Paeds · gastroenterology-hepatology-and-nutrition
Short-bowel syndrome and intestinal failure
Also known as Short bowel syndrome · Intestinal failure · Short gut syndrome · Parenteral nutrition dependence · Intestinal rehabilitation · Teduglutide therapy · Serial transverse enteroplasty · Intestinal failure-associated liver disease · IFALD · Intestinal transplant
Fellowship guide to short-bowel syndrome and intestinal failure in children, built around the rule that the remaining anatomy matters more than the residual length: an intact colon and ileocaecal valve is the strongest predictor that a child will one day feed entirely by mouth. The page covers how massive small-bowel resection from necrotising enterocolitis, volvulus or atresia produces malabsorption, how the remnant gut adapts under the drive of enteral feeding and glucagon-like peptide 2, how to manage parenteral nutrition, advance feeds and balance the sodium-hungry high-output stoma, when to add teduglutide at 0.05 milligrams per kilogram subcutaneously once daily, when to offer serial transverse enteroplasty, and how to recognise and prevent intestinal failure-associated liver disease and central-line bloodstream infection.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Picture a baby born at 28 weeks who develops necrotising enterocolitis, loses most of the small bowel, and comes back to the ward with a high jejunostomy and a central line dripping parenteral nutrition. Or picture a four-year-old who had a mid-gut volvulus as a newborn and now feeds almost entirely by mouth, the gut having reshaped itself over years. These two children are the same diagnosis at opposite ends of its course, and they hold the central idea of this topic: when a child loses most of the small intestine, the body fights to make the remnant do more work, and the job of the paediatric team is to keep that child alive, growing and uninfected long enough for the gut to adapt — or to plan a transplant when it cannot. [1]
The vocabulary matters because examiners use the terms precisely. Short-bowel syndrome is the malabsorption that follows a massive resection of small intestine; it is an anatomical event with a functional consequence. Intestinal failure is the broader state in which the gut cannot sustain growth, hydration and electrolyte balance on its own, so the child needs parenteral support. Every short-bowel child who still needs parenteral nutrition has intestinal failure, but a child can have intestinal failure from mucosal disease or a dysmotile gut with a normal-length bowel. Holding that distinction stops the common viva slip of conflating the two. [1] [2]
The governing principle, then, is to classify the child by what remains, protect the remnant, and buy time for adaptation while preventing the two complications that end the race early — intestinal failure-associated liver disease and central-line bloodstream infection. Almost everything else in management flows from that single strategy. [1]
Classification
Begin classifying the child by what is left after the resection, because that anatomy predicts the future more reliably than any single measurement. The most useful split is whether the colon is in continuity. When the surgeon has been able to join the proximal small bowel to the colon, the child keeps a route for water and sodium absorption and a source of trophic hormones, and the outlook is far better. When the bowel ends in a stoma on the abdominal wall with the colon disconnected, the outlook is harder. [1] [2]
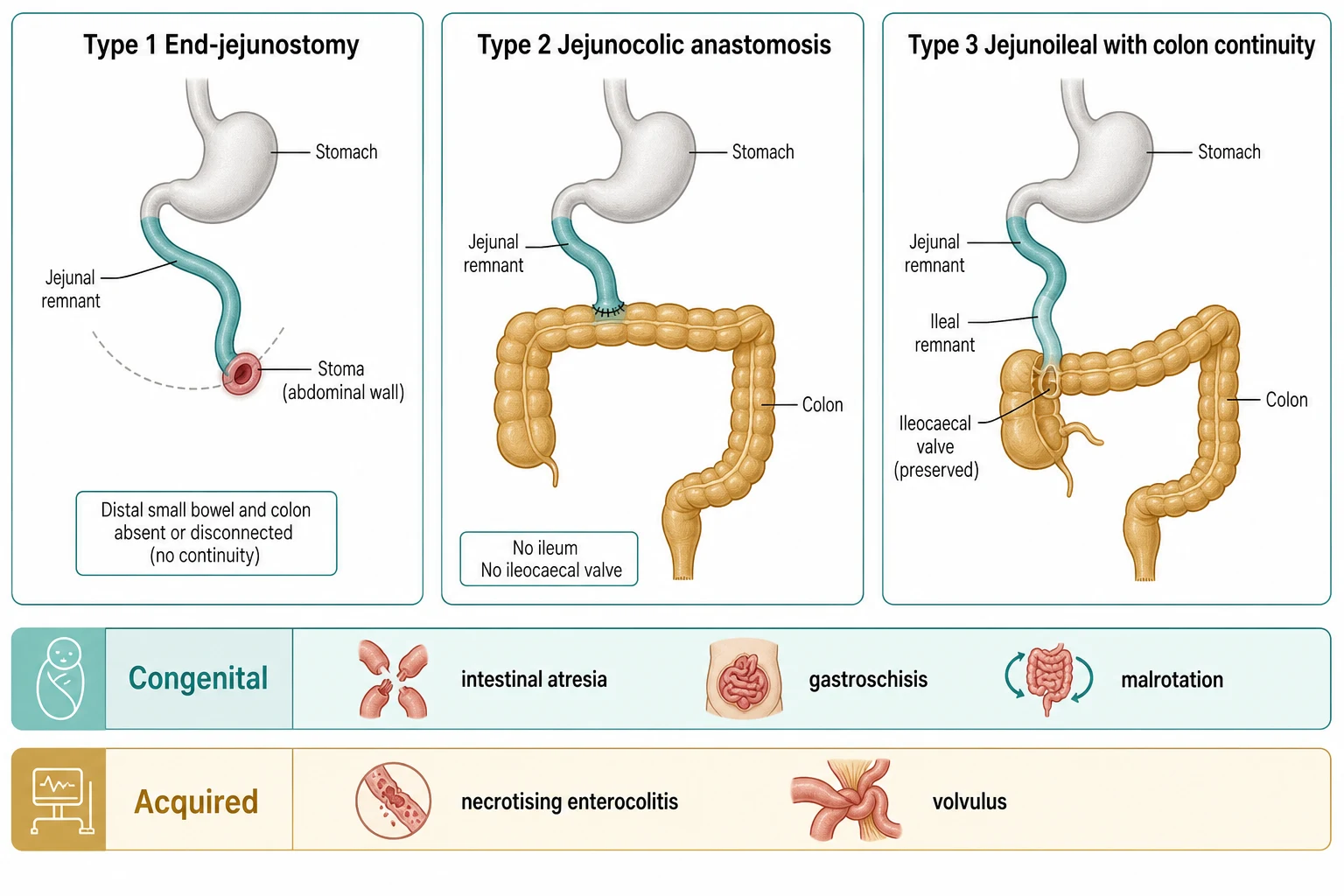
The three classical anatomical types run from worst to best. In type 1, an end-jejunostomy, the small bowel ends in a stoma and the distal bowel and colon are gone or resting disconnected; this child loses almost all water and sodium out of the stoma and is the hardest to wean. In type 2, a jejunocolic anastomosis, the jejunum is joined straight to the colon with the ileum removed, so the ileocaecal valve is lost. In type 3, a jejunoileal anastomosis with colon continuity, the ileocaecal valve and some ileum are preserved together with the colon, and this is the configuration most likely to reach enteral autonomy. [1]
Type 1 End-jejunostomy
- Small bowel ends in a stoma, no colon in continuity
- Highest water and sodium loss, hardest to wean
- Needs early establishment of continuity where possible
- Poorest prognosis for enteral autonomy
Type 2 Jejunocolic
- Jejunum anastomosed directly to colon
- Ileum and ileocaecal valve absent
- Colon recovers some water and sodium
- Intermediate prognosis
Type 3 Jejunoileal with colon
- Jejunum joined to ileal remnant then to intact colon
- Ileocaecal valve preserved
- Colon and ileum supply trophic hormones
- Best prognosis for enteral autonomy
A second, practical axis sorts short bowel by how it arose, because the cause shapes what else you must watch for. The congenital causes, intestinal atresia, gastroschisis and malrotation with mid-gut volvulus, often present in a newborn with a single catastrophic event and a relatively healthy liver. The acquired causes, dominated by necrotising enterocolitis in the premature infant, tend to carry the added burden of prematurity, recurrent sepsis and an immature liver that is already vulnerable to cholestasis. Knowing the cause tells you whether you are fighting one battle or several. [1] [2]
Residual small-bowel length is quoted because it is easy to measure at laparotomy, but it is a weak stand-alone predictor. Adults and older children are often said to have short-bowel syndrome when the remaining jejunoileum is under about 200 centimetres, and the outlook worsens steeply below about 100 centimetres. In infants, who normally have a longer relative bowel, a common teaching threshold is that losing more than three-quarters of the small bowel, or being left with a short remnant such as under about 25 to 40 centimetres with no colon, marks high-risk short bowel. These numbers orient the discussion, but you should always interpret them alongside the anatomy and the functional response to feeding rather than in isolation. [1] [2]
Epidemiology & Risk Factors
Necrotising enterocolitis is the single leading cause of intestinal failure in neonates, and the rise of neonatal intensive care has changed who develops short bowel. As more extremely premature and very-low-birth-weight infants survive, more live long enough to need a major resection, and prematurity itself is the dominant risk factor for the disease that takes their bowel. Volvulus from malrotation, intestinal atresia and gastroschisis follow behind, together accounting for most of the remainder of the paediatric short-bowel population. [1]
The risk of failing to reach enteral autonomy clusters in a recognisable group of anatomical and clinical factors, and an examiner will expect you to list them. The strongest is the absence of colon in continuity, followed by loss of the ileum and ileocaecal valve, an end-jejunostomy, a very short residual jejunal length, and recurrent sepsis that keeps interrupting feeding. A child who combines several of these — for example, a premature baby with necrotising enterocolitis, a 20-centimetre jejunostomy and no colon — sits at the severe end of the spectrum and is the one most likely to need aggressive rehabilitation or transplant. [1] [2]
The arrival of specialised intestinal rehabilitation centres, teduglutide and autologous reconstructive surgery has shifted the epidemiology of outcomes. A generation ago many of these children either died of liver failure or proceeded to transplant; today a substantial proportion wean off parenteral nutrition, and transplant is reserved for those who cannot. Understanding that shift is part of answering a modern question on prognosis, because the numbers a candidate quotes must reflect the current era, not the pre-rehabilitation era. [1] [10]
Pathophysiology
The reason a child can ever recover from losing most of the small bowel is that the remnant gut adapts, and understanding adaptation is the key to understanding every management decision in this topic. Within days of a resection, the remaining small bowel begins to enlarge its absorptive surface. The villi lengthen, the crypts deepen and turn over faster, and the bowel wall thickens and may dilate. Transit slows, which gives the mucosa longer contact with the luminal contents. Over weeks to months this remodelling can multiply absorptive capacity many times, and it is the biological process that rehabilitation aims to protect and amplify. [1] [2]
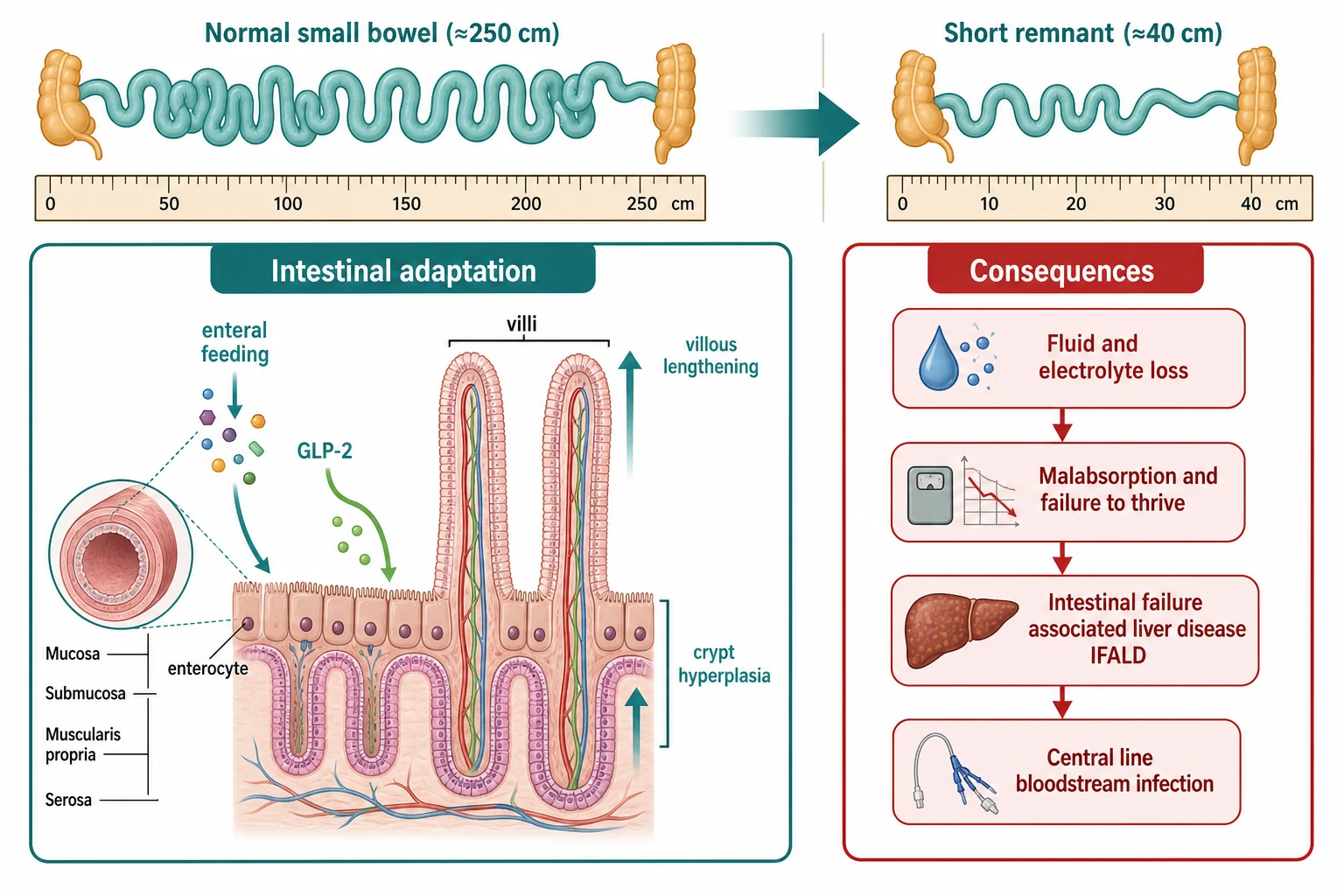
What drives this adaptation is contact between luminal nutrients and the mucosa. Enteral feeding, even in small trophic volumes that contribute little to the calorie count, stimulates the release of gastrointestinal hormones and the direct trophic effects of nutrients on the enterocyte. This is why the rule of every short-bowel service is to feed the child by gut as early and as much as tolerated, even while the bulk of nutrition still comes from the parenteral line. The phrase the examiner rewards is that enteral nutrition is trophic, not merely nutritive. [1]
The most important of the trophic signals is glucagon-like peptide 2, or GLP-2, a peptide secreted by the L-cells of the distal ileum and proximal colon in response to feeding. GLP-2 increases mucosal growth, blood flow and nutrient absorption, and it is the hormone whose synthetic analogue, teduglutide, has been turned into a drug. The biological logic explains the prognosis: when the ileum and colon have been removed, the child loses the very cells that produce GLP-2, so adaptation is blunted and the outlook is worse. This single mechanism ties together anatomy, prognosis and treatment. [3] [4]
Loss of the terminal ileum brings its own specific consequences that an examiner will probe. The terminal ileum is the site of active bile-acid reabsorption and of intrinsic-factor-bound vitamin B12 absorption, so its loss causes bile-acid wastage, steatorrhoea, gallstone formation and eventual B12 deficiency. Bile-acid loss is doubly important because unabsorbed bile salts spill into the colon and worsen secretory diarrhoea, while the depleted bile pool contributes to the cholestasis of intestinal failure-associated liver disease. [5]
The high-output stoma tells the other half of the pathophysiology. A jejunostomy cannot absorb water and sodium the way the colon does, and its output is copious, rich in sodium at roughly 90 to 100 millimoles per litre, and constant. The sodium lost in that output is invisible to the unwary prescriber, because the serum sodium often looks normal while the body is steadily depleting its total body sodium. Chronic sodium depletion suppresses growth independent of calories, which is why measuring and replacing stoma sodium is one of the highest-yield interventions in these children. [2]
Finally, the pathogenesis of intestinal failure-associated liver disease, or IFALD, weaves together several insults that the bedside team can modify. The load of parenteral lipid is central: high and sustained lipid exposure, together with the phytosterols carried in some soy-based emulsions, injure the hepatocyte and impair bile flow. Recurrent sepsis, common because of the long-term central line, multiplies the cholestatic injury. The absence of enteral feeding removes the normal trophic and pro-kinetic stimulus to bile flow. The result is cholestasis that, unchecked, progresses to fibrosis, portal hypertension and liver failure. The encouraging news is that each of these drivers is modifiable, which is why IFALD prevention is now a centrepiece of care. [5] [6]
Clinical Presentation
The child with short bowel usually announces the diagnosis in the history: a premature baby who has had necrotising enterocolitis with bowel resection, or a term infant who underwent surgery for volvulus or atresia. What brings them to ongoing medical attention is the consequence of the shortened gut, not the original event. The neonate with a fresh high jejunostomy produces large volumes of stomal effluent, loses weight despite parenteral nutrition, and develops electrolyte disturbances. The older child may present with chronic diarrhoea, faltering growth, or a specific deficiency. [1]
The high-output stoma is the signature clinical problem of the early phase, and you should define it operationally. Stoma or stoma-plus-stool output above about 40 to 50 millilitres per kilogram per day is high, and above this the child is almost certainly in negative sodium and water balance unless losses are actively replaced. The effluent is liquid, continual and bile-stained, the peristomal skin breaks down from the digestive enzymes, and the child fails to gain weight or actually loses it. Recognising the high output as a sodium-wasting state, rather than simply a volume nuisance, changes the management from fluid restriction to active replacement. [2]
IFALD declares itself in the bloods before it declares itself at the bedside. The first sign is a rising conjugated bilirubin in an infant on parenteral nutrition, followed by raised gamma-glutamyl transferase and alkaline phosphatase. Clinically the child may be jaundiced, with hepatomegaly and, as disease advances, pruritus and splenomegaly from portal hypertension. Because the cholestasis of a sick premature baby has many causes, the key clinical act is to measure the conjugated bilirubin serially and to act on a rising trend early, before fibrosis is established. [5] [6]
Micronutrient deficiencies surface later and follow a predictable pattern that rewards the prepared candidate. Fat-soluble vitamin deficiency causes rickets, easy bruising and night blindness; zinc deficiency causes an acrodermatitis-like rash and worsens diarrhoea; iron deficiency causes anaemia, made worse by frequent blood sampling; and vitamin B12 deficiency appears months after ileal resection as a macrocytic anaemia and neuropathy. Selenium and copper deficiencies are less common but occur in long-term parenteral nutrition and are worth naming at viva. [2]
The atypical presentation to hold in mind is the school-age child with established short bowel who decompensates. Such a child may have been weaning parenteral nutrition well, then loses ground with a viral illness, a line infection or a bout of bacterial overgrowth, and presents with weight loss, worsening diarrhoea or new cholestasis. The principle is that a stable short-bowel child is never cured of vulnerability; a new decompensation demands a search for the precipitant rather than reassurance. [1]
Differential Diagnosis
When a child has chronic diarrhoea, faltering growth and dependence on parenteral nutrition, you must first confirm that the cause is truly a short bowel rather than another form of intestinal failure. The three competing categories are mucosal disease, motility disorder, and the complications of a short bowel itself. Conflating them leads to the wrong operation or the wrong drug. [1]
Intestinal failure from mucosal disease looks like short bowel but has a normal-length gut. The congenital enteropathies, microvillus inclusion disease and tufting enteropathy, present in the first weeks of life with intractable watery diarrhoea and are diagnosed on intestinal biopsy. Autoimmune enteropathy produces a similar picture in an older infant, often with other autoimmune features, and responds to immunosuppression. These children are not candidates for adaptation surgery; their definitive treatment is often transplant, so distinguishing them from a resection short bowel changes the entire plan. [1]
Motility disorders cause intestinal failure through a different mechanism. Chronic intestinal pseudo-obstruction, whether myopathic or neuropathic, leaves a structurally intact but functionally inert bowel, so the child has distension, vomiting and failure to absorb despite adequate length. The contrast study and the manometry findings differ from those of short bowel, and again the management diverges, leaning toward decompression, prokinetics and transplant rather than adaptation surgery. [1]
Even within a confirmed short bowel, several conditions mimic a true deterioration and must be excluded before concluding the gut has failed. Infectious enteritis, from viral or bacterial causes, briefly worsens output and absorption. Lactose overload from an inappropriate feed overwhelms a shortened bowel and causes osmotic diarrhoea. Bile-acid malabsorption after ileal resection produces secretory colonic diarrhoea. Small-intestinal bacterial overgrowth, encouraged by dilated loops and slowed transit, causes bloating, malabsorption and worsened liver function. Each is treatable in its own right, and each is far commoner than a genuine failure of adaptation. [2]
Short-bowel syndrome
- Follows a documented resection or atresia
- Anatomy shortened, biopsy normal
- Adapts over months with feeding
- Responds to teduglutide and STEP
Mucosal disease
- Normal bowel length, abnormal mucosa
- Microvillus inclusion, tufting, autoimmune enteropathy
- Diagnosed on biopsy
- Often needs transplant, not adaptation surgery
Motility disorder
- Structurally intact but non-functioning bowel
- Chronic intestinal pseudo-obstruction
- Distension and vomiting predominate
- Manometry aids diagnosis
A cholestatic short-bowel infant poses a specific differential trap. The same picture of pale stools and a raised conjugated bilirubin could be biliary atresia, and a missed biliary atresia is a catastrophic error. The rule is that any short-bowel infant with conjugated jaundice needs biliary atresia actively excluded by ultrasound, liver biopsy and, if doubt remains, intraoperative cholangiogram, rather than assuming the cholestasis is simply IFALD. Similarly, a febrile child with a central line has a bloodstream infection until blood cultures prove otherwise, and line-related thrombosis must be distinguished from infection when the line stops working. [5] [6]
Clinical & Bedside Assessment
The bedside assessment of a child with short bowel is built around three measurements that tell you more than any single blood test: the weight trend, the stoma or stool output, and the growth centiles. Weigh the child daily in the acute phase and at every clinic visit thereafter, always on the same scale and ideally at the same time relative to the parenteral nutrition cycle, because the weight reflects fluid balance, calorie adequacy and sodium status at once. A child who is not gaining, or who is falling across centiles, has a problem to solve before the clinic visit ends. [2]
Measure and record the output meticulously. Every stoma bag and every nappy is weighed or its volume estimated, and the total is expressed in millilitres per kilogram per day so it can be compared between visits and between children. An output rising above about 40 to 50 millilitres per kilogram per day flags a high-output state. The stool or stoma sodium content can be measured when the source of sodium loss is in doubt, and a sodium concentration above about 70 to 90 millimoles per litre confirms that the output is a major drain on body sodium. These numbers convert an impressionistic 'lots of diarrhoea' into a managed variable. [2]
Hydration is assessed in the usual way, but with two short-bowel refinements. First, the serial weight is the single most reliable guide to hydration in an infant with a high-output stoma, because the signs of dehydration can be subtle until late. Second, chronic mild sodium depletion, invisible in the serum, shows itself as poor weight gain and a flat growth curve long before the child looks clinically dry. Ask about urine output and concentration, examine the mucous membranes and capillary refill, and plot the weight relentlessly. [1]
Examine the stoma and the abdomen directly. Look for stoma prolapse, retraction, ischaemia or a parastomal hernia, any of which may need surgical revision and may be driving the high output. Assess the peristomal skin for enzyme burns and candida, which are painful and undermine feeding. Palpate the abdomen for distension from bacterial overgrowth or an adhesion obstruction, and feel for hepatosplenomegaly and ascites, the stigmata of advancing IFALD and portal hypertension. [5]
Growth plotting is the longitudinal examination that ties everything together. Plot weight, length and head circumference on the appropriate centile chart at every visit, because the trajectory tells you whether the current prescription is working. A child whose weight and length centiles are rising together is adapting and being fed appropriately; a child whose weight is rising but length is falling is being overfed relative to absorption and may be accumulating fluid; a child whose centiles are both falling has an unresolved problem, whether output, infection, overgrowth or inadequate calories. [1]
Investigations
The investigation of a child on long-term parenteral nutrition is a standing panel repeated at defined intervals, because the child is exposed to a therapy that itself causes the complications you are monitoring for. The routine bloods include a full blood count, urea and electrolytes, magnesium, phosphate and calcium, and a liver panel with a conjugated bilirubin, alanine aminotransferase and gamma-glutamyl transferase. Trace elements, including zinc, selenium, copper and manganese, and the fat-soluble vitamins are checked at longer intervals. These panels are not optional; they are how you detect the slow accumulation of toxicity before it becomes clinical. [2]
One investigation deserves special mention because examiners love it: the plasma citrulline. Citrulline is an amino acid produced almost exclusively by the enterocytes of the small bowel, so its plasma level reflects the functional enterocyte mass. A low level correlates with a small absorptive surface, and it is used as a marker of intestinal failure due to enterocyte mass reduction. A level below about 20 micromoles per litre in adults indicates critically reduced enterocyte mass, and rising citrulline over months parallels successful adaptation. In children the absolute thresholds vary with age and renal function, so the trend is more useful than a single value, but the principle of citrulline as a functional enterocyte marker is a high-yield viva point. [11]
Imaging and endoscopy are targeted rather than routine. A contrast study defines the anatomy after resection and is repeated when a stricture, obstruction or dilated dysfunctional segment is suspected before surgery. Abdominal ultrasound looks for gallbladder sludge and stones, which are common in ileal resection and IFALD, and for signs of portal hypertension. Upper endoscopy with aspirates, or hydrogen or glucose breath tests, investigate suspected small-intestinal bacterial overgrowth, which causes bloating and decompensation in the dilated short bowel. [1] [2]
In the cholestatic short-bowel infant, the imperative is to exclude biliary atresia early, because the treatment window is narrow. Abdominal ultrasound may show the absent or abnormal gallbladder and the triangular cord sign, a liver biopsy shows the cholestatic pattern with bile-duct proliferation, and intraoperative cholangiogram confirms the diagnosis. The teaching is that a short-bowel infant with conjugated jaundice needs the same biliary atresia work-up as any cholestatic neonate, in parallel with the management of suspected IFALD, rather than one after the other. [5] [6]
Two audits belong to the transplant assessment, and an examiner may ask for them specifically. The first is the central venous access audit: how many lines has the child had, how many bloodstream infections, and how many venous access sites remain patent, because the loss of central venous access is itself an indication for transplant. The second is the liver assessment, including imaging, endoscopy for varices and a biopsy where indicated, to decide whether the child needs an isolated intestinal graft or a combined liver-intestine graft. Keeping these audits current turns an emergency referral into a planned one. [10]
Management — Resuscitation
The immediate priority after a massive resection is to stabilise the child while the bowel recovers from the operative insult. The neonate is volume-depleted from the illness and the surgery, electrolyte-depleted from the high output, and unable to absorb anything by gut. Resuscitation is therefore intravenous: restore the circulating volume with isotonic crystalloid, correct the acid-base disturbance, and replace the ongoing stoma losses millilitre for millilitre with an appropriate sodium-containing solution. The central line that will carry parenteral nutrition is placed early and with a long-term plan. [1] [2]
Parenteral nutrition is started within the first days and built up carefully as tolerance allows. The prescription delivers fluid, glucose, amino acids and lipid, with electrolytes and micronutrients tailored to the measured losses and the bloods. The energy target is met gradually, because over-rapid escalation risks hyperglycaemia, fluid overload and lipid-driven cholestasis. From the outset the team holds in mind that parenteral nutrition is a bridge to enteral autonomy, not a destination, and every prescription decision is made with the next step of weaning in view. [2]
The high-output stoma demands an active replacement strategy, and this is where the sodium principle becomes operational. The stoma output is measured hourly in the acute phase, and each millilitre is replaced with a solution that contains enough sodium to match the loss, typically using sodium chloride at a concentration near that of the effluent. Restricting free water and failing to replace sodium is the classic error that keeps these children failing to thrive; replacing sodium and water proactively is the intervention that turns them around. [2]
Central-line bloodstream infection is the commonest acute threat, and preventing it begins at resuscitation. Strict aseptic line care, a dedicated lumen for parenteral nutrition, and a low threshold for culturing and treating fever are the cornerstones. When infection recurs, ethanol or taurolidine line locks are added to suppress intraluminal organisms, and a tunnelled or infected line is removed and replaced at a new site rather than salvaged at the cost of recurring sepsis. Each line infection is also an IFALD risk, so infection prevention is liver protection. [5] [6]
Management — Definitive & Stepwise
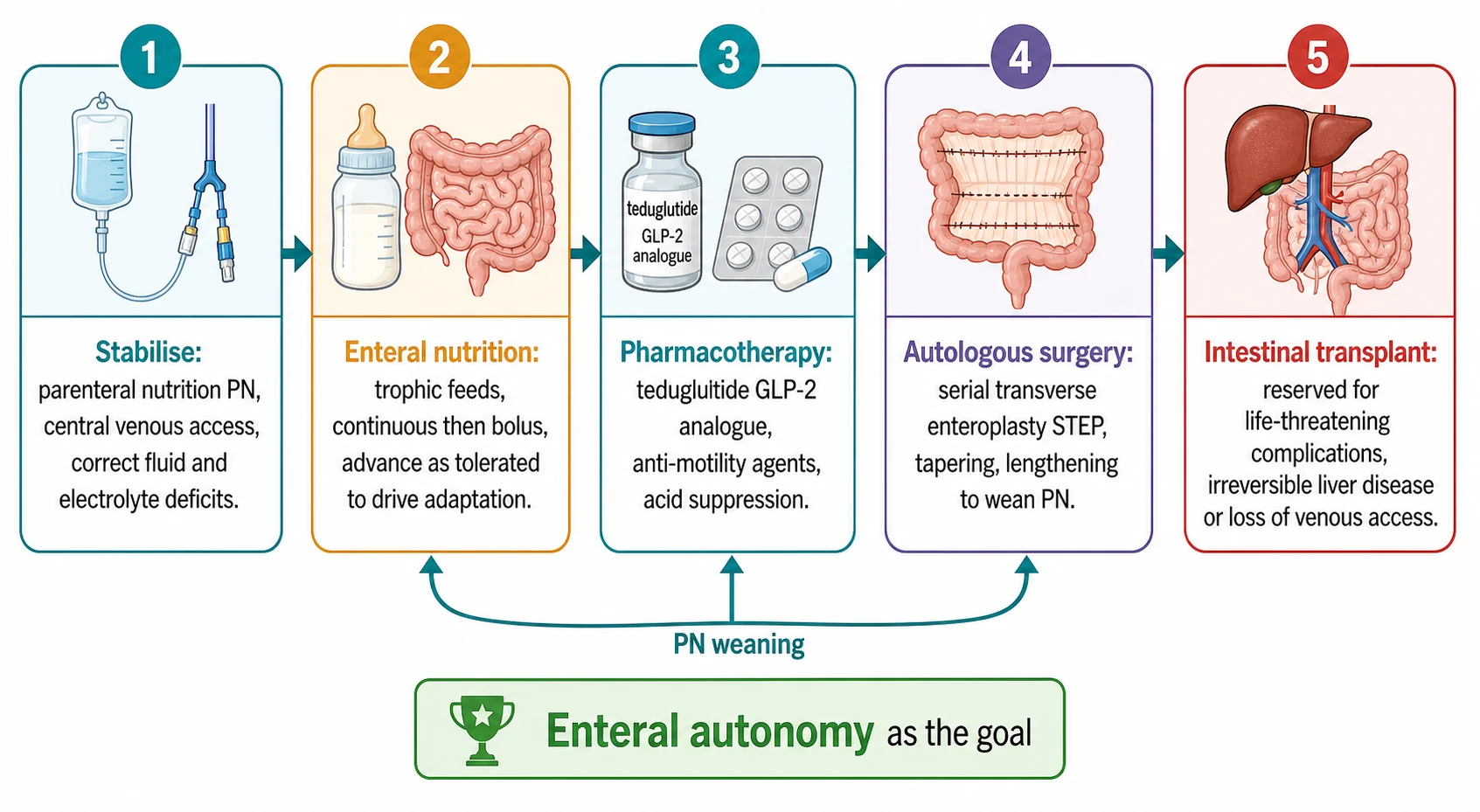
Definitive management is the work of a multidisciplinary intestinal rehabilitation programme, and the guiding aim is always the same: to advance enteral nutrition, to wean parenteral nutrition, and to reach enteral autonomy, defined as full nutritional sufficiency by the gut without parenteral support. Every intervention in this section exists to move the child up that ladder. The programme concentrates the expertise of gastroenterology, surgery, nutrition, pharmacy and nursing in one team, and there is good evidence that children managed in such centres achieve enteral autonomy more often and survive better than those managed piecemeal. [1]
Enteral nutrition is the engine of the whole process, and it is started early and advanced persistently. The preferred substrate is the child's own mother's milk wherever possible, because human milk is trophic, immune-protective and well tolerated. Feeding begins as a small continuous infusion, which a shortened bowel tolerates better than boluses, and is advanced in volume and concentration as output and growth allow. As the child matures, bolus and oral feeding are introduced to develop oral motor skills and prevent feeding aversion, and the formula is moved toward a semi-elemental or polymeric diet depending on tolerance. The trigger to advance is stable output and rising weight; the trigger to hold is a rising output or falling weight. [2]
Stabilise
Parenteral nutrition, secure central venous access, correct fluid, electrolyte and sodium deficits from the high-output stoma
Feed the gut
Start trophic enteral nutrition, continuous then bolus, advance on output and weight, prefer human milk
Pharmacotherapy
Add the teduglutide GLP-2 analogue plus anti-motility agents and acid suppression to reduce output and drive adaptation
Autologous surgery
Serial transverse enteroplasty, tapering or lengthening to use dilated bowel and wean parenteral nutrition
Transplant
Isolated intestine or liver-intestine transplant for irreversible IFALD, loss of venous access or recurrent life-threatening sepsis
Pharmacotherapy now includes teduglutide, and a candidate must know it precisely. Teduglutide is an analogue of glucagon-like peptide 2 that is resistant to breakdown, so it delivers a sustained trophic signal to the intestinal mucosa. It is given by subcutaneous injection at a dose of 0.05 milligrams per kilogram once daily. The paediatric evidence has grown: the Chiba 2023 study reported the efficacy and safety of teduglutide in infants and children with short-bowel syndrome dependent on parenteral support, showing meaningful reductions in parenteral support volume, and adult trials had earlier established increased absorption and enteral autonomy in some patients. Monitoring includes growth, stoma output, fluid balance and surveillance for the recognised concerns of intestinal obstruction and neoplasia with long-term use, so teduglutide is prescribed within a specialist programme, not started casually. [3] [4]
Teduglutide
Dose
0.05 mg/kg
Several other agents work alongside teduglutide to reduce the output that the shortened bowel produces. Loperamide, given orally, slows transit and increases mucosal contact time; higher than standard doses are often needed in the short-bowel child and are used under specialist supervision. Acid suppression is given early because the shortened bowel mounts a transient gastric acid hypersecretion that worsens output and damages the peristomal skin. Octreotide is reserved for refractory high-output states and reduces secretion, though its effect on the pancreas and gallbladder must be weighed. None of these replaces feeding; all of them make feeding more tolerable. [2]
When the medical ladder has done what it can and the child remains dependent, autologous gastrointestinal reconstruction comes into view. The signature operation is the serial transverse enteroplasty, or STEP, in which the surgeon applies alternating stapler firings across a dilated segment of small bowel to narrow and lengthen it, converting a wide, dysfunctional loop into a longer, more functional channel. The procedure was designed to exploit the dilatation that adaptation itself produces, and it improves peristalsis and absorption. The multicentre long-term data show that STEP achieves meaningful reduction in parenteral nutrition in a substantial proportion of children, and repeat STEP can further reduce dependence when the bowel dilates again. The role of the paediatrician is to recognise the dilated, dysfunctional segment — the child whose output rises and whose weight stalls despite maximal medical therapy — and to refer for surgical assessment. [8] [9]
Surgical strategy also includes the early establishment of colon continuity. A jejunostomy that empties onto the abdominal wall wastes water and sodium that a colon would absorb, so when the child is stable the surgeon closes the stoma and joins the proximal bowel to the distal remnant or colon. Restoring continuity is one of the single most effective steps toward weaning parenteral nutrition, because it returns the colon to its absorptive and trophic role. The timing is individualised, but the principle is firm: a stoma is not a permanent destination in a child who can be given a colon. [1]
Intestinal transplantation is the final rung, and the indications are now well defined and deliberately narrow. Transplant is considered when one of three doors is reached: irreversible intestinal failure-associated liver disease that threatens the child's life, progressive loss of central venous access so that parenteral nutrition can no longer be delivered safely, and recurrent life-threatening central-line bloodstream infection. When the liver is failing, the child receives a combined liver-intestine graft; when the liver is spared, an isolated intestinal graft may suffice. Survival in the current era is good, with the majority of children alive at five years, but transplant carries its own burden of immunosuppression and rejection, so it is reserved for those who cannot be rescued by rehabilitation alone. The decision is made in a specialist centre weighing the audit of lines, liver and sepsis. [10]
Specific Subtypes & Scenarios
The end-jejunostomy child is the hardest case and the one most likely to feature in a viva. With no colon in continuity, this child loses water and sodium relentlessly, almost always needs full parenteral nutrition, and is the group least likely to reach enteral autonomy without aggressive intervention. The management priority is to establish continuity as soon as the child is fit, by joining the jejunum to the distal remnant or colon, because nothing else so reliably improves absorption. Until then, meticulous sodium and water replacement, teduglutide and infection control buy the time needed for surgery. A child who remains an end-jejunostomy with a very short remnant despite all of this is a candidate for early transplant assessment. [1]
The neonate after necrotising enterocolitis with a high jejunostomy is the scenario most paediatricians will actually meet. The child is premature, the liver is immature and cholestatic-prone, and the output is enormous. The staged plan is to stabilise and grow the child on parenteral nutrition, to advance trophic enteral feeds into whatever proximal bowel remains, to prevent IFALD by lipid strategy and infection control, and to plan surgical continuity when the child has grown and stabilised. Each step is paced by the weight, the liver function and the output, and the team treats the liver with the same urgency as the gut. [6]
The older child with short bowel from mid-gut volvulus represents a more favourable scenario and a different set of decisions. Such a child often has a short jejunal remnant joined to colon, has grown well, and is partway through weaning parenteral nutrition. The role of the team is to keep advancing enteral feeding, to consider teduglutide to push the last stretch of weaning, and to assess for STEP if the bowel has dilated and output has stalled. These children often reach enteral autonomy over months to a few years, and the long-term question becomes nutritional and developmental follow-up rather than rescue. [8]
The child with established IFALD being assessed for transplant sits at the junction of this topic and the liver topics. The decision between continued intestinal rehabilitation and liver-intestine transplant turns on the trajectory of the liver disease: a child whose bilirubin is falling with lipid modification and infection control can continue rehabilitation, while a child with rising bilirubin, portal hypertension and worsening synthetic function needs urgent transplant assessment. The principle is to escalate to transplant before the liver fails irreversibly, because a child transplanted too late has a worse outcome. [5] [10]
Complications & Pitfalls
The complications of short bowel fall into two families: those of the disease and those of its treatment. The disease complications are the malabsorption, dehydration and electrolyte loss already discussed, together with renal stones from chronic dehydration and fat malabsorption, and gallstones from the loss of the bile-acid pool after ileal resection. The treatment complications are intestinal failure-associated liver disease, central-line bloodstream infection and venous thrombosis, micronutrient toxicity and deficiency, and the developmental and psychosocial burden of a technology-dependent childhood. An examiner will expect both families named. [1] [5]
IFALD deserves its own scrutiny as the complication most likely to end the race. Its two dominant modifiable drivers are the parenteral lipid load and recurrent sepsis. A sustained high lipid dose, particularly from soy-based emulsions rich in phytosterols, drives cholestasis; recurrent line infection multiplies the hepatic injury. The prevention and treatment strategy is therefore to reduce or modify the lipid, using lipid restriction, a mixed emulsion such as SMOFlipid, or an omega-3 fish-oil-based emulsion, while controlling line infection and advancing enteral feeding. The lipid approach varies by region, but the principle of lowering the cholestatic load is universal. [5] [6] [7] [12]
The lipid strategy to prevent and treat IFALD differs between regions. In North America the omega-3 fish-oil emulsion Omegaven has been widely adopted, often with lipid restriction, reflecting the evidence that fish-oil-based emulsions reverse cholestasis in many infants. In Europe and Australasia mixed emulsions such as SMOFlipid, and lipid-sparing regimens that keep the soy load down, are more commonly used. The shared aim is to lower the phytosterol load and the total cholestatic burden; the preferred emulsion is the regional variation. [5] [7]
Central-line bloodstream infection is the day-to-day complication that brings these children to hospital repeatedly, and it has its own pitfalls. The line must be handled with strict asepsis by everyone, the family included, and a single lumen is dedicated to parenteral nutrition. A fever in a child with a central line is a line infection until cultures prove otherwise, and treatment is prompt systemic antibiotics with gram-positive and gram-negative cover, often with a line lock. The pitfall is repeatedly salvaging an infected line until the child has septicaemia; sometimes the correct act is to remove and replace the line at a new site. [1] [5]
The classic management pitfalls are worth naming because they recur. Underestimating the sodium cost of a high-output stoma keeps children failing to thrive. Failing to advance enteral feeds out of caution denies the gut the trophic stimulus it needs to adapt. Over-restricting oral feeding or never offering it produces an oral aversion that is far harder to undo than to prevent. Delaying stoma closure forfeits the absorptive benefit of the colon. And carrying a child on full parenteral nutrition without a weaning plan normalises dependence and delays the move toward autonomy. Each pitfall is avoidable with a clear, paced rehabilitation plan. [1] [2]
The psychosocial complications are real and under-recognised. The family of a child on home parenteral nutrition carries an enormous burden of line care, night-time infusions, repeated admissions and constant vigilance, and the child is at risk of feeding aversion, developmental delay and disrupted schooling. Acknowledging this burden, connecting the family to peer support and home-care services, and protecting the child's oral feeding and development are part of the management, not an optional extra. [1]
Prognosis & Disposition
The prognosis of paediatric short bowel has improved markedly, and a candidate should quote the modern numbers. With a dedicated intestinal rehabilitation programme, a substantial proportion of children achieve enteral autonomy, many within the first two to three years of life. The anatomical and clinical predictors of success are the same ones that recur throughout this topic: an intact colon and ileocaecal valve, a longer residual small-bowel length, the absence of IFALD and recurrent sepsis, and a child who tolerates advancing enteral nutrition. A child who has all of these is likely to wean; a child who has none is the one to assess early for transplant. [1]
Once enteral autonomy is reached, the long-term outlook is generally good, but it is not a return to normal. Growth usually catches up, though some children remain small, and neurodevelopment is usually preserved in children who avoided the extremes of prematurity and recurrent critical illness. Quality of life improves markedly once the central line is gone, though some children are left with feeding aversion, a restricted diet or surgical complications that need ongoing follow-up. The paediatrician's role after autonomy is nutritional, developmental and psychosocial surveillance. [1]
For the child who reaches transplant, survival in the current era is reasonable, with the majority of children alive at five years, though outcomes depend on the era of transplantation, the indication, and the centre. Graft and patient survival are best for elective isolated intestinal grafts in a stable child, and worst for emergency liver-intestine grafts in a child with multi-organ failure, which is exactly why the assessment is timed to avoid the emergency. The paediatrician supports the family through immunosuppression, rejection surveillance and the transition to a post-transplant life. [10]
Disposition is governed by the principle that these children do best in a specialist centre. A child with new intestinal failure is referred to a quaternary intestinal rehabilitation programme for assessment and management, while the local team continues supportive care and shared follow-up. For the child established on home parenteral nutrition, the safety-net is a clear escalation plan: families know when to present, the local hospital has a protocol for the febrile central-line child, and retrieval to the specialist centre is arranged early rather than late. The child who is decompensating, whether from sepsis, dehydration or liver failure, is retrieved, not observed. [1]
Special Populations
The extremely preterm neonate with short bowel is the population in whom every complication is amplified. The immature liver is exquisitely sensitive to the cholestatic injury of parenteral lipid and sepsis, so IFALD develops faster and more severely than in the older child. The immature gut has a longer road to adaptation, and the immature kidney struggles with the sodium and water shifts of the high-output state. Management is therefore more conservative and more intensive at once: gentler lipid strategies, tighter infection control, more cautious advancement of feeds, and a lower threshold for specialist referral. [6]
The child who is technology-dependent on home parenteral nutrition in a rural or remote community faces a genuine equity gap. The expertise, the lipid preparations, the teduglutide and the surgical options cluster in quaternary centres, often hundreds of kilometres from where the child lives. Telehealth, shared-care partnerships with regional hospitals, funded travel and accommodation, and home-care nursing support are the mechanisms that narrow this gap, and advocating for them is part of the paediatrician's role. A child's postcode should not decide whether they reach enteral autonomy. [1]
The adolescent transitioning from paediatric to adult intestinal-failure services is a special population in their own right. The young person who has grown up with a central line and a short bowel is taking on the management of a complex chronic condition at exactly the age when adherence is hardest and the risk of line sepsis and decompensation is highest. A structured transition programme, with joint clinics, education on line care and emergency signs, and a named adult provider, reduces the risk of a faltering transition. The principle is that transition is prepared over years, not handed over in a single appointment. [1]
Children with additional vulnerabilities, whether from disability, socioeconomic disadvantage or being in out-of-home care, carry a heavier burden still. The demands of parenteral nutrition at home presuppose a stable environment, reliable power, a competent carer and funding for supplies, none of which can be assumed. The team assesses the home and social context as part of the discharge planning, arranges the equipment and funding, and builds a back-up plan for when the primary carer is unavailable. Failing to do this sends a child home to fail. [1]
Evidence, Guidelines & Regional Differences
The evidence base for paediatric intestinal failure has matured rapidly. The 2017 New England Journal of Medicine review by Duggan and Jaksic set out the modern framework of intestinal rehabilitation, adaptation physiology and the narrowing indications for transplant, and it remains the orienting reference for the topic. The Premkumar 2022 review of nutritional management codified the feeding and electrolyte strategy, particularly the sodium principle and the advancement of enteral nutrition. These two references together carry most of the framework an examiner expects. [1] [2]
The teduglutide evidence is the area that has moved fastest and that a candidate should know in outline. The adult STEPS trials established that teduglutide at 0.05 milligrams per kilogram daily reduced parenteral support volume and achieved enteral autonomy in a proportion of adults with short-bowel syndrome. The paediatric evidence grew with the Chiba 2023 study, which reported the efficacy and safety of teduglutide in infants and children dependent on parenteral support, showing reductions in parenteral support that supported its use in carefully selected children within specialist programmes. The controversy is real: long-term use raises surveillance questions around neoplasia and obstruction, and teduglutide is not a first-line drug for every short-bowel child. [3] [4]
The STEP evidence has matured from single-centre series to multicentre long-term data. The Dagorno 2025 multicentre study of STEP in children reported the long-term outcomes, confirming that STEP achieves meaningful reduction in parenteral nutrition in a substantial proportion of children, while Mercer 2021 showed that repeat STEP can further reduce dependence when the bowel dilates again. The controversy is which child benefits: STEP works best for the dilated, dysfunctional segment, and the risk is that a poorly selected operation creates a segment that still does not propel. The role of careful preoperative assessment is emphasised throughout the literature. [8] [9]
The regional differences in this topic cluster around the management of IFALD and the organisation of care. The lipid strategy varies by region, as noted, with omega-3 fish-oil emulsions prominent in North America and mixed or lipid-sparing strategies in Europe and Australasia. The professional guidelines, from ESPGHAN in Europe and ASPEN in North America, agree on the principles of multidisciplinary intestinal rehabilitation, strict catheter-care standards for home parenteral nutrition, and the early identification and modification of the lipid load to prevent liver disease, while differing in their preferred emulsion. A candidate who names the principle and acknowledges the regional variation is well placed. [5] [7]
Exam Pearls
COLON
The single highest-yield fact in this topic is that an intact colon and ileocaecal valve predicts enteral autonomy more strongly than the residual small-bowel length alone. The mechanism is that the colon absorbs water and sodium, salvages carbohydrate as short-chain fatty acids, and the preserved ileum secretes the trophic hormone glucagon-like peptide 2 that drives adaptation. State this clearly and you anchor the whole topic. [1] [3]
The drug fact to own precisely is the teduglutide dose: 0.05 milligrams per kilogram subcutaneously once daily, as an analogue of glucagon-like peptide 2 resistant to breakdown, prescribed within a specialist intestinal rehabilitation programme with monitoring for obstruction and long-term neoplasia risk. The paediatric trial to cite is Chiba 2023. [3]
The biomarker fact is citrulline: a plasma level reflecting functional enterocyte mass, low in intestinal failure due to enterocyte mass reduction, with a level below about 20 micromoles per litre in adults indicating critical loss, and a rising trend paralleling successful adaptation. [11]
The classic one-liner for the severe end of the spectrum: a jejunostomy with under about 50 centimetres of residual small bowel and no colon almost never achieves enteral autonomy on its own and usually needs a rehabilitation programme and often a transplant. Necrotising enterocolitis is the leading cause of intestinal failure in neonates. Serial transverse enteroplasty was popularised by Kim and converts a dilated, dysfunctional segment into a longer, more functional channel, with the multicentre long-term data supporting its role in PN reduction. [1] [8]
The disposition pearl is that these children do best in a specialist intestinal rehabilitation centre, and early referral is the rule rather than the exception. The prognostic pearl is that a substantial proportion of children now reach enteral autonomy with a modern programme, so the conversation with a family is one of measured optimism grounded in a clear, paced plan, with transplant reserved for those who cannot be rescued. [1] [10]
References
- [1]Duggan CP; Jaksic T Pediatric Intestinal Failure. N Engl J Med, 2017.PMID 28813225
- [2]Premkumar MH Nutritional Management of Short Bowel Syndrome. Clin Perinatol, 2022.PMID 35659103
- [3]Chiba M; Arnon R; Kori M; et al Efficacy and Safety of Teduglutide in Infants and Children With Short Bowel Syndrome Dependent on Parenteral Support. J Pediatr Gastroenterol Nutr, 2023.PMID 37364133
- [4]Rosete BE; de Paula CB; Sperber LF; et al Teduglutide for pediatric short bowel syndrome patients. Expert Rev Gastroenterol Hepatol, 2021.PMID 33798402
- [5]Lee WS; Teo EH; Chong PY; Poh LK Intestinal failure-associated liver disease (IFALD): insights into pathogenesis and advances in management. Hepatol Int, 2020.PMID 32356227
- [6]Fundora J; Lopez M; Saps M; et al Intestinal Failure-Associated Liver Disease in Neonates. Neoreviews, 2020.PMID 32873652
- [7]Goulet OJ Lipid Emulsion Use in Pediatric Patients Requiring Long-Term Parenteral Nutrition. JPEN J Parenter Enteral Nutr, 2020.PMID 32049395
- [8]Dagorno C; Breton A; Lamireau T; et al Serial Transverse Enteroplasty (STEP) for Short Bowel Syndrome (SBS) in Children: A Multicenter Study on Long-term Outcomes. J Pediatr Surg, 2025.PMID 39368852
- [9]Mercer DF; Sutton DG; Pinschewer DD; et al Repeat serial transverse enteroplasty leads to reduction in parenteral nutrition in children with short bowel syndrome. J Pediatr Surg, 2021.PMID 32736789
- [10]Lee EJ; Iyer KR; Horslen S Pediatric intestinal transplantation. Semin Pediatr Surg, 2022.PMID 35725057
- [11]Crenn P; Messing B; Cynober L Citrulline as a biomarker of intestinal failure due to enterocyte mass reduction. Clin Nutr, 2008.PMID 18440672
- [12]Fallon EM; Le HD; Puder M Prevention of parenteral nutrition-associated liver disease: role of omega-3 fish oil. Curr Opin Organ Transplant, 2010.PMID 20503524