Paeds · gastroenterology-hepatology-and-nutrition
Viral, autoimmune and metabolic hepatitis
Also known as Hepatitis in children · Paediatric hepatitis · Viral hepatitis A to E · Autoimmune hepatitis · Wilson disease hepatitis · Alpha-1-antitrypsin deficiency hepatitis
Fellowship guide to hepatitis in children across viral, autoimmune and metabolic causes: the faecal-oral acute viruses hepatitis A and E that never become chronic, the parenteral and perinatal chronic viruses hepatitis B, C and D with their serological panels and phases, the birth-dose vaccine and hepatitis B immunoglobulin within twelve hours that prevents perinatal chronicity of ninety per cent, the direct-acting antiviral era for hepatitis C with sustained virologic response above ninety-five per cent, the simplified and revised International Autoimmune Hepatitis Group scoring systems and the type 1 and type 2 antibody profiles treated with prednisolone and azathioprine, the Leipzig score and Kayser-Fleischer ring of Wilson disease with ceruloplasmin under zero point two grams per litre and urinary copper over forty micrograms, and the PiZZ phenotype and PAS-positive diastase-resistant globules of alpha-1-antitrypsin deficiency managed supportively with transplant for end-stage liver disease.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
Hepatitis is inflammation of the liver with hepatocellular injury, and in a child it presents most often as a triad of jaundice, dark urine and pale stools, or more subtly as fatigue and raised liver transaminases on a routine blood test. The whole task of the clinician is to move from the inflamed liver to its cause, because the cause alone sets the prognosis and the treatment. A child with hepatitis A will recover fully in weeks and needs supportive care and hygiene advice, whereas a child with perinatally acquired hepatitis B carries a lifelong chronic infection that may end in cirrhosis or hepatocellular carcinoma, and a child with Wilson disease will progress to liver failure unless copper is removed for life. Sorting these outcomes is what the aetiological framework is for. [1] [2]
The causes fall into three families that the title names. Viral hepatitis is caused by the five hepatotropic viruses A to E, which divide cleanly by how they spread and whether they persist. Autoimmune hepatitis is a loss of immune tolerance to liver antigens that drives chronic interface hepatitis and responds to immunosuppression. The metabolic hepatitides are Wilson disease, where a copper-transport defect poisons the liver and the brain, and alpha-1-antitrypsin deficiency, where a misfolded protein accumulates inside hepatocytes. Drug-induced, ischaemic and systemic causes belong in the differential, but the three named families are the ones a fellowship candidate must hold as a model. [8] [11]
The single most important dividing line is acute against chronic. Hepatitis A and E are acute-only infections spread by the faecal-oral route that the immune clears and that never become chronic, whereas hepatitis B and C are parenteral and perinatal viruses that evade clearance and become chronic in a large fraction of children, especially the perinatally infected. Autoimmune and metabolic hepatitis are chronic by nature. This acute-versus-chronic split is the first question to answer at the bedside, because it decides whether the child needs only supportive care or a lifetime of surveillance and treatment. [1] [5]
Classification
The most useful clinical way to hold paediatric hepatitis is by cause, because the cause sets everything else. The viral hepatitides are grouped by their transmission route and their tendency to persist. Hepatitis A and E are faecal-oral, enteric infections that produce acute hepatitis and are cleared by the immune system, so they are acute-only. Hepatitis B and C are blood-borne and perinatal viruses that establish chronic infection in a substantial minority of adults and a large majority of infants infected at birth, so they are the chronic viral hepatitides. Hepatitis D is a defective virus that needs hepatitis B surface antigen to replicate, so it joins B as a parenteral and perinatal chronic virus, and it worsens whatever B is doing. [1] [2]
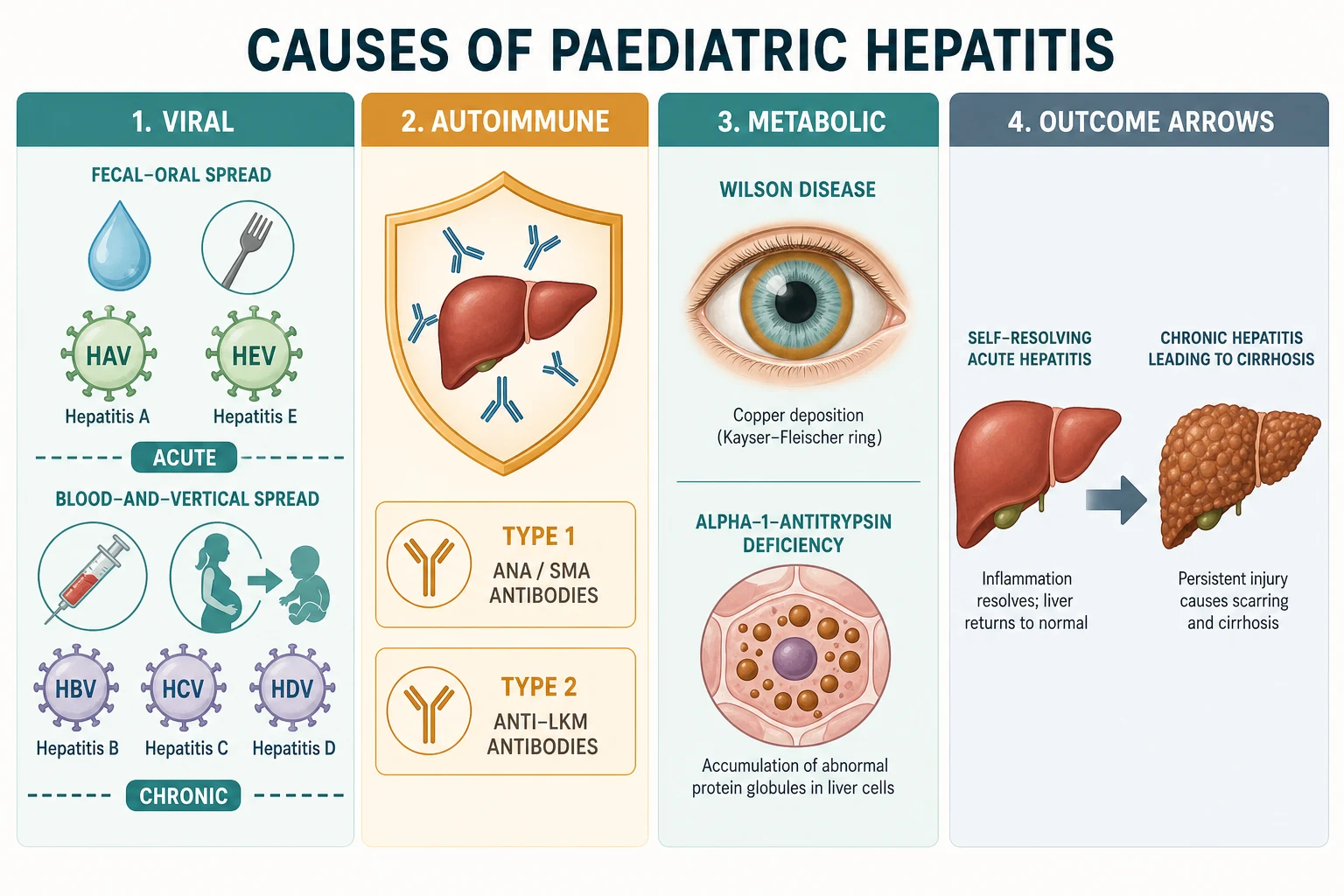
The autoimmune and metabolic causes complete the framework. Autoimmune hepatitis is typed by its autoantibody profile: type 1 carries antinuclear antibody and anti-smooth-muscle antibody and tends to present in adolescence, while type 2 carries anti-liver-kidney-microsomal type 1 antibody and anti-LC1 and tends to present in younger children and to run a more aggressive course. The metabolic hepatitides are Wilson disease, from a copper-transport defect, and alpha-1-antitrypsin deficiency, from a protein-misfolding disorder. Both are autosomal recessive, both demand a high index of suspicion in a child with otherwise unexplained hepatitis, and both can present from infancy to adolescence. [8] [12]
A second axis is the tempo and the severity. Acute hepatitis, whether viral, drug-induced or the first flare of an autoimmune or metabolic disease, ranges from a mild anicteric illness to fulminant liver failure. Chronic hepatitis, the province of the persistent viruses, autoimmune hepatitis and the metabolic diseases, runs silently for years and then declares itself as cirrhosis, portal hypertension or hepatocellular carcinoma. Holding both axes at once lets the clinician frame any child: a febrile jaundiced traveller is likely acute hepatitis A; an asymptomatic screened infant is likely chronic hepatitis B; a tired adolescent girl with amenorrhoea is likely autoimmune hepatitis. [1] [2]
[1] [8]Epidemiology & Risk Factors
The epidemiology of the viral hepatitides follows their transmission routes. Hepatitis A is acquired from contaminated food and water, so it clusters in travellers, in household contacts and in areas with poor sanitation, and most infections in young children are mild or silent, which is why they spread the virus without being noticed. Hepatitis E is likewise faecal-oral and waterborne, the commonest cause of acute viral hepatitis in many endemic regions, and in children it usually runs a mild self-limited course though it can cause acute liver failure. [1] [7]
Hepatitis B and C are the chronic viral hepatitides, and their burden is set by perinatal and blood-borne spread. Without immunoprophylaxis, around ninety per cent of infants infected at birth develop chronic hepatitis B, compared with under ten per cent of immunocompetent adults, so the perinatal route is the engine of the global chronic burden. The vertical transmission rate of hepatitis C is around five per cent, there is no vaccine, and the children who acquire it are usually identified when their mothers are known to be infected. Hepatitis D travels with B, requiring hepatitis B surface antigen to replicate, and it supercharges the liver injury wherever B is endemic. [2] [5]
The autoimmune and metabolic diseases are rarer but disproportionately important in a child with unexplained chronic hepatitis. Autoimmune hepatitis is the most common chronic hepatitis of children in many series after the viral causes, with a female predominance and a peak in adolescence for type 1 and in younger children for type 2, and it travels with other autoimmune diseases in the child and the family. Wilson disease and alpha-1-antitrypsin deficiency are autosomal recessive, with an incidence of around one in thirty thousand for Wilson and a PiZZ frequency of around one in two thousand to five thousand for alpha-1-antitrypsin deficiency, so consanguinity and a family history of liver or neuropsychiatric disease raise the index of suspicion. [8] [12]
Pathophysiology
The viral hepatitides injure the liver through the immune response more than through direct viral killing. In hepatitis A and E the virus replicates in hepatocytes and the cytolytic T-cell response clears it, producing the acute hepatocellular injury and the rise in transaminases that signals the illness, and clearance is the end of the story because the virus does not persist. In chronic hepatitis B the virus is not directly cytopathic in the chronic phase; the liver damage is driven by CD8-positive cytotoxic T cells that recognise hepatitis B surface antigen on the hepatocyte membrane and kill the infected cell, so the degree of inflammation tracks the immune activity rather than the viral load. [1] [3]
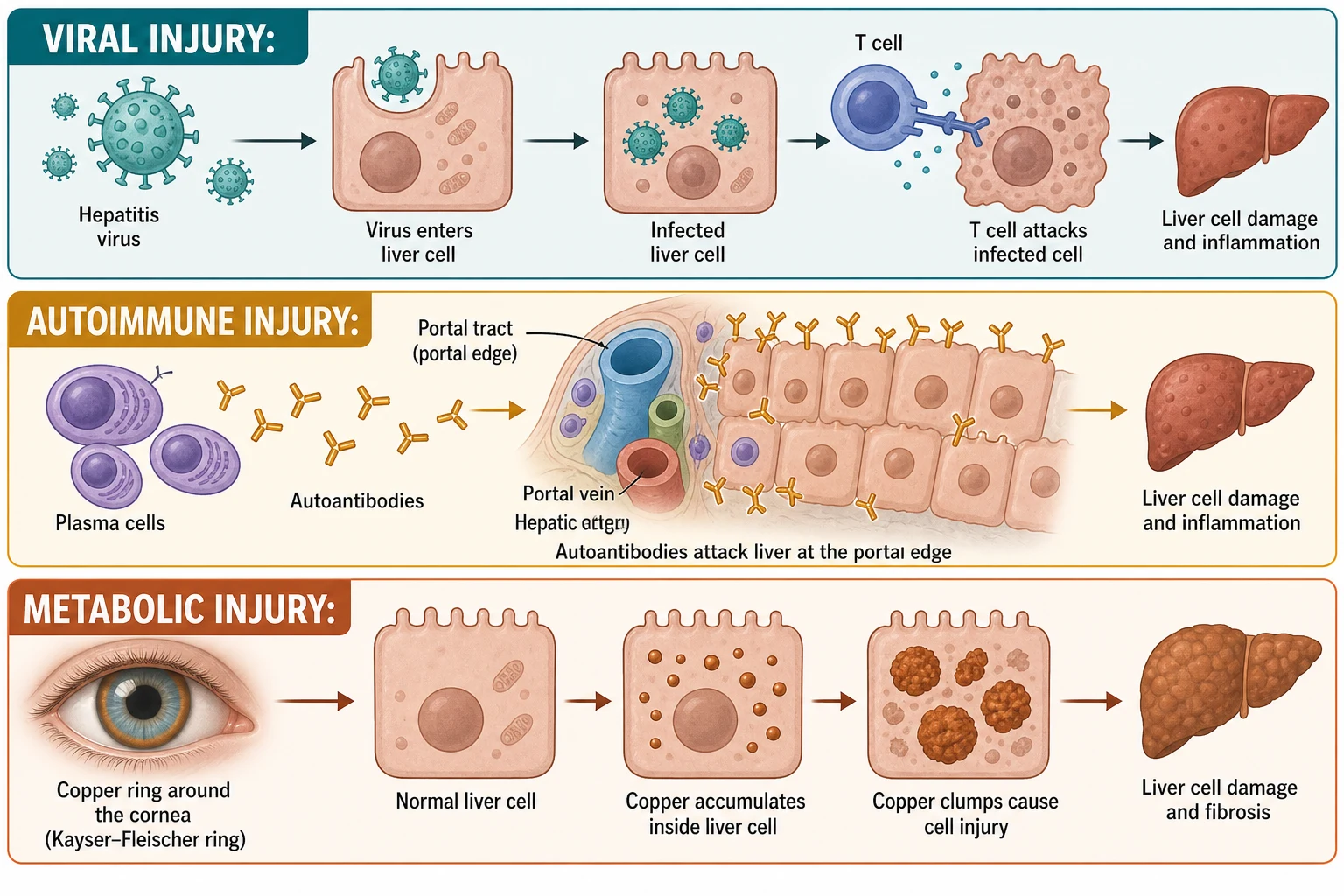
Autoimmune hepatitis is the loss of tolerance to liver antigens at the portal limiting plate. An antigen-presenting cell presents a liver autoantigen on a disease-associated HLA class 2 molecule to CD4-positive T-helper cells, which drive a T-helper-1 inflammatory cascade, recruit plasma cells and autoreactive B cells, and produce the characteristic autoantibodies. The histological signature is interface hepatitis, inflammation spilling from the portal tract across the limiting plate into the lobule, accompanied by a dense plasma-cell infiltrate, and the serological signature is a polyclonal rise in immunoglobulin G and the type-specific autoantibodies. The disease is fully steroid-responsive because the inflammation is immune-driven, which is the rationale for treatment. [8] [9]
Wilson disease is a copper-handling defect and alpha-1-antitrypsin deficiency is a protein-handling defect, and each poisons the hepatocyte in a different way. In Wilson disease, loss of the ATP7B copper-transporting ATPase blocks the excretion of copper into bile and its incorporation into ceruloplasmin, so toxic free copper accumulates in the hepatocyte, generates reactive oxygen species in the mitochondria, and deposits in the corneal Descemet membrane as the golden-brown Kayser-Fleischer ring. In alpha-1-antitrypsin deficiency, the PiZZ mutant protein misfolds in the endoplasmic reticulum and polymerises into chains that the cell cannot export, so the retained polymer aggregates as the PAS-positive, diastase-resistant globules that are the histological hallmark, and the proteotoxic stress drives the liver injury while the loss of circulating antiprotease leaves the lung undefended. [11] [12]
Clinical Presentation
The acute viral hepatitis syndrome is the face the enteric and the flared chronic viruses show. After an incubation that runs from two weeks for hepatitis A to several months for hepatitis B, the child develops a prodrome of fever, fatigue, anorexia, nausea, vomiting and right-upper-quadrant discomfort, often mistaken for a flu-like illness, and this is followed within days by the icteric phase of jaundice, dark urine from bilirubinuria, pale stools from cholestasis, and tender hepatomegaly. Young children often have anicteric or silent acute hepatitis A, which is the trap that lets them spread the virus in households and daycare. [1] [7]
The chronic viral hepatitides are usually silent and are found by screening. The perinatally infected hepatitis B child may carry the virus for decades in an immune-tolerant phase with normal transaminases and a high viral load, feeling entirely well, until the immune system begins to attack infected hepatocytes and raises the transaminases, or until cirrhosis or hepatocellular carcinoma declares the disease. Vertically acquired hepatitis C is similarly quiet in childhood, so the typical child is an asymptomatic sibling or the infant of a known HCV-positive mother found on testing. The presentation of decompensation, with jaundice, ascites, bruising or variceal bleeding, is a late and preventable event. [2] [5]
Autoimmune hepatitis has an insidious and a fulminant face. Most often an adolescent girl presents over months with fatigue, jaundice, acne, amenorrhoea, arthralgia and a rash, and the extrahepatic autoimmune associations of autoimmune thyroid disease, ulcerative colitis and type 1 diabetes are the company it keeps. A smaller number present with an acute hepatitis that mimics a viral infection, and a few present in acute liver failure. Type 2 autoimmune hepatitis, with anti-liver-kidney-microsomal antibody, tends to affect younger children and to run a more aggressive, relapsing course. The growth chart and the pubertal stage matter, because chronic inflammation and steroid treatment both affect growth. [8]
The metabolic hepatitides declare themselves across childhood. Wilson disease presents most often in school-age children or adolescents, with hepatic features ranging from incidental raised transaminases through chronic hepatitis and cirrhosis to the dramatic acute liver failure with a Coombs-negative haemolytic anaemia, or with neuropsychiatric features of tremor, dystonia, dysarthria, deteriorating schoolwork and the Kayser-Fleischer ring on slit-lamp examination. Alpha-1-antitrypsin deficiency presents most often as prolonged neonatal cholestasis that mimics biliary atresia, or later as an unexplained chronic hepatitis or cirrhosis, and the lung disease appears only in adulthood. [11] [12]
[1] [11]Differential Diagnosis
The differential for acute hepatitis in a child is wide, and the aetiological framework narrows it. The viral causes A to E come first, but drug-induced liver injury from paracetamol overdose, anticonvulsants or antibiotics, an autoimmune flare, an ischaemic or hypoxic hepatitis, and a metabolic decompensation in a galactosaemic or tyrosinaemic infant all belong on the list. The 2022 outbreak of acute hepatitis of unknown cause in young children, associated with adenovirus and later with adeno-associated virus type 2, is a recent addition that a fellowship candidate should name as a recognised entity now characterised rather than an idiopathic mystery. Sepsis, Epstein-Barr virus, cytomegalovirus and leptospirosis complete the infective differential. [1] [8]
For chronic hepatitis the differential is narrower and more rewarding. Chronic hepatitis B and C, autoimmune hepatitis, Wilson disease and alpha-1-antitrypsin deficiency are the five to hold, and the discriminating tests are the serology panels and the metabolic screen. Non-alcoholic fatty liver disease is now the commonest cause of raised transaminases in an overweight adolescent, and it must be distinguished from the genuine inflammatory hepatitides by its steatotic picture, normal immunoglobulins and absent autoantibodies. The inborn errors of metabolism, including galactosaemia, hereditary fructose intolerance and tyrosinaemia type 1, present in infancy with hepatitis that the metabolic screen is designed to catch. [2] [12]
Autoimmune hepatitis must be separated from autoimmune sclerosing cholangitis and primary sclerosing cholangitis, because the treatments differ. Autoimmune sclerosing cholangitis shares the autoantibodies and the interface hepatitis of autoimmune hepatitis but adds bile-duct injury visible on cholangiography, and it overlaps strongly with inflammatory bowel disease; primary sclerosing cholangitis is the stricturing, beaded duct disease of older children and adolescents, again strongly linked to inflammatory bowel disease. Magnetic-resonance cholangiopancreatography is the discriminator, performed after the autoimmune and metabolic workup is underway. [8]
The chronic hepatitis differential — 'B-COW-A'
Clinical & Bedside Assessment
Assessment begins, as in all of paediatrics, with the growth chart and the tempo. Plot the weight and height, look for the faltering that signals a chronic inflammatory or metabolic disease, and stage the puberty that autoimmune hepatitis and its steroid treatment will disturb. The history then frames the aetiology: ask about travel and food exposure for the enteric viruses, about the mother's hepatitis B and C status and any household contact for the blood-borne viruses, about consanguinity and a family history of liver or neuropsychiatric disease for the metabolic causes, and about drugs and supplements for a toxin. A girl with amenorrhoea and arthralgia points to autoimmune hepatitis; a child with deteriorating handwriting points to Wilson disease. [2] [11]
Examination focuses on the liver and the company it keeps. Look for jaundice, excoriations from pruritus, spider naevi, palmar erythema, clubbing and the peripheral stigmata of chronic liver disease; palpate for the tender hepatomegaly of acute hepatitis and the hard, nodular edge of cirrhosis, and for splenomegaly and ascites that signal portal hypertension. The Kayser-Fleischer ring is the single most rewarding physical sign in hepatology, a golden-brown copper deposit at the periphery of the cornea best seen on slit-lamp examination, and its presence in a child with hepatitis is Wilson disease until excluded. The neurological examination adds the tremor, dystonia and dysarthria of Wilson disease that the family may have attributed to clumsiness. [11]
The extrahepatic signs carry diagnostic weight because each aetiology keeps its own company. Autoimmune hepatitis travels with thyroid disease, vitiligo, arthritis, ulcerative colitis and the stigmata of type 1 diabetes, so the skin, the joints, the thyroid and the fundus are part of the examination. The child with chronic hepatitis B or C may carry the stigmata of the perinatal or household exposure, and the child with alpha-1-antitrypsin deficiency may have the dyspnoea of early lung disease in an affected adult relative, though this is rare in childhood. The combination of a careful history and a directed examination usually narrows the aetiological list to two or three possibilities before any blood is drawn. [8] [12]
Investigations
The investigation of hepatitis is built on the serology and metabolic panels, and the first question is whether the disease is acute or chronic and viral or otherwise. The hepatitis A serology is straightforward: IgM anti-HAV confirms acute infection and IgG anti-HAV confirms past immunity or vaccination, and there is no chronic state to test for. The hepatitis E serology parallels it, with IgM anti-HEV for acute infection and the recognition that chronic infection can occur in the immunosuppressed child, where HCV-style RNA testing is adapted. Transaminases are raised in the hundreds to thousands during acute viral hepatitis, and the prothrombin time is the single most important prognostic test, because a rising or prolonged prothrombin time signals synthetic failure and impending liver failure. [1] [7]
The hepatitis B serology panel is the one examiners probe, because it distinguishes the phases that decide management. Hepatitis B surface antigen marks infection, and antibody to surface antigen marks immunity; hepatitis B e antigen marks high infectivity and immune tolerance, and antibody to e antigen marks the transition to lower infectivity; antibody to core antigen in the IgM class marks acute infection and in the IgG class marks past exposure. The viral load by HBV DNA quantifies replication, and the four phases follow from these markers: the immune-tolerant phase of high viral load with normal transaminases seen in perinatally infected children; the immune-active HBeAg-positive phase of high viral load with raised transaminases and active inflammation; the inactive carrier phase of low viral load with normal transaminases after e-antigen seroconversion; and the HBeAg-negative immune-active phase of fluctuating transaminases with precore-mutant virus. The AASLD 2018 guidance defines when to treat. [3] [2]
Hepatitis C is simpler because it has no carrier phase. Antibody to HCV screens for exposure, and because maternal antibody passively transfers and persists for up to eighteen months, the diagnosis in infancy is confirmed by HCV RNA, which is also the test of active infection at any age. Once chronic infection is confirmed, the genotype and the viral load are taken before the direct-acting antiviral regimen is chosen, and the sustained virologic response, defined as undetectable HCV RNA twelve weeks after completing treatment, is the cure endpoint that exceeds ninety-five per cent with modern regimens. [5] [6]
Autoimmune hepatitis is diagnosed on the autoantibodies, the immunoglobulins, the histology and the exclusion of viral causes, combined in the International Autoimmune Hepatitis Group scoring systems. The simplified score adds four components: autoantibody titre, with antinuclear or anti-smooth-muscle antibody at one in forty scoring one point and at one in eighty or anti-liver-kidney-microsomal antibody scoring two; immunoglobulin G above the upper limit of normal scoring one and above one point one times the limit scoring two; histology compatible with autoimmune hepatitis scoring one and typical scoring two; and the absence of viral hepatitis scoring two. A total of seven or more defines definite autoimmune hepatitis and six defines probable, a threshold the examiner rewards for naming. The revised original score is broader, including the sex, the alcohol intake and the response to treatment, with a pretreatment score of fifteen or more defining definite disease and ten to fifteen defining probable disease. A liver biopsy confirms the interface hepatitis and stages the fibrosis. [9] [10]
The simplified autoimmune hepatitis score — the detail examiners probe
The simplified International Autoimmune Hepatitis Group score (Hennes 2008) sums four components. Autoantibodies: antinuclear antibody or anti-smooth-muscle antibody at one in forty scores one point, and at one in eighty or above, or anti-liver-kidney-microsomal antibody type 1 at one in forty or above, or soluble liver antigen positive, scores two points. Immunoglobulin G: above the upper limit of normal scores one, above one point one times the upper limit scores two. Histology: compatible with autoimmune hepatitis scores one, typical of autoimmune hepatitis scores two. Absence of viral hepatitis: confirmed scores two. The maximum is eight points; a total of seven or more is definite autoimmune hepatitis and a total of six is probable. The revised original score (Alvarez 1999) is broader and gives a pretreatment score, with fifteen or more defining definite disease. [9] [10]
Wilson disease is diagnosed on the Leipzig score, a point system that combines the clinical and biochemical features. A serum ceruloplasmin under zero point two grams per litre scores two points; the presence of a Kayser-Fleischer ring scores two points with a count of one; a twenty-four-hour urinary copper over forty micrograms per day scores one point and over one hundred scores two, with the higher cut-off very suggestive of the disease; and the neurological presentation and the Coombs-negative haemolytic anaemia add further points. A Leipzig score of four or more establishes the diagnosis, two to three prompts further testing, and the score is the framework the examiner expects. The diagnosis is anchored on the combination of low ceruloplasmin, the Kayser-Fleischer ring, a high urinary copper and the ATP7B genotype, never on any single test, because ceruloplasmin is an acute-phase reactant and can be normal in early or inflammatory disease. [11]
Alpha-1-antitrypsin deficiency is diagnosed on the serum level and the phenotype. The serum alpha-1-antitrypsin level is low, and the phenotype by isoelectric focusing identifies the PiZZ protease-inhibitor variant that causes the disease, distinguishing it from the normal PiMM and the intermediate PiMZ carrier states that do not cause liver disease in their own right. A liver biopsy shows the PAS-positive, diastase-resistant globules of retained polymer in periportal hepatocytes, the histological hallmark, and it stages any fibrosis. The genetic test confirms the SERPINA1 mutation. The important pitfall is that a normal serum level does not exclude the deficiency if the phenotype is abnormal, because the misfolded protein is made but not secreted. [12]
Management — Resuscitation
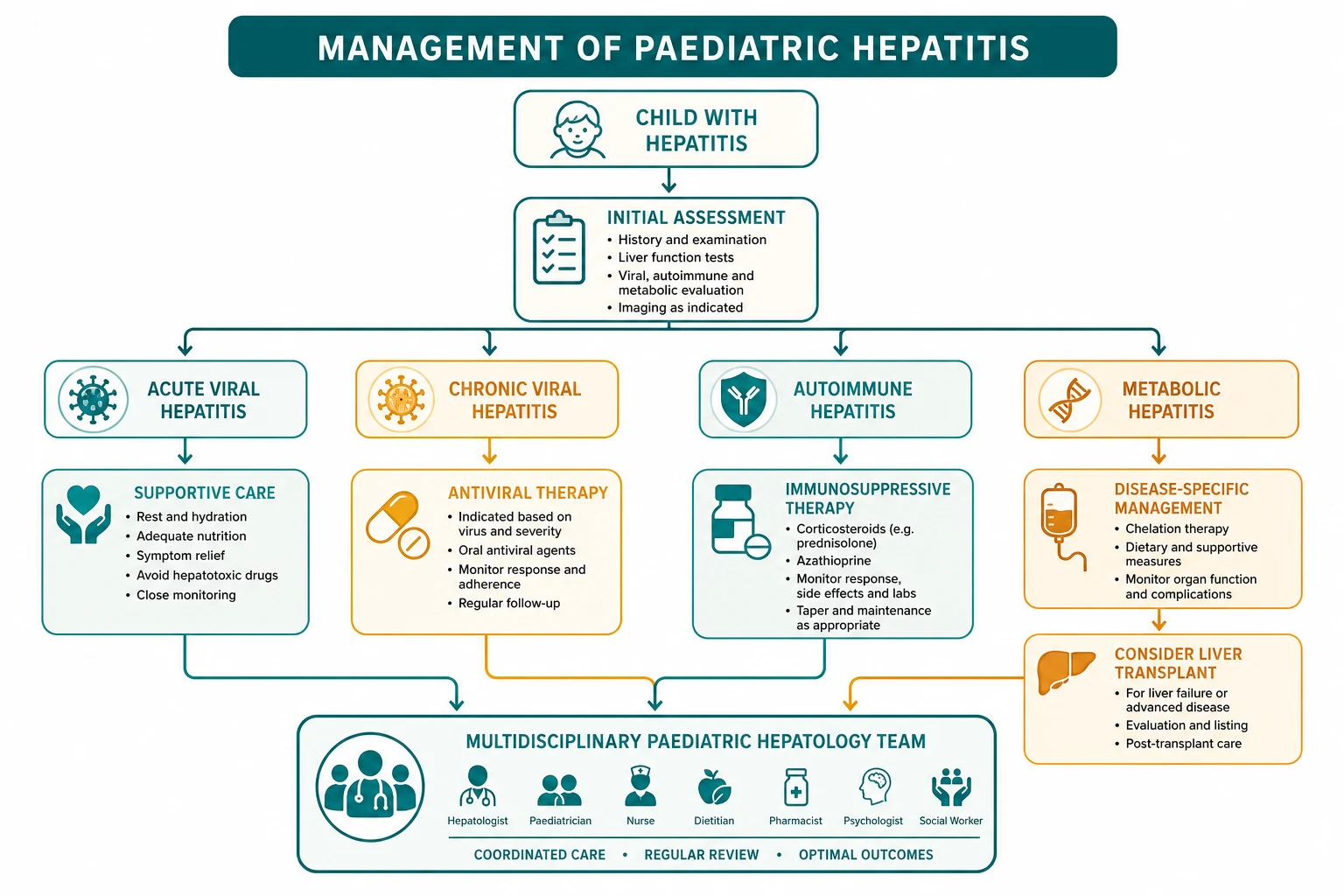
Most hepatitis is managed electively, but the child who presents in acute liver failure from any cause is a time-critical emergency, and the resuscitation is the same regardless of the aetiology. Assess the airway, breathing and circulation, secure intravenous access, and check the glucose immediately because hypoglycaemia from synthetic failure is both common and harmful. Correct the coagulopathy with vitamin K and, for bleeding or before procedures, fresh frozen plasma, recognising that the prothrombin time is the single most sensitive marker of hepatic synthetic function and a rising value is an ominous sign. Treat the encephalopathy with lactulose and rifaximin to reduce ammonia, identify and remove any precipitant such as an infection or a sedating drug, and refer early to a paediatric liver transplant centre, because the window for transfer closes once multi-organ failure develops. [2] [11]
The fulminant Wilsonian crisis is a specific emergency that the examiner rewards for recognising. The child presents in acute liver failure with a Coombs-negative haemolytic anaemia, a low urate, a low alkaline phosphatase relative to bilirubin, and a high urinary copper, and the only curative treatment is liver transplantation. Chelation with penicillamine or trientine and plasmapheresis or albumin dialysis to remove copper are used as a bridge to transplant, but they do not rescue the failing liver, so the referral to a transplant centre must be made at the moment the diagnosis is suspected. The King's College criteria for liver transplantation in Wilson disease, including a combined bilirubin and INR threshold, guide the listing decision. [11]
Severe autoimmune hepatitis presenting with acute liver failure is the third resuscitation scenario. Corticosteroids at high dose may be started once infection is excluded, because the inflammation is immune-mediated and steroid-responsive, but a child who does not improve promptly is heading for transplant, and the listing decision is made on the clinical course and the King's College criteria rather than on a single blood test. The post-exposure prophylaxis scenarios complete the acute picture: the infant of an HBsAg-positive mother receives hepatitis B vaccine and hepatitis B immunoglobulin within twelve hours of birth, and a close contact of a hepatitis A case receives the hepatitis A vaccine within two weeks, with immunoglobulin reserved for the immunocompromised and the under-twelve-months infant. [4] [8]
Immediate management of the child with acute liver failure from hepatitis
Assess airway, breathing and circulation; secure access; check capillary glucose and treat hypoglycaemia
Send coagulation, full blood count, ammonia, a drug screen including paracetamol level, viral serology, autoimmune and metabolic panels including ceruloplasmin and urinary copper
Give vitamin K and correct coagulopathy; transfuse for bleeding only
Treat encephalopathy with lactulose and rifaximin; identify and remove precipitants
Monitor prothrombin time, glucose and ammonia hourly; the rising prothrombin time signals worsening synthetic failure
Refer early to a paediatric liver transplant centre and apply the King's College criteria for listing
Start aetiology-specific treatment once the cause is identified: chelation for Wilson disease, corticosteroids for autoimmune hepatitis, antivirals for chronic viral hepatitis
Management — Definitive & Stepwise
The definitive management follows the cause. Acute hepatitis A and E need no specific antiviral treatment: supportive care with hydration, nutrition and rest, attention to the pruritus and the nausea, and monitoring for the small minority who develop fulminant failure, with hospital admission for the child with a prolonged prothrombin time or encephalopathy. Prevention is the public-health companion: the hepatitis A vaccine from twelve months of age and before travel, good hand and food hygiene, and post-exposure vaccine within two weeks for close contacts. There is no specific antiviral for hepatitis E, and chronic hepatitis E in the immunosuppressed is treated by reducing immunosuppression where possible and, for the persistent infection, ribavirin. [1] [7]
Chronic hepatitis B is managed by phase, and the AASLD 2018 guidance sets the rules. The immune-tolerant child, common in perinatally acquired disease, has a high viral load with normal transaminases and minimal inflammation, and is monitored rather than treated, because the risk of treatment is not justified by the benefit in this low-inflammation phase. The immune-active child, with raised transaminases and active inflammation, is treated with a nucleoside analogue, entecavir in children aged two years and older or tenofovir disoproxil fumarate in those aged twelve years and older, or with pegylated interferon, to suppress viral replication and halt the progression to cirrhosis and hepatocellular carcinoma. Lifelong surveillance for hepatocellular carcinoma with six-monthly ultrasound and alpha-fetoprotein begins once chronic infection is established. [3] [2]
Chronic hepatitis C is now curable in children, and the direct-acting antiviral era has transformed it from a slowly progressive disease into a short medical treatment. Regimens such as ledipasvir-sofosbuvir, sofosbuvir-velpatasvir and glecaprevir-pibrentasvir are approved in children aged three years and older, given as a single daily tablet for eight to twelve weeks, and they achieve a sustained virologic response in over ninety-five per cent of children regardless of genotype and without the toxicity of the old interferon regimens. The treatment is so safe and effective that the modern approach is to treat every child with chronic infection, and the remaining challenge is case-finding in the vertically infected cohort. [6] [5]
Hepatitis C direct-acting antiviral regimens in children
Loading dose
Ledipasvir-sofosbuvir is approved for children aged three years and older, dosed by weight or as a fixed-dose dispersible tablet; sofosbuvir-velpatasvir and glecaprevir-pibrentasvir are options from three years of age
Maintenance dose
An eight-to-twelve-week course achieves sustained virologic response in over ninety-five per cent of children regardless of genotype, replacing the toxic interferon-based regimens of the past
Autoimmune hepatitis is treated with immunosuppression, and the standard induction is oral prednisolone with azathioprine. Prednisolone is started at one to two milligrams per kilogram per day, to a maximum of around sixty milligrams per day, and tapered over weeks to months as the transaminases and immunoglobulin G fall, while azathioprine is introduced at one to two milligrams per kilogram per day as a steroid-sparing maintenance agent, with thiopurine methyltransferase activity checked before the full dose to avoid fatal myelosuppression. Budesonide is an alternative steroid with less systemic absorption for those without cirrhosis. The goal is biochemical remission, and the maintenance continues for years, often to adulthood, because relapse off treatment is the rule. Liver transplantation is reserved for those who present in fulminant failure or who progress to cirrhosis despite treatment. [8]
Autoimmune hepatitis induction in children
Loading dose
Prednisolone one to two milligrams per kilogram per day by mouth, to a maximum of around sixty milligrams per day, combined with azathioprine one to two milligrams per kilogram per day by mouth
Maintenance dose
Taper the prednisolone over weeks to months as transaminases and immunoglobulin G normalise; maintain azathioprine for years. Check thiopurine methyltransferase activity before introducing the full azathioprine dose to avoid myelosuppression.
Wilson disease is treated with lifelong chelation or zinc, and the choice depends on the presentation. Chelation with penicillamine, at around twenty milligrams per kilogram per day in divided doses, or trientine, drives the urinary excretion of copper and is the standard initial treatment for the symptomatic child, with the caveat that penicillamine can worsen the neurological presentation at the start and requires pyridoxine supplementation and monitoring for proteinuria and skin reactions. Zinc, at the age-adjusted elemental dose, blocks the intestinal absorption of copper by inducing metallothionein in the enterocyte, and it is used for maintenance and for the presymptomatic child. Treatment is lifelong, because stopping copper removal leads to rapid hepatic and neurological deterioration, and adherence is the single most important determinant of outcome. Liver transplantation rescues the fulminant or end-stage case. [11]
Wilson disease chelation and zinc in children
Loading dose
Penicillamine around twenty milligrams per kilogram per day in divided doses, or trientine, with pyridoxine twenty-five to fifty milligrams daily for penicillamine; alternatively zinc as elemental zinc by age for maintenance and presymptomatic disease
Maintenance dose
Lifelong treatment is mandatory; monitor twenty-four-hour urinary copper to titrate the dose, aiming for a body copper balance, and screen for proteinuria on penicillamine and for skin reactions
Alpha-1-antitrypsin deficiency has no specific medical treatment for the liver disease, because the injury is the retained misfolded polymer and augmentation therapy raises the circulating level without addressing the retention. Management is supportive and surveillance-based: avoidance of hepatotoxins including alcohol in later life, hepatitis A and B vaccination to remove additional insults, monitoring of liver function and liver synthetic markers, and nutritional support. Augmentation therapy with weekly intravenous alpha-1-antitrypsin is used for the lung disease in adults with established emphysema, but it does not help and does not prevent the liver disease, and to offer it for the hepatic phenotype is the classic pitfall. Liver transplantation replaces the failing liver and the abnormal genotype with donor tissue that makes normal protein, and it is curative for the end-stage liver disease, though it does not address any established lung disease. [12]
Specific Subtypes & Scenarios
The perinatally infected hepatitis B child is the commonest chronic hepatitis scenario, and the test of breadth is knowing the phase and when to treat. Acquired at birth, the child enters a prolonged immune-tolerant phase that may last two to three decades, with a high HBV DNA, positive hepatitis B e antigen, normal transaminases and minimal histological inflammation, and the correct management is surveillance, not treatment, because treating here gains little and risks resistance. Treatment begins when the child enters the immune-active phase, signalled by rising transaminases and active inflammation, with a nucleoside analogue to suppress replication. The hepatocellular carcinoma surveillance with six-monthly ultrasound begins once chronic infection is confirmed, because childhood-onset hepatocellular carcinoma, though rare, occurs in chronic hepatitis B. [2] [3]
The adolescent with vertically acquired hepatitis C is the curable scenario. Identified by the maternal history or by screening, confirmed by HCV RNA, and treated at age three years or older with a short direct-acting antiviral course, the adolescent is cured in the great majority, and the burden shifts from managing a chronic disease to finding and treating the infected cohort. The remaining challenge is the infant under three years, in whom treatment is deferred until the licensed age, with monitoring to confirm the persistence of infection rather than spontaneous clearance, which occurs in a quarter of vertically infected infants in the first years of life. [6] [5]
The adolescent girl with type 1 autoimmune hepatitis is the immunosuppression scenario. She presents insidiously with fatigue, jaundice, acne and amenorrhoea, a raised immunoglobulin G and a positive antinuclear or anti-smooth-muscle antibody, and the simplified score confirms the diagnosis. The induction with prednisolone and azathioprine brings her into remission, the steroid is tapered, and she is maintained on azathioprine for years, with attention to the bone density, the growth and the psychological burden of a chronic disease and its treatment in adolescence. The overlap with autoimmune sclerosing cholangitis and inflammatory bowel disease is sought by cholangiography and colonoscopy, because the overlap changes the prognosis and the surveillance. [8] [10]
The young child with Wilson disease in acute liver failure is the transplant scenario, and the infant with alpha-1-antitrypsin-deficiency neonatal cholestasis is the diagnostic-trap scenario. The Wilsonian child presents with a Coombs-negative haemolytic anaemia, acute liver failure and a low urate, the diagnosis is confirmed by a high urinary copper and a Kayser-Fleischer ring, and the treatment is urgent transplant listing. The alpha-1-antitrypsin infant presents with prolonged conjugated jaundice and pale stools that mimic biliary atresia, and the phenotype test is what distinguishes the two, because the intraoperative cholangiogram that confirms biliary atresia is an avoidable operation in the alpha-1-antitrypsin child. [11] [12]
[11] [12]Complications & Pitfalls
The complications of untreated or poorly controlled hepatitis flow from chronic inflammation and hepatocellular injury. The chronic viral hepatitides, autoimmune hepatitis and the metabolic diseases all converge on the same endpoint of fibrosis, cirrhosis and portal hypertension, with the risks of variceal bleeding, ascites, spontaneous bacterial peritonitis and hepatorenal syndrome. Hepatocellular carcinoma is the feared late complication of chronic hepatitis B at any stage and of cirrhosis from any cause, and it is the reason for the six-monthly surveillance ultrasound in chronic hepatitis B. Acute liver failure, with encephalopathy and synthetic failure, is the immediate life-threatening complication of fulminant viral hepatitis, the Wilsonian crisis and the severe autoimmune flare. [2] [11]
The drug-treatment complications matter because each regimen carries its own. Penicillamine for Wilson disease causes an initial worsening of the neurological presentation in a minority, skin reactions and proteinuria, so it is introduced slowly and with pyridoxine and monitoring; the early nephrotic syndrome and the late elastosis perforans serpiginosa are the recognised effects. Azathioprine for autoimmune hepatitis can cause fatal myelosuppression in the thiopurine methyltransferase-deficient child, so the enzyme is checked before the full dose, and pancreatitis and hepatotoxicity are the other toxicities. The corticosteroid burden on growth, bone density, glucose and mood is the everyday price of autoimmune hepatitis treatment in childhood. [8] [11]
The diagnostic pitfalls are the failures the examiner rewards for naming. The first is relying on a single normal ceruloplasmin to exclude Wilson disease, because it is an acute-phase reactant and can be normal in early or inflammatory disease, so the urinary copper and the Kayser-Fleischer ring must always accompany it. The second is mistaking the immune-tolerant phase of paediatric hepatitis B for active disease and treating unnecessarily, because the high viral load with normal transaminases is not inflammation. The third is offering alpha-1-antitrypsin augmentation therapy for the hepatic phenotype, because it treats the lung and does nothing for the liver. The fourth is omitting the alpha-1-antitrypsin phenotype in the infant with neonatal cholestasis and missing the diagnosis. [11] [12]
Prognosis & Disposition
The prognosis follows the cause and the treatment. Acute hepatitis A and E carry an excellent prognosis with full recovery in the great majority within weeks, and fulminant failure is rare in children. Chronic hepatitis B is a lifelong infection whose outcome depends on the phase, the treatment and the surveillance, with the perinatally infected child facing decades of monitoring and a real risk of cirrhosis and hepatocellular carcinoma in adulthood; treatment of the immune-active phase and lifelong hepatocellular carcinoma surveillance are the modifiers. Chronic hepatitis C is now curable in the great majority with a short direct-acting antiviral course, so the prognosis is excellent once the diagnosis is made and the treatment given. [2] [6]
Autoimmune hepatitis has a high remission rate on immunosuppression, but it is a relapsing disease and many children require maintenance treatment into adulthood, with the prognosis set by the adherence and the avoidance of relapse. The child who presents in fulminant failure or who progresses to cirrhosis despite treatment faces a worse outcome, and a minority come to liver transplantation. The long-term risks of corticosteroid and azathioprine treatment, including impaired growth, reduced bone density and the small excess of skin cancer and lymphoma on long-term azathioprine, are the price of the disease control, and they demand structured monitoring. [8]
Wilson disease has an excellent prognosis with early diagnosis and lifelong chelation or zinc, because removing copper halts the hepatic and neurological injury, and the treated child can expect a normal life. The prognosis is poor once cirrhosis is established or once the neurological damage has occurred, because the latter is only partly reversible, and the child who presents in fulminant Wilsonian crisis survives only with transplantation. Alpha-1-antitrypsin deficiency has a variable prognosis: many PiZZ children have mild or subclinical liver disease, but a minority progress to cirrhosis and liver failure in childhood, and the only curative treatment for the end-stage liver is transplantation. The lung disease appears in adulthood and is the major long-term driver of morbidity in the smoker. [11] [12]
Disposition follows the diagnosis and the response. The child with acute viral hepatitis is managed at home or briefly in hospital with supportive care, with referral for the fulminant case. The child with chronic viral hepatitis is shared between the general paediatrician and a paediatric hepatology service for phase assessment, treatment and surveillance. The child with autoimmune hepatitis or a metabolic hepatopathy is managed in a tertiary centre with paediatric gastroenterology and hepatology, dietetics, psychology and, where needed, transplant surgery, and transition to adult services is planned in mid-adolescence. The child in acute liver failure is transferred to a liver transplant centre as soon as the diagnosis is suspected, because the window for listing closes with multi-organ failure. [2] [11]
Special Populations
The pregnant adolescent and the prevention of perinatal hepatitis B is the first special population, because the chronicity rate of ninety per cent without immunoprophylaxis makes prevention the single most effective intervention in all of hepatology. Every pregnant woman is screened for hepatitis B surface antigen, and the infant of a positive mother receives the hepatitis B vaccine and hepatitis B immunoglobulin within twelve hours of birth, followed by the full vaccine course, with post-vaccination serology at nine to twelve months to confirm protection. For the mother with a high viral load in the third trimester, maternal antiviral prophylaxis with tenofovir from twenty-eight weeks further reduces the residual transmission risk, and this two-pronged prevention is now the standard of care in most guidelines. [4] [3]
The immunocompromised child is the second special population, because the rare chronic hepatitis E occurs here. In the solid-organ transplant recipient or the child on intensive immunosuppression, hepatitis E can persist as a chronic infection with progressive fibrosis, the diagnosis requires HEV RNA testing, and the treatment is reduction of immunosuppression where possible and ribavirin for the persistent infection. The immunocompromised child is also vulnerable to the reactivation of chronic hepatitis B, so screening before chemotherapy or biologic therapy and prophylaxis during and after treatment are the safeguards. [7] [3]
The Indigenous, remote and migrant child is the third, because the burden of chronic viral hepatitis falls unevenly. Higher rates of vertical hepatitis B transmission, poorer access to antenatal screening and birth-dose vaccine, and the late presentation of cirrhosis and hepatocellular carcinoma are the signature of under-resourced settings, and the response is catch-up vaccination, culturally safe shared-care pathways and telehealth support for the local team. In many regions the screening and treatment of hepatitis C in the vertically infected cohort is the unfinished task, and the direct-acting antiviral cure is the tool to finish it. [2] [5]
Across Australia, New Zealand and the United Kingdom, universal hepatitis B vaccination from birth is the cornerstone of prevention, with antenatal screening of every pregnant woman and birth-dose vaccine and immunoglobulin for the infants of HBsAg-positive mothers. The direct-acting antivirals for hepatitis C are funded and available in paediatric hepatology centres from age three years. Autoimmune hepatitis is managed in tertiary paediatric gastroenterology and hepatology services with the standard prednisolone-and-azathioprine regimen, and Wilson disease is managed with chelation and lifelong monitoring. In many low- and middle-income settings the birth-dose vaccine coverage is incomplete, the direct-acting antivirals and the metabolic workup are harder to access, and the burden of chronic viral hepatitis and undiagnosed metabolic disease is carried silently, so the global agenda is vaccine equity and case-finding. [3] [8] [11]
Evidence, Guidelines & Regional Differences
The evidence base for the viral hepatitides is anchored on the AASLD and ESPGHAN guidance and the ACIP prevention recommendations. The AASLD 2018 hepatitis B guidance defined the phases of chronic infection and the criteria for treatment with entecavir and tenofovir, and the ESPGHAN paediatric hepatitis B guideline adapted these to children, emphasising the prolonged immune-tolerant phase and the surveillance for hepatocellular carcinoma. The ACIP 2018 recommendations set the universal birth-dose vaccine and the immunoprophylaxis of infants of HBsAg-positive mothers, the single most effective intervention in all of hepatology. [3] [4]
The hepatitis C evidence is the direct-acting antiviral revolution. The Indolfi 2019 reviews set out the epidemiology and the natural history of hepatitis B and C in children, and the Indolfi 2018 paper on direct-acting antivirals reported the sustained virologic response rates above ninety-five per cent that transformed hepatitis C from a slowly progressive disease into a short curable treatment, leading to the licensing of ledipasvir-sofosbuvir and related regimens in children aged three years and older and to the modern approach of treating every infected child. [5] [6]
The autoimmune hepatitis evidence is the International Autoimmune Hepatitis Group scoring systems and the ESPGHAN paediatric position paper. The Alvarez 1999 revised report set the comprehensive scoring system that integrates the autoantibodies, the immunoglobulins, the histology and the response to treatment, and the Hennes 2008 simplified score distilled this into the four-component score, with seven or more defining definite disease, that is the everyday clinical tool. The Mieli-Vergani 2018 ESPGHAN position paper set the paediatric management, the type 1 and type 2 distinction and the overlap with autoimmune sclerosing cholangitis. [8] [9] [10]
The metabolic hepatitides are anchored on the Wilson disease practice guidance and the alpha-1-antitrypsin reviews. The 2022 Wilson disease practice guidance codified the Leipzig score, the choice between chelation and zinc, and the role of liver transplantation, and it is the current reference for the diagnosis and management of the disease. The alpha-1-antitrypsin reviews set the gain-of-toxic-function mechanism of the liver disease, the PAS-positive diastase-resistant globules, the PiZZ phenotype and the absence of a specific medical treatment for the liver, with transplantation as the curative option. The Verghese 2014 systematic review set the epidemiology and the natural history of hepatitis E in children. [11] [12] [7]
Exam Pearls
The hepatitis B serology panel — 'SAg, SAb, eAg, eAb, cAb, DNA'
Wilson disease at the bedside — 'COPPER'
References
- [1]Abutaleb A; Kottilil S; Wilson E Hepatitis A: Epidemiology, Natural History, Unusual Clinical Manifestations, and Prevention. Gastroenterol Clin North Am, 2020.PMID 32389358
- [2]Indolfi G; Gramenzi A; Degasperi E; Lorini FL; De Santis A; Saccardi F Hepatitis B virus infection in children and adolescents. Lancet Gastroenterol Hepatol, 2019.PMID 30982722
- [3]Terrault NA; Lok ASF; McMahon BJ; Chang KM; Hwang JP; Jonas MM Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology, 2018.PMID 29405329
- [4]Schillie S; Vellozzi C; Reingold A; Harris A; Haber P; Ward JW Prevention of Hepatitis B Virus Infection in the United States: Recommendations of the Advisory Committee on Immunization Practices. MMWR Recomm Rep, 2018.PMID 29939980
- [5]Indolfi G; Easterbrook P; Dusheiko G; El-Sayed MH; Leser M; Thorne C Hepatitis C virus infection in children and adolescents. Lancet Gastroenterol Hepatol, 2019.PMID 30982721
- [6]Indolfi G; Hierro L; Debray D; D'Antiga L; Nossova N; Arenas JI Direct-acting antivirals for children and adolescents with chronic hepatitis C. Lancet Child Adolesc Health, 2018.PMID 30169301
- [7]Verghese VP; Rongsen-Chandola T; Sridhar S; Chitambar SD; Goyal S; Jana A A systematic review of hepatitis E virus infection in children. Clin Infect Dis, 2014.PMID 24846637
- [8]Mieli-Vergani G; Vergani D; Baumann U; Czubkowski P; Debray D; Dezsofi A Diagnosis and Management of Pediatric Autoimmune Liver Disease: ESPGHAN Hepatology Committee Position Paper. J Pediatr Gastroenterol Nutr, 2018.PMID 29356770
- [9]Hennes EM; Zeniya M; Czaja AJ; Parés A; Dalekos GN; Krawitt EL Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology, 2008.PMID 18537184
- [10]Alvarez F; Berg PA; Bianchi FB; Bianchi L; Burroughs AK; Cancado EL International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol, 1999.PMID 10580593
- [11]Schilsky ML; Roberts EA; Ala A; Allen KR; Almeida MP; Anand MK A multidisciplinary approach to the diagnosis and management of Wilson disease: 2022 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology, 2025.PMID 36151586
- [12]Suri A; Altraja A; Mäkelä S; Krams A; Mindikoglu AL; Galvin Z Alpha-1 Antitrypsin Deficiency Liver Disease. Clin Liver Dis, 2022.PMID 35868681