
- [2]Abu-El-Haija M; Kumar S; Quiros JA; Balzer B; Durie PR; Elinoff B Management of Acute Pancreatitis in the Pediatric Population: A Clinical Report From the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition Pancreas Committee. J Pediatr Gastroenterol Nutr, 2018.PMID 29280782
- [3]Párniczky A; Abu-El-Haija M; Husain S; Lowe M; Amir L; Brodzicki J EPC/HPSG evidence-based guidelines for the management of pediatric pancreatitis. Pancreatology, 2018.PMID 29398347
- [4]Taylor CJ; Chen K; Horvath K; Hughes J; Rothbaum R; Shun-Shin M ESPGHAN and NASPGHAN Report on the Assessment of Exocrine Pancreatic Function and Pancreatitis in Children. J Pediatr Gastroenterol Nutr, 2015.PMID 25915425
- [5]Kumar S; Ooi CY; Werlin S; Abu-El-Haija M; Barth B; Bellin MD Risk Factors Associated With Pediatric Acute Recurrent and Chronic Pancreatitis: Lessons From INSPPIRE. JAMA Pediatr, 2016.PMID 27064572
- [6]Uc A; Cress GA; Wang F; Abou-Resh N; Barth BA; Bellin MD Analysis of INSPPIRE-2 Cohort: Risk Factors and Disease Burden in Children With Acute Recurrent or Chronic Pancreatitis. J Pediatr Gastroenterol Nutr, 2022.PMID 35976273
- [7]Abu-El-Haija M; Uc A; Werlin SL; Freeman AJ; Geiger A; Homme J Nutritional Considerations in Pediatric Pancreatitis: A Position Paper From the NASPGHAN Pancreas Committee and ESPGHAN Cystic Fibrosis/Pancreas Working Group. J Pediatr Gastroenterol Nutr, 2018.PMID 29927872
- [8]Scheers I; Palermo JJ; Freedman S; Wilschanski M; Smith G; Ali MF Recommendations for Diagnosis and Management of Autoimmune Pancreatitis in Childhood: Consensus From INSPPIRE. J Pediatr Gastroenterol Nutr, 2018.PMID 29746340
- [9]Husain SZ; Morinville V; Pohl J; Rabinowitz S; Arsenescu R; Barth BA Toxic-metabolic Risk Factors in Pediatric Pancreatitis: Recommendations for Diagnosis, Management, and Future Research. J Pediatr Gastroenterol Nutr, 2016.PMID 26594832
- [10]Troendle DM; Fishman DS; Barth BA; Giefer MJ; Lin TK; Palermo JJ Therapeutic Endoscopic Retrograde Cholangiopancreatography in Pediatric Patients With Acute Recurrent and Chronic Pancreatitis: Data From the INSPPIRE Study. Pancreas, 2017.PMID 28609364
- [11]Dike CR; Zimmerman B; Zheng Y; Lin TK; Hornung L; Bisgaard SG Clinical and Practice Variations in Pediatric Acute Recurrent or Chronic Pancreatitis: Report From the INSPPIRE Study. J Pediatr Gastroenterol Nutr, 2020.PMID 32079978
- [12]Heinzman C; Hornung L; Lin TK; Stevens J; Spence AB; Bunch K Total pancreatectomy with islet autotransplantation reduces opioid use and improves nutritional support in children with debilitating pancreatitis. PLoS One, 2023.PMID 37540665