Paeds · gastroenterology-hepatology-and-nutrition
Polyps and inherited gastrointestinal cancer syndromes
Also known as Juvenile polyposis syndrome · Familial adenomatous polyposis · Peutz-Jeghers syndrome · Inherited polyposis · Hamartomatous polyposis · MUTYH-associated polyposis · PTEN hamartoma tumour syndrome
Fellowship guide to gastrointestinal polyps in children: distinguishing the benign isolated juvenile polyp from the inherited cancer syndromes juvenile polyposis, familial adenomatous polyposis, Peutz-Jeghers syndrome and MUTYH-associated polyposis, with the genes, histology, cancer risks, red-flag cutaneous clues, and the surveillance and prophylactic surgery that prevent early colorectal and extraintestinal cancer.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A four-year-old who passes a few drops of bright red blood with an otherwise normal stool, a teenager whose father had a colectomy for hundreds of colon polyps, and a boy with dark freckles on his lips and recurrent belly pain are very different presentations that all turn on the same question: is this child's bowel growth a harmless one-off, or the first sign of a cancer syndrome that runs through the family? Gastrointestinal polyps are abnormal outgrowths of the bowel lining into the lumen, and in children they span a spectrum from the common, benign isolated juvenile polyp to rare inherited syndromes that carry a high lifetime risk of cancer. [7]
The single most useful division is histological. Hamartomatous polyps are disorganised overgrowths of the tissue that normally belongs there, mucus-filled and lobulated, and an isolated juvenile polyp of this kind is benign. Adenomatous polyps are true neoplasms in which the glandular epithelium has begun to proliferate and accumulate genetic damage, placing them on the adenoma-to-carcinoma pathway that produces colorectal cancer. The inherited syndromes cluster around these two histologies: juvenile polyposis syndrome and Peutz-Jeghers syndrome are hamartomatous, while familial adenomatous polyposis and MUTYH-associated polyposis are adenomatous. [7]
What makes these syndromes dangerous, and what makes them examinable, is that the polyps are only the visible tip of an inherited defect that touches many organs and raises cancer risk across a lifetime. The clinician's job is to recognise when a child's polyp is not an isolated event, to confirm the syndrome with histology and germline genetics, and to enrol the child and family in structured, lifelong surveillance that prevents cancer. [9]
Classification
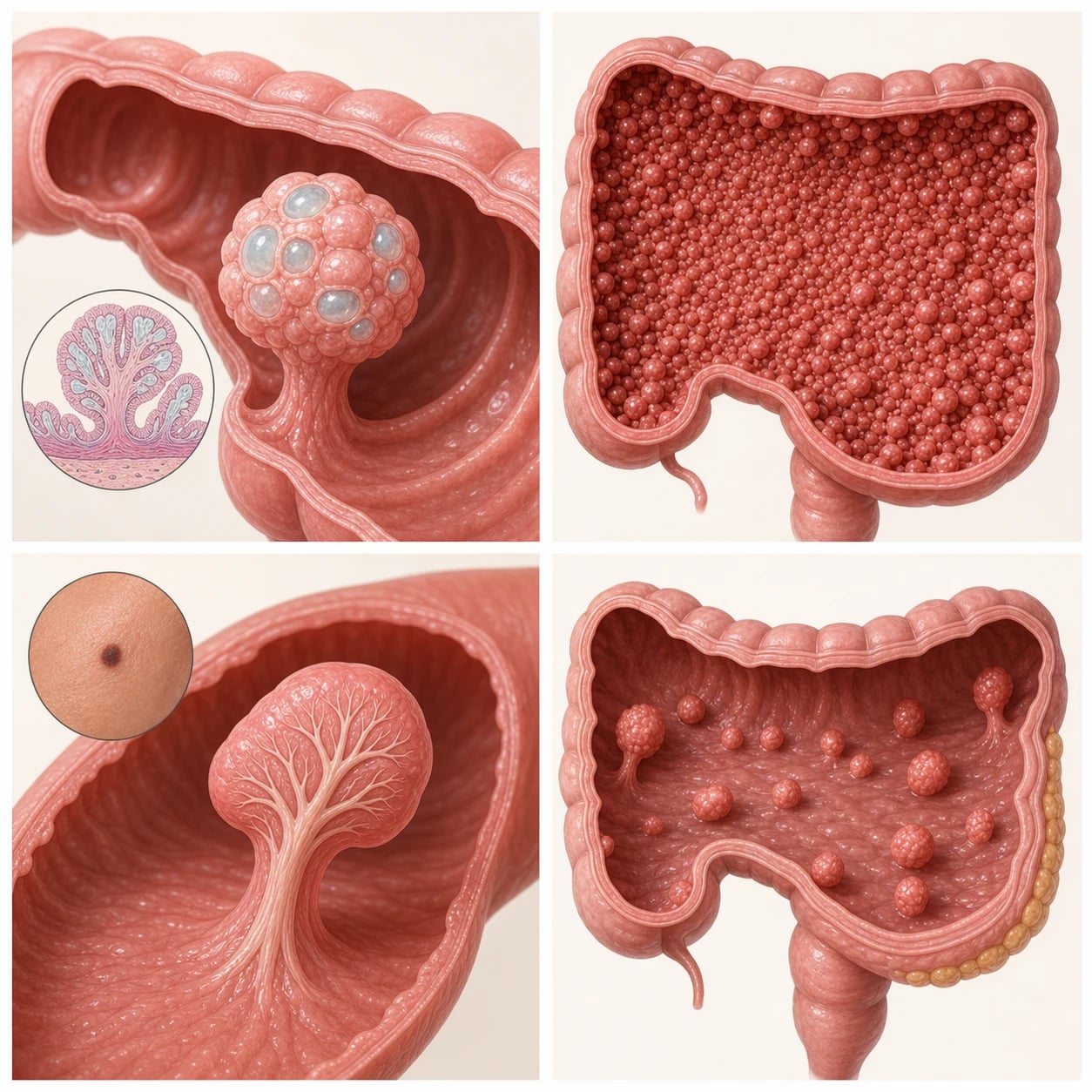
The clearest way to hold the syndromes in mind is to split them first by histology and then by inheritance, because histology decides the cancer mechanism and inheritance decides who else in the family is at risk. The hamartomatous syndromes are juvenile polyposis syndrome and Peutz-Jeghers syndrome; the adenomatous syndromes are familial adenomatous polyposis, its attenuated form, and MUTYH-associated polyposis. The PTEN hamartoma tumour syndromes, such as Cowden syndrome, form a separate hamartomatous group with their own extraintestinal cancer profile. [7]
[9]A practical consequence of this table is that almost every syndrome is autosomal dominant except MUTYH-associated polyposis, which is recessive. That single fact changes the counselling: a dominant syndrome puts parents, siblings, and children at one in two risk and makes predictive testing of relatives central, whereas a recessive syndrome puts siblings at one in four risk and rarely affects the parents directly. [8]
Epidemiology & Risk Factors
Isolated juvenile polyps are common in general paediatric practice and are the most frequent gastrointestinal polyp of childhood, typically presenting with painless rectal bleeding between two and ten years of age. By contrast, the inherited syndromes are individually rare. Juvenile polyposis syndrome affects roughly one in 100,000 people, classic familial adenomatous polyposis about one in 7,000 to one in 10,000, and Peutz-Jeghers syndrome about one in 200,000. [7]
Because the major syndromes are autosomal dominant with high penetrance, the strongest risk factor by far is a first-degree affected relative. A family history that should trigger referral to a familial cancer service includes early colorectal or gastric cancer, pancreatic or breast cancer at a young age, gynaecological or testicular tumours, desmoid tumours, hepatoblastoma, or any known polyposis gene mutation. Many affected children are identified not because they are symptomatic but because a relative's diagnosis prompts predictive genetic testing. [9]
A specific and easily missed risk is hepatoblastoma in infants with familial adenomatous polyposis, which is several times more common than in the general population and is the reason some guidelines consider screening affected infants with alpha-fetoprotein and abdominal ultrasound. Recognising this links a liver tumour in infancy to an underlying APC mutation and vice versa. [2]
Pathophysiology
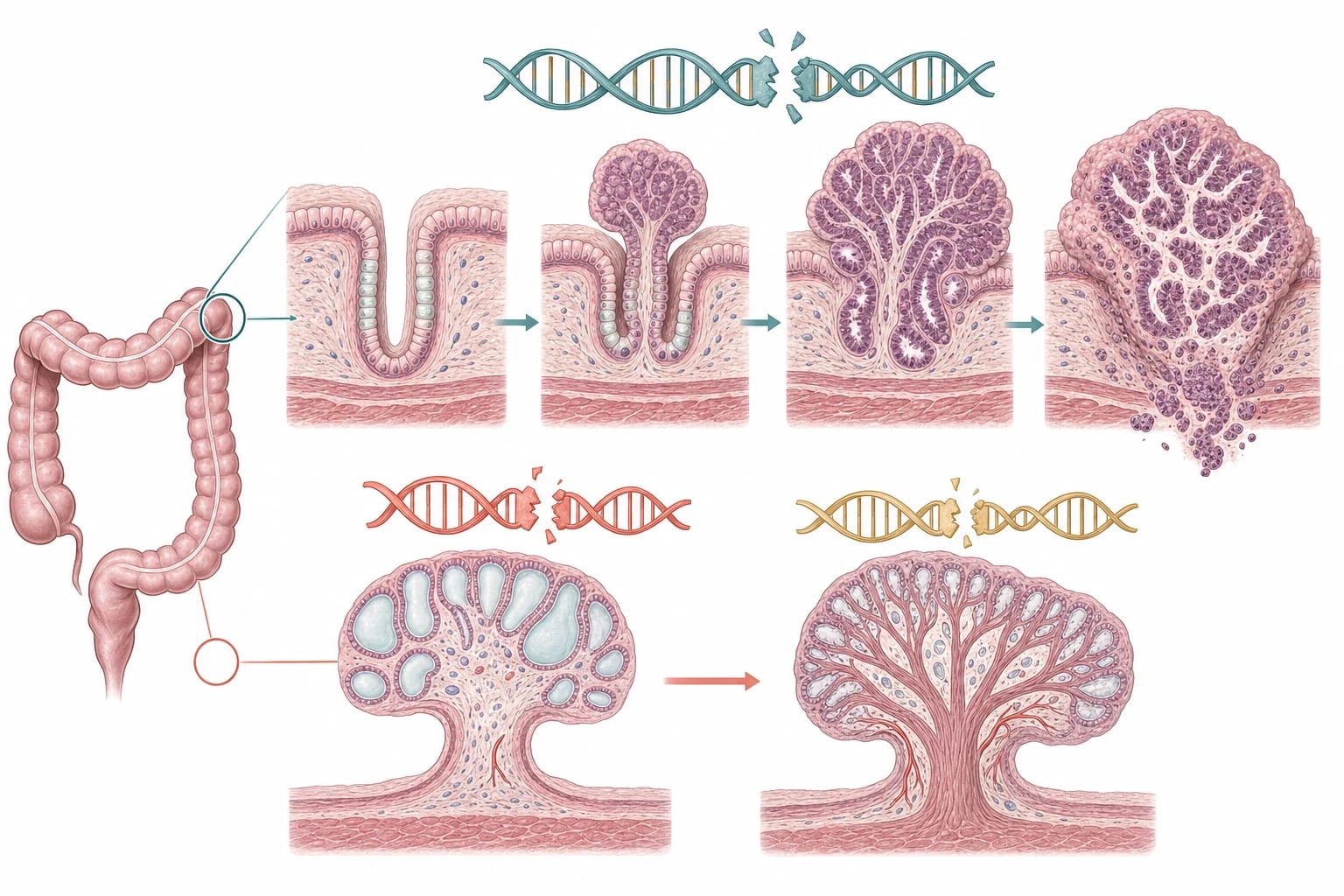
Although the syndromes look different, each ultimately follows the loss of a tumour-suppressor pathway that normally restrains how gut cells grow. In familial adenomatous polyposis, a child inherits one broken copy of the APC gene on chromosome 5q. The second, healthy copy is then lost by chance in individual colonic crypts, and once both copies are gone the brake on the Wnt and beta-catenin signalling pathway is released, so the crypt proliferates into an adenoma. Because this two-hit loss happens independently in hundreds of crypts, hundreds of adenomas appear, and over years the cells in any one of them can accumulate further damage and progress to invasive carcinoma. [2]
The hamartomatous syndromes fail by a different route. In juvenile polyposis syndrome, a germline mutation in SMAD4 or BMPR1A disrupts the BMP and transforming growth factor beta pathway that tells gut stroma and epithelium when to stop growing, producing the swollen, mucus-filled juvenile polyps. In Peutz-Jeghers syndrome, loss of STK11, also called LKB1, dysregulates energy sensing through AMP-activated kinase and mTOR signalling, producing hamartomatous polyps with their characteristically tree-like arborising core of smooth muscle. These hamartomatous polyps are not neoplasms at birth, but they are not inert either, and over many years a proportion acquire dysplasia and progress to cancer. [5]
MUTYH-associated polyposis is the odd one out because it is recessive. The MUTYH protein repairs oxidative DNA damage through the base excision repair pathway, and when both copies are faulty, guanine mispairs accumulate and seed adenomas. It typically produces an attenuated adenomatous polyposis that declares itself in adulthood, which is why it is a differential to name rather than a paediatric diagnosis to chase. [8]
Clinical Presentation
The presentation of a polyp in a child is usually simple, and the art lies in spotting when it is not. An isolated juvenile polyp causes painless bright red blood coating the outside of an otherwise normal stool, sometimes with anaemia or, occasionally, a fleshy mass prolapsing at the anus during defaecation, in a well child between two and ten years old. This story is so characteristic that the first task is simply to confirm it and remove the polyp. [7]
The syndromes reframe that story in three ways. Juvenile polyposis syndrome presents like an isolated juvenile polyp but with multiple polyps, so the child has recurrent bleeding, chronic iron-deficiency anaemia, diarrhoea, protein-losing enteropathy with hypoalbuminaemia, or failure to thrive, sometimes beginning in infancy. Familial adenomatous polyposis is usually silent in childhood and surfaces through family screening, so any child who is actually symptomatic with bleeding, diarrhoea, or abdominal pain already has a significant polyp burden. Peutz-Jeghers syndrome shows its hand on the skin before it shows it in the bowel. [3]
Age-related and syndrome-specific clues
Toddler or preschool child: painless rectal bleeding from an isolated juvenile polyp, well between episodes
Any age with multiple juvenile polyps: anaemia, diarrhoea, enteropathy, or failure to thrive favour juvenile polyposis syndrome
Screened asymptomatic teenager with an affected parent: think familial adenomatous polyposis until proven otherwise
Lip and buccal freckling with cramping pain or a palpable mass: Peutz-Jeghers polyps and possible intussusception
Boy with gynaecomastia or early puberty: Peutz-Jeghers Sertoli-cell tumour
Infant with hepatoblastoma: consider underlying familial adenomatous polyposis
The cutaneous pigmentation of Peutz-Jeghers syndrome is its signature and is worth knowing in detail. Small dark brown to blue-grey freckles appear in infancy on the lips, around the mouth, on the buccal mucosa, and on the fingers and toes, and they characteristically fade during adolescence and adulthood even as the cancer risk persists. Their appearance, combined with cramping mid-abdominal pain and anaemia from small-bowel polyps, or with acute intussusception in late childhood, makes Peutz-Jeghers one of the few syndromes a clinician can suspect at the bedside before any test. [6]
Differential Diagnosis
The first fork in the differential is whether a juvenile polyp is truly isolated or syndromic, and three findings reclassify it. Three or more juvenile polyps, any juvenile polyp outside the colon, or any juvenile polyp in a child with a family history move the diagnosis from a benign one-off to juvenile polyposis syndrome. The histology, the polyp count, the distribution along the gut, and the family history together settle this, which is why every polyp removed must be sent for full histological typing. [1]
Once the colon is examined, the polyp burden and its histology point to the remaining syndromes. Innumerable small adenomas carpeting the colon mean familial adenomatous polyposis or, when there are only tens of adenomas with biallelic MUTYH, MUTYH-associated polyposis. Hamartomatous polyps with an arborising smooth-muscle core and the characteristic pigmentation mean Peutz-Jeghers syndrome. Macrocephaly with hamartomas and a PTEN mutation points to a PTEN hamartoma tumour syndrome such as Cowden syndrome. [9]
[10]In a child who simply presents with rectal bleeding, the non-syndromic causes must not be forgotten. An anal fissure, infectious colitis, Meckel diverticulum, and inflammatory bowel disease all produce bleeding and each has distinguishing features. Lymphoid nodular hyperplasia and the rare Cronkhite-Canada syndrome are less common mimics. The point is that a single bleeding polyp removed at colonoscopy in a well child with a normal family history closes the question, while anything atypical opens the syndromic differential. [7]
Clinical & Bedside Assessment
The highest-yield instrument in this topic is a carefully drawn three-generation family tree. Ask specifically about colorectal and gastric cancer at a young age, pancreatic, breast, endometrial, and ovarian cancer, medulloblastoma, desmoid tumours, hepatoblastoma, and testicular tumours, and about anyone who has had polyps removed or a colectomy. A pedigree that shows dominant transmission of early cancer across generations reframes an isolated polyp as part of a syndrome and drives referral to a familial cancer service. [9]
The physical examination is where several syndromes declare themselves. Inspect the lips, buccal mucosa, and the fingers and toes for the dark freckles of Peutz-Jeghers syndrome. Look for epidermoid cysts, osteomas, and dental abnormalities, which mark the Gardner variant of familial adenomatous polyposis. Measure head circumference for the macrocephaly of a PTEN hamartoma syndrome, and assess for the developmental features that accompany it. Examine the breasts and genitalia of boys with Peutz-Jeghers for gynaecomastia or signs of feminisation from a Sertoli-cell tumour. [10]
Growth and puberty must always be plotted, because failure to thrive, short stature, delayed puberty, or precocious puberty are clues to chronic disease or a hormone-secreting tumour. Palpate the abdomen for a mass or a palpable loop of intussuscepted bowel, and assess for the pallor of iron-deficiency anaemia. A careful bedside assessment, combining a focused pedigree with a search for cutaneous, dysmorphic, and endocrine signs, frequently makes the diagnosis before the endoscope or the gene panel is ordered. [7]
Investigations
Diagnosis combines three streams: histopathology of every polyp, endoscopic mapping of the polyp burden, and germline genetic testing. Any polyp removed must be fully characterised by an experienced pathologist to distinguish juvenile, Peutz-Jeghers-type hamartomatous, and adenomatous types and to grade any dysplasia, because this single read determines whether a child has a benign isolated polyp or a cancer syndrome. [7]
Once a syndrome is suspected, colonoscopy and oesophagogastroduodenoscopy define how many polyps are present and where, and allow endoscopic clearance of accessible lesions. For Peutz-Jeghers syndrome the small bowel must also be assessed, because its polyps cluster in the jejunum and cause intussusception; magnetic resonance enterography or video capsule endoscopy are the preferred tools, with capsule used cautiously in a child with obstructive symptoms. [3]
Germline genetic testing confirms the diagnosis and unlocks predictive testing for the rest of the family. APC is tested for familial adenomatous polyposis, SMAD4 and BMPR1A for juvenile polyposis syndrome, STK11 for Peutz-Jeghers syndrome, PTEN for the PTEN hamartoma syndromes, and biallelic MUTYH for MUTYH-associated polyposis. A child found to carry a SMAD4 mutation must also be screened for hereditary haemorrhagic telangiectasia, because juvenile polyposis and hereditary haemorrhagic telangiectasia overlap in the JP-HHT syndrome with its risk of vascular malformations and bleeding. [9]
Blood tests target the consequences of chronic polyp disease: a full blood count and ferritin for iron-deficiency anaemia, and albumin and a protein level when protein-losing enteropathy is suspected in juvenile polyposis. In an infant at risk of familial adenomatous polyposis, the question of hepatoblastoma screening with serial alpha-fetoprotein and abdominal ultrasound is considered, balancing the modest absolute risk against the value of early detection of an aggressive tumour. [2]
Management — Resuscitation

Most children with polyps or an inherited syndrome are haemodynamically stable and present electively, but two situations demand immediate action. Acute, ongoing lower gastrointestinal bleeding causing shock is rare in childhood polyposis but, when it occurs, requires airway, breathing, and circulation assessment, secure intravenous access, fluid or blood transfusion for haemodynamic instability, and urgent gastroenterology and surgical review. [6]
The more common emergency is intussusception of a Peutz-Jeghers small-bowel polyp. A child with known or suspected Peutz-Jeghers who develops severe colicky abdominal pain, bilious vomiting, and a palpable abdominal mass may have a polyp acting as the lead point for intussusception, which can obstruct or, less commonly, ischaemia the bowel. This is a surgical emergency: resuscitate with intravenous fluids, keep the child fasted, obtain imaging, and arrange urgent surgical or endoscopic reduction. A large polyp that prolapses and strangulates at the anus likewise needs urgent surgical review. [6]
For the far more common elective presentation, the resuscitation phase is really a diagnostic and counselling phase. The immediate priorities are to establish the histological diagnosis, assess the polyp burden, and begin genetic counselling, so that surveillance and family testing can be planned deliberately rather than in a panic. The duty to recognise intussusception and significant haemorrhage is constant, but most decisions in this topic are planned and longitudinal. [9]
Management — Definitive & Stepwise
Definitive care is lifelong and built on three pillars that apply, in different proportions, to every syndrome: endoscopic surveillance with polypectomy, prophylactic surgery when the polyp burden can no longer be controlled endoscopically, and extraintestinal cancer screening coordinated through a familial cancer service. The isolated juvenile polyp sits outside this framework entirely: one or two typical juvenile polyps in a well child with a normal family history are removed once at colonoscopy and need no further surveillance. [7]
Juvenile polyposis syndrome is managed by endoscopic surveillance and polypectomy rather than prophylactic surgery. The ESPGHAN position paper recommends oesophagogastroduodenoscopy and colonoscopy beginning at the onset of symptoms or in the early to mid-teens, repeated every one to three years, with all polyps removed to control bleeding and anaemia and to reduce cancer risk. Affected children are enrolled in lifelong surveillance because of the substantial cumulative colorectal and gastric cancer risk. [1]
Familial adenomatous polyposis is the syndrome in which prophylactic surgery dominates. At-risk children undergo flexible sigmoidoscopy from the early to mid-teens, moving to colonoscopy once adenomas appear, and once the polyp burden or dysplasia can no longer be controlled endoscopically a prophylactic colectomy is planned, usually in the late teens to mid-twenties. The surgical options are a total colectomy with ileorectal anastomosis or a proctocolectomy with ileal pouch-anal anastomosis, chosen according to the rectal polyp burden, the family desmoid risk, and the wishes of the young person, with lifelong upper gastrointestinal surveillance for duodenal adenomas added in adulthood. [2]
Familial adenomatous polyposis surveillance and surgery
Dose
Flexible sigmoidoscopy from the early to mid-teens, colonoscopy once adenomas appear, annual to biennial; prophylactic colectomy in the late teens to mid-twenties once polyp burden or dysplasia is not endoscopically controllable
Peutz-Jeghers syndrome is managed mainly by surveillance and preventive polypectomy. The ESPGHAN position paper recommends small-bowel surveillance from late childhood, around eight years, using magnetic resonance enterography or video capsule endoscopy, together with oesophagogastroduodenoscopy and colonoscopy, with large or symptomatic small-bowel polyps removed to prevent intussusception and bleeding. Because of the very high extraintestinal cancer risk, surveillance extends across the lifetime to the breast, pancreas, gynaecological organs, and testes under a familial cancer service. [3]
[3]Underpinning all of this is the genetic service. Every child with a confirmed syndrome is referred for genetic counselling, predictive testing of at-risk relatives, and coordination of lifelong, age-based screening. The general paediatrician usually holds the coordinating role, linking gastroenterology, surgery, genetics, oncology, and psychology, and planning a structured transition to adult care as adolescence approaches. [9]
Specific Subtypes & Scenarios
Juvenile polyposis syndrome is defined by its criteria rather than by a single polyp: more than five juvenile polyps in the colorectum, any juvenile polyp elsewhere in the gastrointestinal tract, or any number of juvenile polyps in a child with a family history. Roughly half of affected families carry a germline SMAD4 or BMPR1A mutation, and SMAD4 carriers overlap with hereditary haemorrhagic telangiectasia in the JP-HHT syndrome, which adds vascular malformations, epistaxis, and visceral arteriovenous malformations to the cancer risk. [1]
Familial adenomatous polyposis is the prototypical adenomatous syndrome. Classic disease shows hundreds to thousands of adenomas from an APC mutation, while attenuated familial adenomatous polyposis has fewer than one hundred adenomas and a later onset. The Gardner variant adds desmoid tumours, osteomas, and epidermoid cysts, and the Turcot variant associates familial adenomatous polyposis with medulloblastoma. The desmoid tumour deserves special emphasis: it is a locally invasive fibroproliferative mass, often triggered by surgery, that can encase mesenteric vessels and is a leading cause of death in familial adenomatous polyposis, which is why desmoid risk shapes the timing and type of colectomy. [2]
Peutz-Jeghers syndrome is the syndrome with the broadest reach. The STK11 mutation produces the characteristic pigmentation and small-bowel hamartomatous polyps, but its burden extends to a very high lifetime cancer risk across gastrointestinal, breast, pancreatic, gynaecological, and testicular sites. Boys are at risk of feminising Sertoli-cell tumours that cause gynaecomastia and precocious puberty, and girls of sex-cord tumours with annular tubules and other gynaecological neoplasms. The childhood complications are anaemia, intussusception, and obstruction, and the lifelong task is multicancer surveillance. [4]
MUTYH-associated polyposis is the recessive exception and is included chiefly as a differential. Biallelic MUTYH mutations cause an attenuated adenomatous polyposis with a high colorectal cancer risk that usually declares itself in adulthood, so it rarely presents to paediatric services but must be named when an adolescent or young adult has tens of adenomas and no APC mutation. The PTEN hamartoma tumour syndromes, including Cowden syndrome, present with macrocephaly, gastrointestinal hamartomas, and a distinct profile of breast, thyroid, and endometrial cancer risk managed mainly in adult familial cancer services. [8]
Complications & Pitfalls
Complications divide into those generated by the polyps themselves and those that follow treatment. The polyps cause chronic iron-deficiency anaemia, hypoproteinaemia and protein-losing enteropathy, and intussusception with obstruction, which is especially characteristic of Peutz-Jeghers small-bowel polyps. Over years, the polyps of every syndrome can progress to adenocarcinoma, and that inexorable progression is the entire rationale for surveillance and prophylactic surgery. [5]
In familial adenomatous polyposis the dominant complication is the desmoid tumour, a locally invasive mass that is provoked by surgical trauma, recurs aggressively, and can compress mesenteric vessels and ureters. Because desmoid disease is a leading cause of death in this syndrome, a strong family history of desmoids pushes the surgical choice towards the option that minimises further operations and away from premature surgery. Surgery itself brings pouchitis, reduced fertility, and altered bowel function that weigh heavily on a young person's quality of life. [2]
The classic pitfalls are all avoidable. Dismissing three or four juvenile polyps as benign one-offs misses juvenile polyposis syndrome. Failing to take a three-generation family history, or to examine the lips, mouth, and head circumference, misses Peutz-Jeghers and the PTEN syndromes at the bedside. Removing a polyp without sending it for full histological typing throws away the decisive diagnostic step. Performing prophylactic colectomy too early in familial adenomatous polyposis, or without assessing desmoid risk, trades one danger for another. And ordering genetic testing on a child without proper counselling, or forgetting to offer predictive testing to at-risk relatives, wastes the single most valuable outcome of making the diagnosis. [9]
Prognosis & Disposition
With early genetic diagnosis and disciplined surveillance, colorectal cancer is largely preventable and life expectancy approaches normal, but each syndrome carries its own burden. Untreated classic familial adenomatous polyposis progresses to colorectal cancer at a median age in the late thirties to early forties, which is why prophylactic colectomy is timed before this point. Juvenile polyposis carries a cumulative colorectal cancer risk of roughly thirty-eight to thirty-nine per cent and a meaningful gastric cancer risk. Peutz-Jeghers carries the heaviest overall burden, with a lifetime cancer risk that may exceed eighty per cent across many sites. [4]
Disposition is shared and lifelong. The general paediatrician usually coordinates care, linking paediatric gastroenterology for surveillance and polypectomy, paediatric surgery for prophylactic and emergency operations, clinical genetics for diagnosis and predictive family testing, oncology for extraintestinal cancer screening, and psychology for the substantial burden of living with a cancer diagnosis. Regular review, adherence to the surveillance schedule, and clear communication with the family and the young person are what turn a frightening diagnosis into a managed, predictable condition. [9]
Adolescence is the pivotal transition. The young person takes increasing ownership of surveillance, faces major decisions about prophylactic surgery that affect fertility and body image, and must move from paediatric to adult familial cancer services. A structured, planned transition that hands over a complete record, a clear surveillance plan, and the genetic information the adult service needs is essential, because disengagement in the gap between services is a well-recognised cause of late cancer presentation. [8]
Special Populations
Children identified only through an affected relative face the particular question of predictive genetic testing. Testing is generally deferred until a pathogenic variant has been confirmed in the family and is offered with genetic counselling and the child's assent, timed so that the result changes management rather than simply labelling a child. In familial adenomatous polyposis, testing guides when to start surveillance; in juvenile polyposis and Peutz-Jeghers, it confirms who does and does not need endoscopy. [9]
Infants and toddlers with familial adenomatous polyposis are a distinct group because of the hepatoblastoma risk, and some programmes consider screening with alpha-fetoprotein and abdominal ultrasound in the first years of life. Boys with Peutz-Jeghers need surveillance for feminising Sertoli-cell tumours and precocious puberty, and girls enter gynaecological surveillance as they grow. Each age band brings its own surveillance emphasis, and the plan is revisited at every stage. [3]
Families from lower-resource settings, migrant and refugee backgrounds, and Indigenous communities may face real barriers to genetic testing, surveillance endoscopy, and specialist multidisciplinary care. Practical measures help to close this gap: clear explanation, primary care coordination, telehealth genetic counselling and specialist review, and attention to culturally safe communication about cancer and inheritance. The biology is the same everywhere, but access is not, and equity of surveillance is part of the standard of care. [9]
[9]Evidence, Guidelines & Regional Differences
The three ESPGHAN Polyposis Working Group position papers from 2019 are the paediatric reference standards for this topic. Cohen and colleagues address juvenile polyposis syndrome, Hyer and colleagues familial adenomatous polyposis, and Latchford and colleagues Peutz-Jeghers syndrome, each giving age-based recommendations for surveillance, polypectomy, and prophylactic surgery in children and adolescents. Together they define the framework within which most paediatric practice and examination answers are built. [1]
The supporting evidence defines the cancer risks and the management consequences. The Beggs 2010 systematic review consolidated Peutz-Jeghers management and the very high cumulative cancer risk, while the Blatter 2020 cohort of over two hundred European patients characterised the disease expression and cancer risk of juvenile polyposis syndrome in SMAD4 and BMPR1A carriers. The joint European Hereditary Tumour Group and European Society of Coloproctology 2024 guideline updates the management of familial adenomatous polyposis, MUTYH-associated polyposis, and the rare adenomatous polyposis syndromes, mainly from an adult perspective. [4]
[8]Controversies and regional differences persist despite the broad framework. The optimal age to begin and the best modality for small-bowel surveillance in Peutz-Jeghers remain debated, as do the timing and type of prophylactic surgery in familial adenomatous polyposis when balanced against desmoid risk. Whether to screen infants with familial adenomatous polyposis for hepatoblastoma divides programmes, and the management of the SMAD4 JP-HHT overlap is still being refined. Gene-panel and exome sequencing are increasingly reshaping diagnosis when the clinical picture is atypical, and access to genetic services and surveillance varies widely between and within countries. [9]
Exam Pearls
POLYP for the paediatric polyp decision
References
- [1]Cohen S, Hyer W, Mas E, Auth M, Attard TM, Spalinger J, Latchford A, Durno C Management of Juvenile Polyposis Syndrome in Children and Adolescents: A Position Paper From the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr, 2019.PMID 30585890
- [2]Hyer W, Cohen S, Attard T, Vila-Miravet V, Pienar C, Auth M, Septer S, Hawkins J, Durno C, Latchford A Management of Familial Adenomatous Polyposis in Children and Adolescents: Position Paper From the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr, 2019.PMID 30585891
- [3]Latchford A, Cohen S, Auth M, Scaillon M, Viala J, Daniels R, Talbotec C, Attard T, Durno C, Hyer W Management of Peutz-Jeghers Syndrome in Children and Adolescents: A Position Paper From the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr, 2019.PMID 30585892
- [4]Beggs AD, Latchford AR, Vasen HF, Moslein G, Alonso A, Aretz S Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut, 2010.PMID 20581245
- [5]Blatter R, Tschupp B, Aretz S, Bernstein I, Colas C, Evans DG, Genuardi M, Hes FJ, Huneburg R, Jarvinen H, Lalloo F, Moeslein G, Renkonen-Sinisalo L, Resta N, Spier I, Varvara D, Vasen H, Latchford AR, Heinimann K Disease expression in juvenile polyposis syndrome: a retrospective survey on a cohort of 221 European patients and comparison with a literature-derived cohort of 473 SMAD4/BMPR1A pathogenic variant carriers. Genet Med, 2020.PMID 32398773
- [6]Hinds R, Philp C, Hyer W, Fell JM Complications of childhood Peutz-Jeghers syndrome: implications for pediatric screening. J Pediatr Gastroenterol Nutr, 2004.PMID 15269641
- [7]Durno CA Colonic polyps in children and adolescents. Can J Gastroenterol, 2007.PMID 17431512
- [8]Zaffaroni G, Mannucci A, Koskenvuo L, de Lacy B, Maffioli A, Bisseling T Updated European guidelines for clinical management of familial adenomatous polyposis (FAP), MUTYH-associated polyposis (MAP), gastric adenocarcinoma, proximal polyposis of the stomach (GAPPS) and other rare adenomatous polyposis syndromes: a joint EHTG-ESCP revision. Br J Surg, 2024.PMID 38722804
- [9]MacFarland SP, Zelley K, Rojas I, Durno C Genetic Testing in Gastrointestinal Polyposis Syndromes: Considerations in Pediatrics. Genes (Basel), 2026.PMID 42353760
- [10]Wang MX, Shi A, Shetty D, Mousa M, Diab M, Taher A Cowden Syndrome: Imaging Review and Cancer Surveillance. Radiographics, 2026.PMID 41746821