Paeds · genetics-dysmorphology-and-metabolism
22q11.2 deletion syndrome
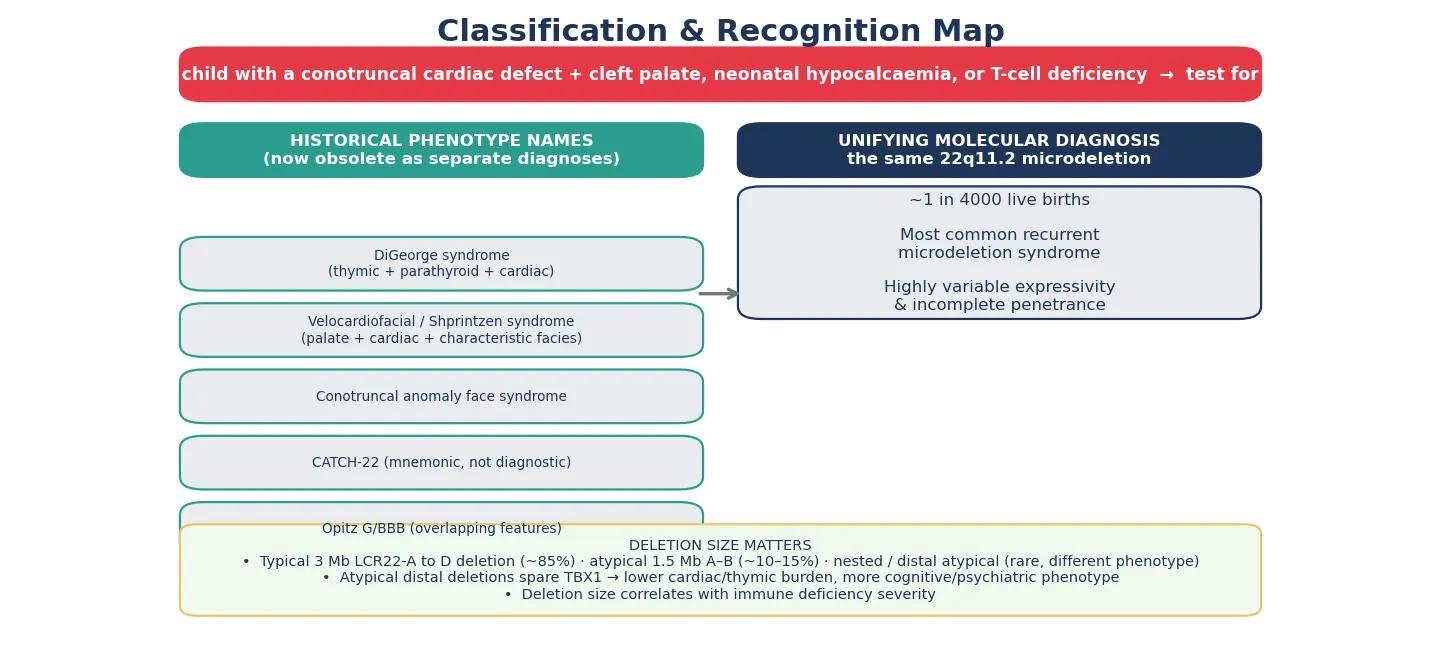
Also known as 22q11.2 deletion syndrome · DiGeorge syndrome · velocardiofacial syndrome · Shprintzen syndrome · conotruncal anomaly face syndrome · CATCH-22
A fellowship approach to 22q11.2 deletion syndrome: recognise the multisystem fingerprint (conotruncal cardiac defect, cleft palate, neonatal hypocalcaemia, thymic and T-cell deficiency, characteristic facies) that earns a chromosomal microarray, confirm the deletion with a microarray rather than a karyotype, stage every organ system at diagnosis, defer live vaccines until T-cell deficiency is excluded, and run an age-based multidisciplinary surveillance plan through to adult transition.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A neonate in the intensive care unit is grunting and duset at twenty-four hours of age, the duct-dependent heart closing in front of you; or a new baby is seized, and the ionised calcium is undetectable; or a three-year-old speaks through her nose, catches every virus at preschool, and carries a repaired tetralogy of Fallot on her chart. The fellowship task is to see that each of these is the same chromosome, to send the test that will find it, and to lay a surveillance net across heart, calcium, palate, immunity, development, and mental health before any one of them causes harm. [1] [11]
CATCH-22 — and the sixth finger
Overview & Definition
A baby born with a hemizygous loss of genetic material at chromosome 22q11.2 carries the most common recurrent microdeletion in humans, and the phenotype that follows is so variable that it gathered a handful of eponyms before the molecular unification. What Angelo DiGeorge described in the 1960s as congenital absence of the thymus and parathyroids, Robert Shprintzen later called velocardiofacial syndrome for the cleft palate, cardiac defect, and characteristic face, and cardiologists labelled the conotruncal anomaly face syndrome. Each is the same deletion, and the modern name — 22q11.2 deletion syndrome — is the one that unifies them and sets the genetic counselling. [1] [11]
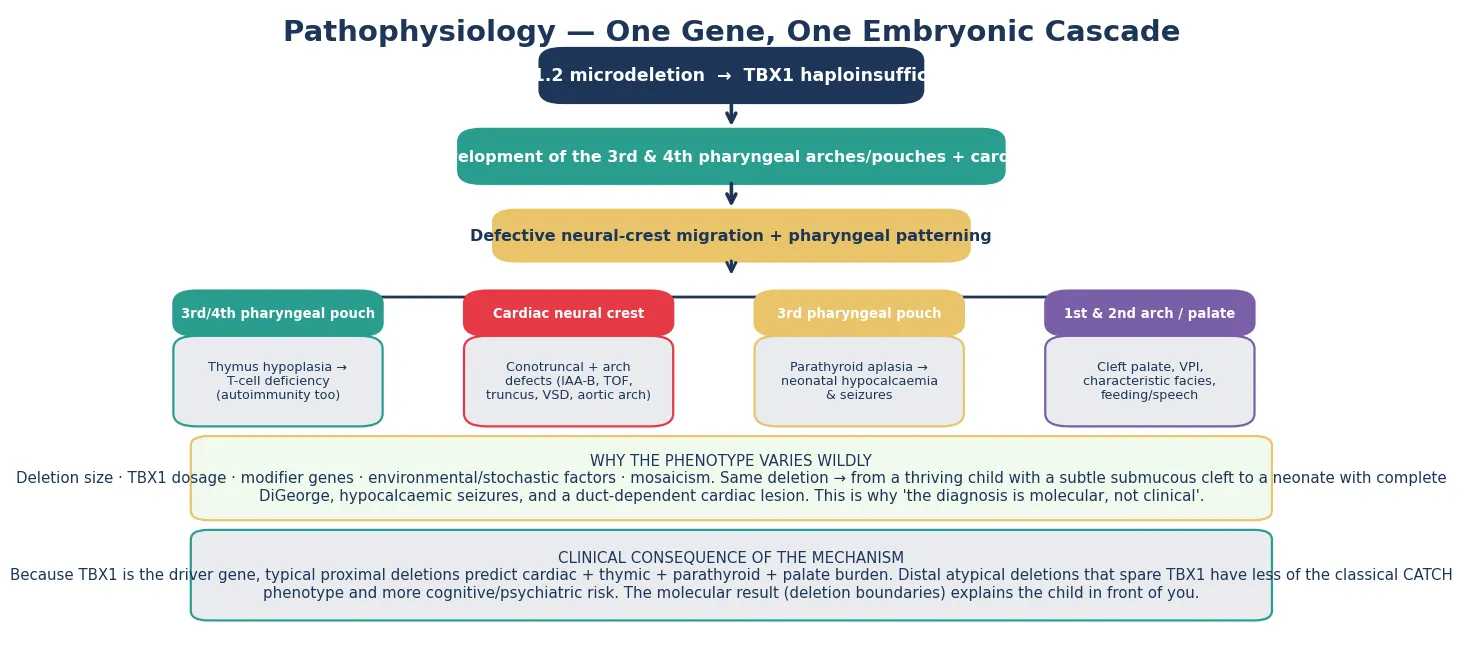
The deletion removes around 30 to 40 genes, and the loss of a single one — TBX1, a transcription factor that patterns the third and fourth pharyngeal arches and pouches — accounts for the cardiac, thymic, parathyroid, palatal, and facial features. Expressivity is wildly variable and penetrance incomplete, so the same deletion can produce a neonate with complete DiGeorge syndrome and an adult whose only clue is a repaired heart lesion and a late diagnosis of schizophrenia. That variability is the central teaching point: the microdeletion is the unifying fact, but the patient in front of you is one point on a wide spectrum. [1] [3]
The lifespan task has shifted as survival has improved. A generation ago the diagnosis centred on the neonate with severe immunodeficiency or a complex heart; today the population includes school-age children with learning differences, adolescents with emerging anxiety, and adults living with thyroid disease, arthritis, and psychiatric illness. The general paediatrician sits at the centre of this trajectory, coordinating cardiology, immunology, cleft and craniofacial surgery, endocrinology, and psychiatry from diagnosis to transition. [1] [4]
Classification
The deletion has a characteristic anatomy, and the size of the lost segment predicts some of the phenotype. The typical deletion spans about three megabases between two blocks of low-copy repeats called LCR22-A and LCR22-D, and it accounts for most cases. A smaller deletion of about one and a half megabases between LCR22-A and LCR22-B makes up most of the remainder, and rare atypical distal deletions that spare the TBX1 gene produce a different, milder cardiac and thymic picture with a heavier cognitive and psychiatric burden. Mapping the boundaries is part of what a chromosomal microarray does, and it explains why two children with the same label behave so differently. [1] [9]
Separate from deletion size, the immune phenotype runs its own spectrum from the common partial form to the rare complete DiGeorge syndrome. Most children have a mild to moderate T-cell lymphopenia that is clinically silent or causes only recurrent respiratory infection; a small minority have profound thymic aplasia with near-absent T-cells — complete DiGeorge — which behaves like a severe combined immunodeficiency and demands urgent transplant evaluation. Recognising where a child sits on this immune spectrum is what governs the live-vaccine rule and the infection-management plan. [10] [9]
The reason the historical names collide is that the molecular lesion is identical across them, and severity is set by modifier genes, by mosaicism, and by stochastic factors that are still being mapped. A fellowship answer that lists the eponyms and then unifies them under the microdeletion reads as mature, because it shows that the candidate understands the literature has converged on one diagnosis while the bedside presentations remain kaleidoscopic. [1] [11]
Epidemiology & Risk Factors
The deletion occurs in roughly one in every four thousand live births, which makes it the most common recurrent microdeletion syndrome and a condition every general paediatrician will meet. The recurrence is driven by the genome itself: the 22q11.2 region is flanked by blocks of low-copy repeats that misalign during meiosis and recombine unequally, producing the same deletion over and over through non-allelic homologous recombination. That genomic architecture is why the deletion is recurrent rather than random, and why the prevalence is stable across populations. [1] [2]
Most deletions — around 85 to 90 per cent — arise de novo, but 10 to 15 per cent are inherited from a parent who carries the deletion, often with a milder phenotype that was never diagnosed. That inherited fraction is why a family history matters: a parent with the deletion has a 50 per cent recurrence risk in each pregnancy, and cascade testing of parents and siblings identifies the mildly affected adults whose own cardiac, thyroid, and psychiatric surveillance has been missed. The expanding population of late-diagnosed adults is a direct consequence of cascade testing reaching into families. [1] [5]
From an epidemiology standpoint, the most actionable fact is the cardiac association. Interrupted aortic arch type B is the lesion most strongly linked to 22q11.2 — a substantial proportion of all such infants carry the deletion — and truncus arteriosus and tetralogy of Fallot follow closely behind. Any conotruncal lesion should trigger a microarray, because finding the deletion changes the anaesthetic plan, the live-vaccine schedule, the calcium management, and the lifelong surveillance, none of which the cardiac diagnosis alone reveals. [2] [1]
Pathophysiology
The deletion removes a cluster of genes, but TBX1 is the driver. TBX1 encodes a T-box transcription factor expressed in the pharyngeal apparatus and in the secondary heart field, and its haploinsufficiency deranges the embryonic structures that build the thymus, the parathyroids, the outflow tract and aortic arch, the palate, and the face. Lose one copy of TBX1 and the third and fourth pharyngeal pouches fail to form properly, the cardiac neural crest mis-migrates, and a recognisable set of malformations appears in a single developmental event. [1] [3]
Defective cardiac neural-crest migration explains the conotruncal and aortic arch lesions that dominate the neonatal presentation. The outflow tract of the heart and the pharyngeal-arch arteries share a neural-crest origin, which is why interrupted aortic arch type B, truncus arteriosus, and tetralogy of Fallot cluster together in the same child and in the same syndrome. When a fellow candidate connects the cardiac lesion to its embryological root, the whole phenotype becomes intelligible rather than a list of associations. [1] [2]
The thymic and parathyroid consequences arise because both organs develop from the third and fourth pharyngeal pouches. Thymic hypoplasia produces a variable T-cell deficiency — from trivial lymphopenia to the near-complete absence of complete DiGeorge — and the same immune dysregulation that lowers the threshold for infection also raises the risk of autoimmunity later in life. Parathyroid aplasia or hypoplasia produces neonatal hypocalcaemia, which may declare itself as seizures in the first days of life and can recur during adolescence or intercurrent illness when calcium demand shifts. [8] [10]
Why the same deletion produces a spectrum from an asymptomatic adult to a critically ill neonate is one of the live questions in the field, and the answer is multifactorial. Modifier genes beyond TBX1, somatic mosaicism, the stochastic shaping of the immune repertoire, and environmental factors each bend the phenotype, and the large international cohorts now map the genetic contributors to the psychiatric risk in particular. The fellowship teaching is that variability is the rule, so the surveillance plan must be staged to the individual, not assumed from the label. [1] [12]
Clinical Presentation
The presentation tracks the life stage, and a fellowship answer earns depth by separating the neonate from the infant, the school-age child, and the adult. The neonate declares the diagnosis through the most dangerous elements — duct-dependent cardiac disease, hypocalcaemic seizures, severe immunodeficiency, or airway and feeding difficulty from a cleft — and these are the presentations that bring the child to the intensive care unit before the microarray returns. [1] [8]
The characteristic facial gestalt is a tubular nose with a bulbous, pinched tip, small and structurally anomalous ears, narrow palpebral fissures, a relatively long face, and retrognathia. The face is most reliable in later infancy and childhood and is easily missed in the neonate, where oedema and the severity of the acute illness obscure it; this is one reason the facies should never be the gate for the microarray. A cardiac lesion, a cleft, or a low calcium in a baby who does not look typical should still be tested. [1] [11]
In infancy and the preschool years, the presentation shifts to velopharyngeal insufficiency and feeding. Nasal regurgitation, hypernasal speech, and feeding difficulty with poor growth dominate, alongside recurrent respiratory and middle-ear infection driven by the immune deficiency and the palatal anomaly. Submucous cleft palate and occult submucous cleft are easy to miss on casual inspection and are a classic reason the diagnosis is delayed until a speech pathologist or a cleft clinic flags the nasal speech. [2] [3]
The school-age child presents with learning and behaviour. The cognitive profile is a non-verbal learning pattern — relative strength in rote language and verbal memory against weakness in visuospatial skill, mathematics, and abstract reasoning — accompanied by attention deficits, anxiety, and social difficulty. The fellowship trap is to attribute the school struggle to low intelligence when the profile is actually one of uneven strengths, and the management is targeted educational support rather than a global deficit label. [7]
Adolescence and adulthood are the high-risk phases for emergent illness, and they are where missed childhood diagnoses resurface. Depression and anxiety are common, and the lifetime risk of schizophrenia-spectrum psychosis is around 25 per cent — the largest known single-locus risk for schizophrenia. Thyroid disease, juvenile idiopathic arthritis, immune cytopenia, and aortic and skeletal problems accumulate through the adult years, which is why the surveillance plan must transfer intact into adult care. [12] [4]
Differential Diagnosis
The differential splits into two questions: which other syndromes produce a cardiac-plus-palate-plus-immune-plus-developmental phenotype, and when an isolated finding still warrants a microarray. The must-not-miss mimics are CHARGE syndrome, the Noonan and related RASopathies, Williams syndrome, Alagille syndrome, and Smith-Lemli-Opitz syndrome, each with a discriminating feature that separates it from 22q11.2. [1] [11]
CHARGE syndrome, caused by CHD7 mutations, overlaps on the coloboma, the cardiac defect, the cleft, and the developmental delay, but it adds choanal atresia, characteristic ear anomalies with vestibular abnormality, and genital hypoplasia that 22q11.2 does not carry. The Noonan and RASopathy family shares short stature, a characteristic face, and congenital heart disease, but the heart lesion is typically pulmonary stenosis or hypertrophic cardiomyopathy rather than a conotruncal defect, and the karyotype and microarray are normal. Williams syndrome pairs supravalvular aortic stenosis with the hypercalcaemic, over-friendly elastin phenotype — the opposite calcium problem from 22q11.2. [1]
The more common diagnostic error is not confusing the syndromes but failing to test an isolated finding. An isolated conotruncal cardiac lesion, an isolated cleft palate or velopharyngeal insufficiency, or an isolated neonatal hypocalcaemia can each be the presenting feature of 22q11.2, and the safe habit is to send a microarray for every conotruncal defect and for cleft palate with any syndromal feature. Transient hypocalcaemia of prematurity or vitamin D deficiency can mimic the neonatal 22q11.2 presentation, and a persistent low calcium with a normal vitamin D status, or a concomitant cardiac or palatal lesion, resolves the question toward the microarray. [2] [8]
A final discriminator at the severe end is the distinction between complete DiGeorge syndrome and severe combined immunodeficiency. Both present in the neonate or young infant with severe infection, failure to thrive, and profound lymphopenia, and both are emergencies. The TREC — T-cell receptor excision circle — newborn screen and flow cytometry separate them, and both demand urgent immunology referral, because complete DiGeorge may require thymic transplant while severe combined immunodeficiency requires haematopoietic stem cell transplant. [10] [9]
| Feature | 22q11.2 deletion | CHARGE syndrome | Noonan / RASopathy |
|---|---|---|---|
| Cardiac lesion | Conotruncal: IAA-B, truncus, TOF | Conotruncal and other | Pulmonary stenosis, HCM |
| Palate | Cleft, submucous cleft, VPI | Cleft, choanal atresia | Usually normal |
| Distinguishing sign | Hypocalcaemia, T-cell deficiency | Coloboma, ear/vestibular, genital | Webbed neck, bleeding diathesis |
| Genetics | 22q11.2 deletion (microarray) | CHD7 (sequencing) | RASopathy, normal microarray |
Clinical & Bedside Assessment
The recognition move is the hinge of the whole topic, and it is simple: any conotruncal cardiac defect, any cleft palate or velopharyngeal insufficiency, any neonatal hypocalcaemia or seizure, and any unexplained T-cell deficiency earns a chromosomal microarray regardless of how typical or atypical the face looks. The face is a clue, not a gate, because the neonatal face is unreliable and the mildly affected child looks normal. Leading with the microarray when the pattern fits is what prevents the late, missed diagnosis. [1] [2]
The history gathers the discriminators that drive the test. Ask about the cardiac lesion type, because a conotruncal lesion changes the probability of the deletion more than any other single feature; about neonatal seizures or a known low calcium; about cleft palate, nasal speech, and feeding difficulty; about recurrent or severe infection; and about a family history of cardiac disease, cleft, learning difficulty, or psychiatric illness that might mark an inherited deletion. Growth, feeding, and the developmental trajectory function as vital signs here, because faltering growth or a plateau often signals an unrecognised cardiac, palatal, or immune problem. [3]
Examination is systems-staging at the bedside. Listen to the heart and feel the pulses, recognising that a duct-dependent arch lesion can collapse as the duct closes. Inspect and palpate the palate for a submucous cleft, listening for hypernasality. Examine for the facial gestalt without over-relying on it, check the hips and spine, and screen the development against age expectations. When the immune phenotype may be severe, isolate the child from live-vaccine contacts and arrange urgent flow cytometry rather than waiting for the outpatient clinic. [2] [9]
The first visit also assesses the family's practical capacity for a lifelong multisystem condition, because the surveillance plan is only as good as the family's ability to attend it. Map the care coordination, the travel distance, the schooling and learning support, the mental-health access, and the interpreter and cultural needs, and identify a named coordinator early. Combining the clinical gestalt with the molecular result — and holding both when they seem to disagree — is what protects against both over-diagnosis of an incidental atypical deletion and under-diagnosis of a classical phenotype. [3] [6]
Investigations
The chromosomal microarray is the first-tier test, and a standard karyotype will miss the deletion — this is the single most testable investigation point in the topic. The microarray both detects the deletion and maps its boundaries, so it tells you the deletion size, which predicts part of the cardiac, thymic, and cognitive phenotype. Fluorescence in-situ hybridisation and quantitative PCR are now reserved for cascade testing of relatives, for rapid prenatal confirmation, and for the occasional targeted confirmation, because the microarray is more comprehensive and informative. [1] [2]
At diagnosis, every system must be staged, and the staging work-up is the backbone of the first months. An echocardiogram defines the cardiac anatomy; a calcium, albumin-corrected calcium, and parathyroid hormone level define the parathyroid axis; lymphocyte subsets and, where available, TREC assays define the T-cell compartment; a palate assessment by a cleft or speech clinician defines the velopharyngeal anatomy; a renal ultrasound screens for structural anomalies; and thyroid function, vision and hearing screening, and a baseline developmental assessment complete the staging. Each item prevents a specific, predictable harm, and omitting any of them is the commonest source of later morbidity. [3] [6]
The immune evaluation is deepened when the initial screen suggests more than mild lymphopenia. Flow cytometry quantifies the naive T-cell compartment and thymic output, vaccine-specific antibody responses are checked when the B-cell compartment is in question, and the criteria for complete DiGeorge — very low T-cell counts with severe hypocalcaemia and a cardiac lesion — are applied to identify the child who needs transplant evaluation. Immune function can mature over the first years, so the work-up is repeated when the clinical picture changes. [9] [10]
Prenatal diagnosis closes the loop. When a fetal cardiac lesion — particularly a conotruncal defect — or a cleft is found on the anatomy scan, a prenatal chromosomal microarray on chorionic villus sampling or amniocentesis confirms or excludes the deletion, and this is increasingly the route by which the diagnosis is made before birth. A prenatal diagnosis allows planning of the delivery at a cardiac centre, the immediate calcium and immune staging, and the early withholding of live vaccines. [1] [3]
Management — Resuscitation
Resuscitation in 22q11.2 deletion means managing the acute neonatal threats that bring the child to attention, and three dominate: duct-dependent cardiac collapse, hypocalcaemic seizures, and severe infection from immunodeficiency. Each has a defined pathway, and recognising which threat is in front of you — often before the microarray returns — is the immediate skill. [1] [2]
A neonate with a duct-dependent arch or outflow lesion — interrupted aortic arch, severe coarctation, or severe tetralogy — collapses as the ductus arteriosus closes, and the resuscitation is prostaglandin E1 to maintain ductal patency, ventilation and metabolic correction, and urgent cardiology and cardiothoracic referral. The 22q11.2 context changes the subsequent plan — the anaesthetic airway risk from a cleft or retrognathia, the calcium management, and the live-vaccine hold — but the immediate cardiac resuscitation follows the standard duct-dependent pathway. [1] [3]
Hypocalcaemic seizures are treated with intravenous calcium gluconate under cardiac monitoring, followed by maintenance oral calcium and active vitamin D in the form of calcitriol, titrated to the albumin-corrected calcium and the parathyroid hormone level. The principle is rapid correction of the symptomatic deficit followed by structured maintenance, because hypocalcaemia can recur during adolescence, pregnancy, or intercurrent illness, and the family is taught the warning signs of tetany. [8] [3]
The live-vaccine rule is a resuscitation-grade safety measure, not a routine scheduling detail. Rotavirus, BCG, measles-mumps-rubella, and varicella vaccines are live attenuated and can cause disseminated disease in a child with significant T-cell deficiency, so they are withheld until the immune work-up clears them. The clearance comes from the immunology team once lymphocyte subsets and, where indicated, vaccine-specific responses are known, and the doses are then given on a planned schedule. Giving a live vaccine first, on the assumption that the immune deficiency is mild, is a preventable catastrophe. [10] [9]
Severe infection in an immunodeficient infant is managed with prompt microbiological assessment and broad-spectrum coverage, with a low threshold for intravenous immunoglobulin when there is a documented antibody deficiency, and urgent immunology involvement. The child with complete DiGeorge syndrome — profound T-cell deficiency with severe hypocalcaemia and a cardiac lesion — is a candidate for thymic transplant, and early recognition is what makes that option available. The family is also given an honest early explanation of the multisystem, lifelong nature of the diagnosis, because a family that understands the plan from the start adheres to it. [9] [1]
Management — Definitive & Stepwise
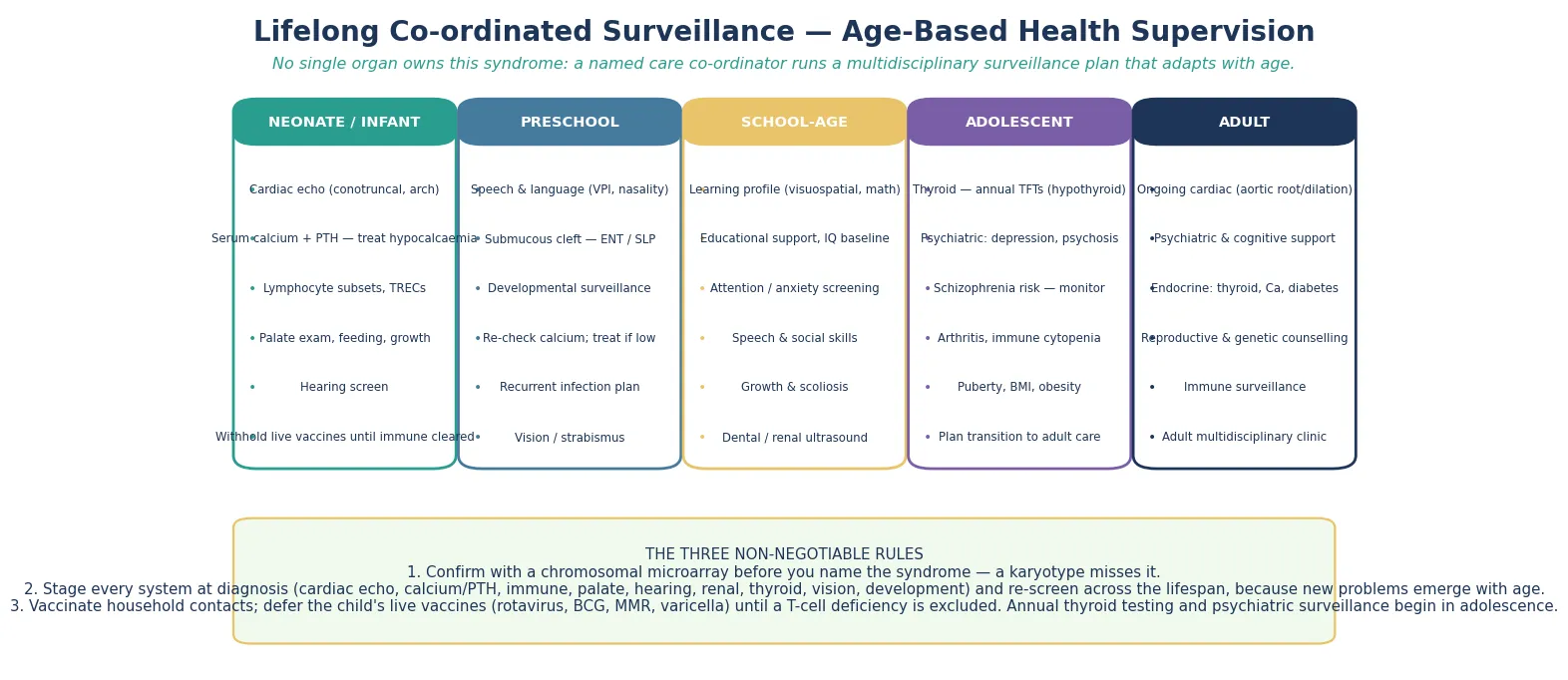
Definitive management is an age-based health-supervision plan that moves with the person from the neonatal period into adult care, and it is the core of fellowship-level competence. The plan is staged by age band, and each band carries system checks that prevent specific harm — cardiac and calcium and immune and palate in the neonate, speech and development in the preschool child, learning and attention in the school years, and thyroid and psychiatric surveillance in adolescence and beyond. [3] [6]
In the neonate and infant, the priorities are the cardiac lesion and its surgical repair, the calcium and vitamin D management, the immune staging and the live-vaccine clearance, and the palate and feeding assessment with early speech and cleft-team involvement. Thyroid function is checked and a baseline developmental assessment is made, hearing and vision are screened, and the family is connected to early intervention and to a 22q support organisation. The first year is dense precisely because every system declares itself then. [3] [2]
Across the preschool and school years, the plan layers speech therapy for velopharyngeal insufficiency, surgical correction of the cleft where indicated, targeted educational support for the non-verbal learning profile, and attention and anxiety management. Calcium is monitored for recurrence, thyroid function is checked periodically, and the immune status is reviewed as the child grows. Growth, development, and school progress are tracked at every visit, because a plateau often signals an unrecognised complication. [7] [3]
Adolescence and adulthood bring the psychiatric and endocrine burden to the foreground. Thyroid disease, arthritis, and immune cytopenia are screened for, calcium is rechecked, and active surveillance for depression, anxiety, and psychosis is built into the schedule, because the schizophrenia-spectrum risk is the single largest psychiatric risk attributable to any one genetic locus. The cardiac surveillance continues, with attention to the aortic root and arch into adult life, and a structured transition hands the documented deletion, the cardiac history, the immune status, and the psychiatric and thyroid surveillance to adult care. [4] [12]
Specific Subtypes & Scenarios
Complete DiGeorge syndrome is the severe end of the immune spectrum, and it carries a distinct management pathway from the common partial form. The child presents with profound T-cell lymphopenia — often flagged by a low or absent TREC on the newborn screen — alongside severe hypocalcaemia and a cardiac lesion, and behaves clinically like a severe combined immunodeficiency. Urgent immunology referral, strict infection-precaution isolation, and evaluation for thymic transplant define the pathway, because the absent thymus cannot generate a T-cell repertoire without a transplant. [9] [10]
The atypical distal deletion that spares TBX1 is a distinct scenario that a fellowship answer should name. Because the driver gene is retained, the conotruncal cardiac and thymic burden is lighter, but the cognitive and psychiatric phenotype is relatively heavier, and the child may present to developmental or mental-health services rather than to cardiology. Recognising the atypical deletion prevents the error of reassuring a family on the cardiac findings while the learning and psychiatric surveillance is neglected. [1] [7]
The inherited deletion from a mildly affected parent shapes both counselling and the extended family. A parent who carries the deletion has a 50 per cent recurrence risk in each pregnancy, and siblings may also carry it, so parental microarray and cascade testing identify the wider family who themselves need cardiac, thyroid, immune, and psychiatric surveillance. The mildly affected adult whose own diagnosis was missed until a child presented severely is a common and poignant scenario, and the genetic counselling addresses both recurrence and the parent's unmet surveillance needs. [5] [2]
The adolescent-onset psychiatric phenotype is the scenario that most often surprises clinicians who met the child only as a cardiac patient. Depression and anxiety emerge first, and a subset progress to schizophrenia-spectrum psychosis, so structured surveillance — screening, early psychological intervention, and access to psychiatric care — is built into the adolescent plan. The pregnant adult with 22q11.2 adds prenatal testing, a 50 per cent recurrence risk, and multisystem pregnancy care, and the 22q11.2 duplication — the reciprocal event on the same locus — is a related but distinct condition that appears in the differential. [12] [4]
Why schizophrenia risk dominates the adult story
The 22q11.2 deletion carries the largest known single-locus risk for schizophrenia-spectrum psychosis, at around 25 per cent over a lifetime. The mechanism is partly genetic — modifier genes across the deleted region and elsewhere shape the risk — and partly neurodevelopmental. The practical consequence is that adolescent and adult surveillance must include active psychiatric screening, early intervention for anxiety and depression, and a low threshold for specialist assessment when thought disorder emerges, because early treatment changes the trajectory. [12] [4]
Complications & Pitfalls
The harm in 22q11.2 deletion comes most often from a late or missed diagnosis, and the mechanism of the miss is consistent: a standard karyotype that reports normal, or an isolated cardiac lesion that was never tested. The fellowship answer names the missed diagnosis as the cardinal pitfall and the microarray as the safeguard, because every downstream surveillance item depends on the label being made. Once the deletion is confirmed, the complications are the predictable, age-dependent problems that the surveillance plan was designed to catch. [1] [2]
The emergent complications that must not be missed cluster in the neonatal period. Hypocalcaemic seizures from an unrecognised parathyroid deficiency, duct-dependent cardiac collapse as the duct closes, severe infection from an unrecognised T-cell deficiency, and airway or feeding failure from a severe cleft each demand prompt recognition and a defined pathway. The pitfall common to all is treating the acute problem without sending the microarray, so that the child is resuscitated but the diagnosis and the surveillance are lost. [8] [9]
The age-dependent complications run through the lifespan and are the substance of long-term surveillance. Thyroid disease, juvenile idiopathic arthritis, immune cytopenia, schizophrenia-spectrum psychosis, and aortic disease each declare themselves at a characteristic age, and the pitfall is letting the surveillance lapse as the child moves from paediatric to adult care. The adolescent who was followed only by cardiology, and whose thyroid and psychiatric surveillance was never established, is the archetypal missed-adult-complication case. [4] [12]
Management pitfalls share a common root: treating before staging. Starting live vaccines before the immune work-up clears them, missing the hypocalcaemia because the calcium was not checked, failing to check the thyroid annually from adolescence, and over-interpreting a variant of uncertain significance or a small atypical deletion as classical 22q11.2 are each preventable errors. The safeguard is the staged work-up at diagnosis and the documented surveillance plan that follows the person across the lifespan. [10] [3]
Prognosis & Disposition
The prognosis spans from a near-normal lifespan with consistent surveillance to severe early morbidity from complex cardiac disease or complete DiGeorge syndrome, and the determinant is less the deletion itself than the quality and timing of the surveillance. Early diagnosis and consistent multisystem care are the single biggest predictors of outcome, because each preventable complication — the unrepaired heart, the untreated hypocalcaemia, the unrecognised psychiatric illness — subtracts from the trajectory the surveillance plan exists to protect. [1] [4]
The general paediatrician owns the coordination, and the disposition is shared, structured care. Cardiology drives the cardiac repair and the arch surveillance, immunology owns the T-cell staging and the live-vaccine clearance, the cleft and craniofacial team manages the palate and speech, endocrinology manages the calcium and the thyroid, and psychiatry and genetics contribute across the lifespan. A named coordinator prevents the fragmentation that is the enemy of a checklist-based plan, and referral to a specialist 22q multidisciplinary clinic at the point of diagnosis — rather than after complications — measurably improves outcomes. [3] [6]
The psychiatric and cognitive trajectory shapes the educational, vocational, and independence planning, and the fellowship answer frames prognosis in those terms rather than in deficit language alone. Many children with 22q11.2 attend mainstream school with targeted support, complete further education, and live independently or semi-independently, while a subset need sustained psychiatric and educational input. The prognosis is therefore a function of the match between the support and the profile, and the plan is built around that match. [7] [4]
Recurrence-risk counselling closes the prognostic picture. When a parent carries the deletion, the recurrence risk is 50 per cent in each pregnancy, and gonadal mosaicism accounts for a small additional risk even when parental testing is normal; prenatal testing and preimplantation genetic testing are options the family is offered. The counselling addresses not only the next pregnancy but the surveillance of the wider family, because the inherited fraction means a positive test opens a cascade of relatives who may themselves have been living undiagnosed. [5] [2]
Special Populations
The deletion interacts with the child's social, cultural, and developmental context, and the same surveillance plan behaves differently across populations — access, adherence, and late presentation each shape the outcome. A plan that is clinically correct but unattainable for a family is no plan at all, and the fellowship answer recognises that the schedule is only as good as the family's ability to engage with it. [3] [6]
Indigenous children in Australia and New Zealand may carry a higher background burden of respiratory infection, otitis media, and sleep-disordered breathing, and the T-cell deficiency of 22q11.2 amplifies that burden. Reduced access to specialist services in remote communities intensifies the need for early, structured surveillance and for a low threshold to investigate symptoms, and telehealth and outreach extend the surveillance net into communities that a clinic-based model would miss. The immune deficiency makes the standard indigenous-health emphasis on early respiratory and audiological surveillance even more urgent. [10] [9]
Migrant, refugee, and asylum-seeking families may arrive with incomplete medical records, an uncertain family history, and no prior genetic testing, and the diagnosis may not have been made in the country of origin. A careful reconstruction of the history, confirmation of the deletion and its inheritance by microarray and parental testing, and an interpreter-mediated explanation of the multisystem plan are the foundations. Vaccination status is reconciled with the live-vaccine hold, and the written schedule is provided in the family's language. [1] [2]
Socioeconomic disadvantage shapes late presentation, care-coordination feasibility, and mental-health access, because the limiting step is often attendance and transport rather than the medicine. Structuring the surveillance around a single coordinated visit, linking the family to a support organisation and to transport and appointment support, and using telehealth to reduce travel all improve engagement. The aim is to fit the surveillance to the family's reality, and a coordinator who knows the local services is the asset that makes that possible. [3] [6]
The school-age child with the non-verbal learning profile is a population in their own right, because the uneven cognitive profile is easily misread as global delay or as behavioural difficulty. Targeted educational and psychosocial support — strength in rote language leveraged against weakness in visuospatial and mathematical skill — is the intervention, and trauma-informed, interpreter-supported communication extends to the genetic-counselling conversation itself. The adolescent in transition, carrying a chronic multisystem label and an emerging psychiatric risk, needs a planned handover that preserves the cardiac, thyroid, immune, and psychiatric surveillance intact. [7] [4]
Evidence, Guidelines & Regional Differences
The evidence base rests on the McDonald-McGinn 2015 Nature Reviews Disease Primers overview, which remains the foundational reference and frames the genetics, the comorbidity map, and the lifespan trajectory that a fellowship answer is built on. The practical structure of care comes from the Bassett 2011 international practical guidelines and from the 2023 updated clinical practice recommendations — Óskarsdóttir for children and Boot for adults — which translate the evidence into an age-stratified schedule. The American Academy of Pediatrics 2025 health-supervision clinical report adds the most current operational guidance for children. [1] [2] [3]
The immune and psychiatric evidence is what distinguishes a mature answer from a checklist. The Crowley 2018 and McLean-Tooke 2007 data define the variable immune deficiency and its relationship to deletion size, anchoring the live-vaccine rule and the surveillance of autoimmunity, while the Cleynen 2021 international consortium study maps the genetic contributors to the schizophrenia-spectrum risk and makes psychiatric surveillance an evidence-based rather than an elective activity. The Cheung 2014 data on neonatal hypocalcaemia and seizures anchor the neonatal calcium pathway, and the Swillen 2018 neurodevelopmental review frames the learning profile and its management. [9] [10] [12] [8] [7]
Where the evidence is weak, a fellowship answer says so honestly. The optimal frequency of cardiac and psychiatric surveillance, the role of prophylactic intravenous immunoglobulin in the milder immune phenotypes, and the long-term outcomes of the atypical distal deletions are areas of genuine uncertainty, and regional practices differ in the availability of specialist 22q clinics and in the TREC newborn-screening programmes that flag severe T-cell deficiency. Naming the uncertainty is a mark of intellectual honesty that examiners reward. [4] [9]
In Australia and New Zealand, care follows the international 22q11.2 clinical practice recommendations, with multidisciplinary 22q clinics in the major centres coordinating cardiology, immunology, cleft and craniofacial, endocrinology, psychiatry, and genetics. The live-vaccine rule operates through the national immunisation programme, with immunology clearance governing rotavirus, BCG, measles-mumps-rubella, and varicella scheduling. Newborn bloodspot screening for severe combined immunodeficiency by TREC varies by jurisdiction and may flag the severe T-cell deficiency of complete DiGeorge. Access to specialist clinics, genetic counselling, and adolescent psychiatric services is uneven across rural and remote communities, which intensifies the need for early referral and for telehealth-supported surveillance. [3] [6]
Exam Pearls
A fellowship candidate answering on 22q11.2 deletion should land five anchor points and avoid three classic traps. The anchors are the framework examiners listen for; the traps are where easy marks are lost. [1] [6]
Anchor one: recognise the pattern. Any conotruncal cardiac lesion — interrupted aortic arch type B, truncus arteriosus, or tetralogy of Fallot — any cleft palate or velopharyngeal insufficiency, any neonatal hypocalcaemia or seizure, and any unexplained T-cell deficiency earns a microarray. The face is a clue, not a gate. [2]
Anchor two: confirm with a microarray. A standard karyotype misses the deletion, and ordering one first is the classic reason the diagnosis is delayed. The chromosomal microarray detects the deletion and maps its boundaries, which predict part of the phenotype. [1]
Anchor three: stage every system at diagnosis. An echocardiogram, a calcium and parathyroid hormone, lymphocyte subsets, a palate assessment, a renal ultrasound, thyroid function, vision and hearing, and a developmental baseline are the staging work-up, and each item prevents a specific harm. [3] [6]
Anchor four: defer live vaccines. Rotavirus, BCG, measles-mumps-rubella, and varicella are withheld until the immune work-up clears them, because a significant T-cell deficiency can turn a live vaccine into disseminated disease. The immunology team clears the schedule. [10] [9]
Anchor five: run an age-based surveillance plan. Cardiac and calcium and immune and palate in the neonate, speech and development in the preschool child, learning and attention in the school years, and thyroid and psychiatric and aortic surveillance in adolescence and adulthood — the schedule does not end at the cardiac repair. [4] [12]
The three traps to avoid are sending a karyotype that misses the deletion, giving live vaccines before the immune clearance, and forgetting the thyroid and psychiatric surveillance in adolescence. Interrupted aortic arch type B is the cardiac lesion most strongly associated with the deletion, TBX1 is the driver gene, around 85 per cent of deletions are de novo and 15 per cent inherited, and the schizophrenia-spectrum lifetime risk is around 25 per cent — the high-yield numbers a candidate holds. Avoid the traps and land the anchors, and the rest of the answer falls into place. [1] [2]
References
- [1]McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, Scambler PJ, Bassett AS. 22q11.2 deletion syndrome. Nat Rev Dis Primers, 2015.PMID 27189754
- [2]Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, Marino B, Oskarsdottir S, Philip N, Sullivan K, Swillen A, Vorstman J, International 22q11.2 Deletion Syndrome Consortium. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr, 2011.PMID 21570089
- [3]Óskarsdóttir S, Boot E, Crowley TB, Loo JCY, Arganbright JM, Armando M, Baylis AL, Breetvelt EJ, Castelein RM, Chadehumbe M, Cielo CM, de Reuver S, Eliez S, Fiksinski AM, Forbes BJ, Gallagher E, Hopkins SE, Jackson OA, Levitz-Katz L, Klingberg G, Lambert MP, Marino B, Mascarenhas MR, Moldenhauer J, Moss EM, Nowakowska BA, Orchanian-Cheff A, Putotto C, Repetto GM, Schindewolf E, Schneider M, Solot CB, Sullivan KE, Swillen A, Unolt M, Van Batavia JP, Vingerhoets C, Vorstman J, Bassett AS, McDonald-McGinn DM. Updated clinical practice recommendations for managing children with 22q11.2 deletion syndrome. Genet Med, 2023.PMID 36729053
- [4]Boot E, Óskarsdóttir S, Loo JCY, Crowley TB, Orchanian-Cheff A, Andrade DM, Arganbright JM, Castelein RM, Cserti-Gazdewich C, de Reuver S, Fiksinski AM, Klingberg G, Lang AE, Mascarenhas MR, Moss EM, Nowakowska BA, Oechslin E, Palmer L, Repetto GM, Reyes NGD, Schneider M, Silversides C, Sullivan KE, Swillen A, van Amelsvoort TAMJ, Van Batavia JP, Vingerhoets C, McDonald-McGinn DM, Bassett AS. Updated clinical practice recommendations for managing adults with 22q11.2 deletion syndrome. Genet Med, 2023.PMID 36729052
- [5]Fung WL, Butcher NJ, Costain G, Andrade DM, Boot E, Chow EW, Chung B, Cytrynbaum C, Faghfoury H, Fishman L, García-Miñaúr S, George S, Lang AE, Repetto G, Shugar A, Silversides C, Swillen A, van Amelsvoort T, McDonald-McGinn DM, Bassett AS. Practical guidelines for managing adults with 22q11.2 deletion syndrome. Genet Med, 2015.PMID 25569435
- [6]Scheuerle AE, Geleske TA, Merchant N, Goldenberg PC, Council on Genetics. Health Supervision for Children With 22q11.2 Deletion Syndrome: Clinical Report. Pediatrics, 2025.PMID 40685150
- [7]Swillen A, Moss E, Duijff S. Neurodevelopmental outcome in 22q11.2 deletion syndrome and management. Am J Med Genet A, 2018.PMID 29696780
- [8]Cheung EN, George SR, Andrade DM, Chow EW, Silversides CK, Bassett AS. Neonatal hypocalcemia, neonatal seizures, and intellectual disability in 22q11.2 deletion syndrome. Genet Med, 2014.PMID 23765047
- [9]Crowley B, Ruffner M, McDonald McGinn DM, Sullivan KE. Variable immune deficiency related to deletion size in chromosome 22q11.2 deletion syndrome. Am J Med Genet A, 2018.PMID 29341423
- [10]McLean-Tooke A, Spickett GP, Gennery AR. Immunodeficiency and autoimmunity in 22q11.2 deletion syndrome. Scand J Immunol, 2007.PMID 17587340
- [11]Shprintzen RJ, Higgins AM, Antshel K, Fremont W, Roizen N, Kates W. Velo-cardio-facial syndrome. Curr Opin Pediatr, 2005.PMID 16282778
- [12]Cleynen I, Engchuan W, Hestand MS, Heung T, Holleman AM, Johnston HR, Monfeuga T, McDonald-McGinn DM, Gur RE, Morrow BE, Swillen A, Vorstman JAS, Bearden CE, Chow EWC, van den Bree M, Emanuel BS, Vermeesch JR, Warren ST, Owen MJ, Chopra P, Cutler DJ, Duncan R, Kotlar AV, Mulle JG, Voss AJ, Zwick ME, Diacou A, Golden A, Guo T, Lin JR, Wang T, Zhang Z, Zhao Y, Marshall C, Merico D, Jin A, Lilley B, Salmons HI, Tran O, Holmans P, Pardinas A, Walters JTR, Demaerel W, Boot E, Butcher NJ, Costain GA, Lowther C, Evers R, van Amelsvoort TAMJ, van Duin E, Vingerhoets C, Breckpot J, Devriendt K, Vergaelen E, Vogels A, Crowley TB, McGinn DE, Moss EM, Sharkus RJ, Unolt M, Zackai EH, Calkins ME, Gallagher RS, Gur RC, Tang SX, Fritsch R, Ornstein C, Repetto GM, Breetvelt E, Duijff SN, Fiksinski A, Moss H, Niarchou M, Murphy KC, Prasad SE, Daly EM, Gudbrandsen M, Murphy CM, Murphy DG, Buzzanca A, Fabio FD, Digilio MC, Pontillo M, Marino B, Vicari S, Coleman K, Cubells JF, Ousley OY, Carmel M, Gothelf D, Mekori-Domachevsky E, Michaelovsky E, Weinberger R, Weizman A, Kushan L, Jalbrzikowski M, Armando M, Eliez S, Sandini C, Schneider M, Béna FS, Antshel KM, Fremont W, Kates WR, Belzeaux R, Busa T, Philip N, Campbell LE, McCabe KL, Hooper SR, Schoch K, Shashi V, Simon TJ, Tassone F, Arango C, Fraguas D, García-Miñaúr S, Morey-Canyelles J, Rosell J, Suñer DH, Raventos-Simic J, International 22q11.2DS Brain and Behavior Consortium, Epstein MP, Williams NM, Bassett AS. Genetic contributors to risk of schizophrenia in the presence of a 22q11.2 deletion. Mol Psychiatry, 2021.PMID 32015465