Paeds · genetics-dysmorphology-and-metabolism
Amino-acid disorders including phenylketonuria and MSUD
Also known as Aminoacidopathies · Phenylketonuria · PKU · Maple syrup urine disease · MSUD · Tyrosinaemia type I · Classical homocystinuria · BH4 deficiency
A fellowship approach to the inherited amino-acid disorders: recognise phenylketonuria, maple syrup urine disease, tyrosinaemia type I and homocystinuria as autosomal-recessive blocks in amino-acid catabolism that share a single dangerous mechanism — a blocked step, an accumulating neurotoxic or tissue-toxic metabolite, and preventable brain injury — distinguish them on newborn-screen, metabolite and clinical signatures, deliver the acute 'switch off catabolism, clear the toxin' protocol for an MSUD crisis, and lock in long-term diet-plus-cofactor-plus-transplant management knowing that outcome tracks time on the toxic metabolite.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who reads these diseases through one shared mechanism rather than as four separate lists. The child in front of you may be a neonate in ketoacidotic coma, an infant with liver failure, or a tall thin child with a dislocated lens. The question is never "which rare disease is this" in isolation; it is "what is accumulating, where is the block, and how fast can I remove the substrate and clear the toxin". The biochemistry is the treatment plan, because the accumulating compound — phenylalanine, leucine, succinylacetone, homocysteine — is itself the disease. [1] [12]
Overview & Definition
The inherited amino-acid disorders are a family of autosomal-recessive diseases in which a single enzyme that breaks down a dietary amino acid is missing or sluggish, so the amino acid — and its downstream metabolites — accumulate in blood and tissue. The accumulation is not a marker of disease; it is the disease. Phenylalanine is neurotoxic to the developing brain, leucine causes cerebral oedema, succinylacetone destroys the liver and kidney, and homocysteine injures the lens, bone and blood vessels. This shared logic is why the management of all four disorders looks the same: restrict the precursor amino acid, supply the missing chemistry with a cofactor or enzyme when possible, and prevent the catabolic states that flood the blocked pathway. [1] [12]
Clinically these conditions sit among the intoxication-type inborn errors of metabolism, alongside the organic acidaemias and the urea cycle disorders. The unifying idea is a blocked catabolic pathway that generates a circulating toxin. That framing matters at the bedside because it tells you the priority: remove the toxin, do not wait for the enzyme name. Phenylketonuria is the prototypical and the most common, and it is the disorder that proved the entire concept — a toxic metabolite, a diet that removes it, and a normal intellect rescued by newborn screening. [1] [5]
Classification
The classification that earns marks is biochemical and positional, because the position of the block determines the accumulating metabolite, the organ injured, and — crucially — the therapy. A block at the first committed step of an amino-acid pathway tends to be the most severe: phenylalanine cannot move past phenylalanine hydroxylase, the branched-chain amino acids cannot move past their shared dehydrogenase complex, tyrosine cannot move past fumarylacetoacetate hydrolase, and homocysteine cannot be condensed into cystathionine. Each block produces a characteristic fingerprint on quantitative amino acids and on the newborn bloodspot, and each has a treatment that targets that fingerprint. [1] [7]
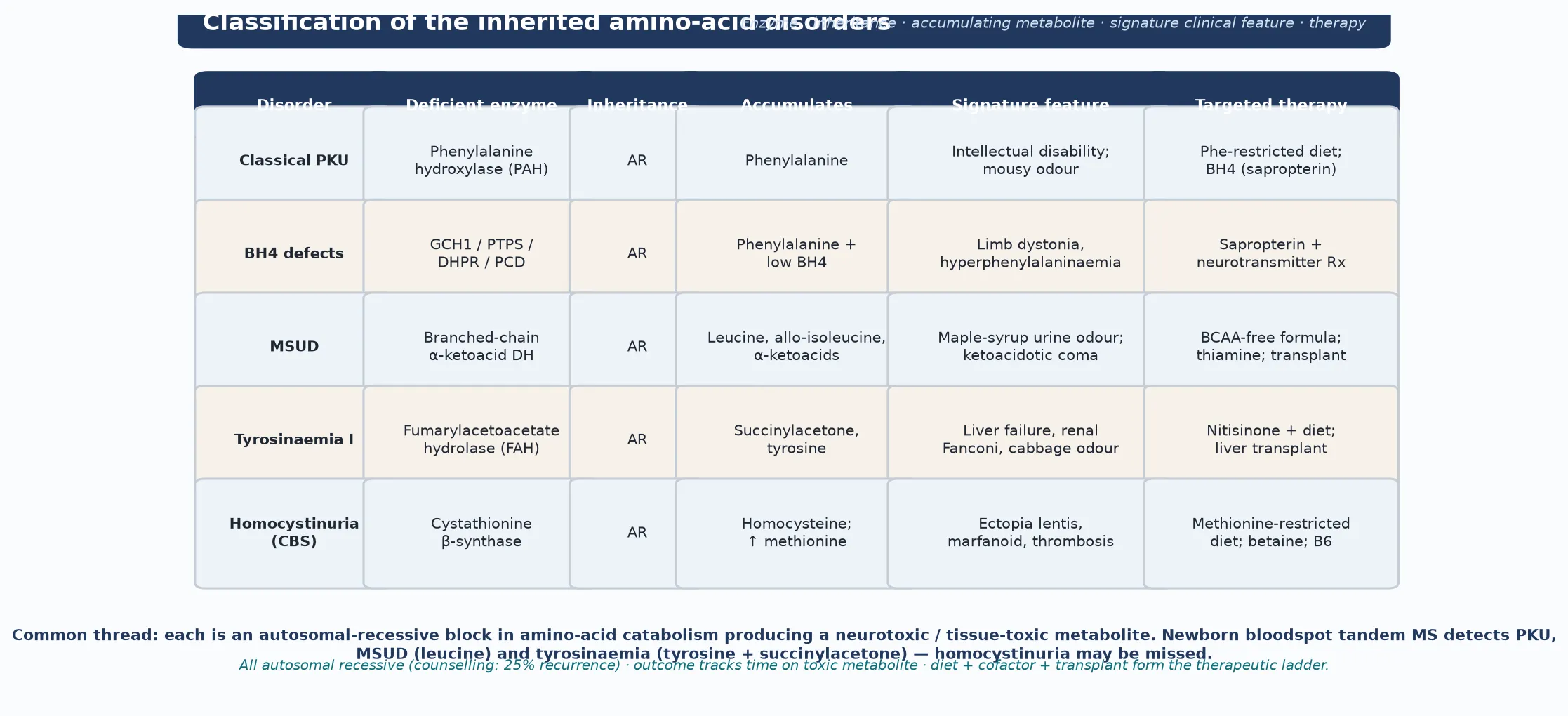
The single most useful discriminator is whether the block sits in the amino acid itself or in its cofactor. Classical phenylketonuria is a defect of the apoenzyme phenylalanine hydroxylase (PAH), and it is treated by removing the substrate. The BH4 cofactor defects — GTP cyclohydrolase (Segawa disease), 6-pyruvoyl-tetrahydropterin synthase (PTPS), dihydropteridine reductase (DHPR) and pterin-4a-carbinolamine dehydratase — share the same hyperphenylalaninaemia but ALSO starve the tyrosine and tryptophan hydroxylases of their shared cofactor, so the child develops dystonia and a neurotransmitter deficiency that diet alone will not fix. Distinguishing apoenzyme from cofactor defect at the first visit is a fellowship-level skill that changes the whole management. [1] [12]
Epidemiology & Risk Factors
Phenylketonuria is the commonest aminoacidopathy detected by newborn screening, with an incidence around one in 10,000 in populations of European descent; it is rarer in African and some Asian populations and higher in others, reflecting founder effects and carrier frequency. Maple syrup urine disease is far rarer, around one in 185,000 overall, but it is dramatically concentrated in the Old Order Mennonite community of North America, where founder effects push the incidence toward one in 380. Tyrosinaemia type I shows the same founder-effect logic in the Saguenay–Lac-Saint-Jean region of Quebec, where it reaches roughly one in 16,000. These founder concentrations matter: a child from one of these communities with the relevant presentation carries a much higher pre-test probability. [1] [7]
The major risk factor for an acute presentation or decompensation is a catabolic trigger. Fasting, intercurrent infection, surgery, trauma, a high-protein load, or corticosteroids all increase endogenous protein breakdown, flooding a blocked pathway with substrate. A previously stable child with maple syrup urine disease who develops a febrile illness can tip into cerebral oedema within hours, which is why every family carries a written emergency regimen that anticipates catabolism before it arrives. Consanguinity and a sibling history of neonatal or unexplained death are the highest-yield historical clues for any autosomal-recessive aminoacidopathy. [7] [8]
Access shapes outcome as much as biology. Delayed or missed newborn screening — more common in remote, migrant and refugee families — means a child presents with established brain injury. The cost of medical formula, sapropterin, pegvaliase and nitisinone is substantial and unevenly funded, so the child whose family can afford and access therapy does better than the child whose family cannot. The general paediatrician is often the advocate who closes that gap. [2] [4]
Pathophysiology
The molecular story begins with dietary and endogenous protein, which is broken into amino acids that the body either uses for synthesis or oxidises for energy. Oxidation proceeds through a series of enzyme steps, each of which requires both a protein apoenzyme and, often, a cofactor. When a step is blocked, the amino acid upstream accumulates and the product downstream is depleted. In phenylketonuria, phenylalanine cannot be converted to tyrosine, so phenylalanine rises and tyrosine falls; the falling tyrosine is itself part of the injury, because tyrosine is the precursor of dopamine, the melanins and thyroid hormone, which explains the fair skin and hair. [1] [12]
Elevated phenylalanine damages the brain through the large neutral amino acid (LNAA) transporter at the blood–brain barrier. Phenylalanine competes with tyrosine, tryptophan and the other large neutral amino acids for this shared transporter, so as phenylalanine rises, the brain is selectively starved of the very amino acids it needs to make neurotransmitters and myelin. The downstream consequences — impaired myelination, reduced dopamine and serotonin synthesis, disrupted brain protein synthesis, and altered white matter on imaging — are proportional to the lifetime, time-integrated exposure to phenylalanine. This is the biochemical reason that early and sustained control protects intellect, and that maternal phenylketonuria devastates an otherwise unaffected fetus. [1] [5]
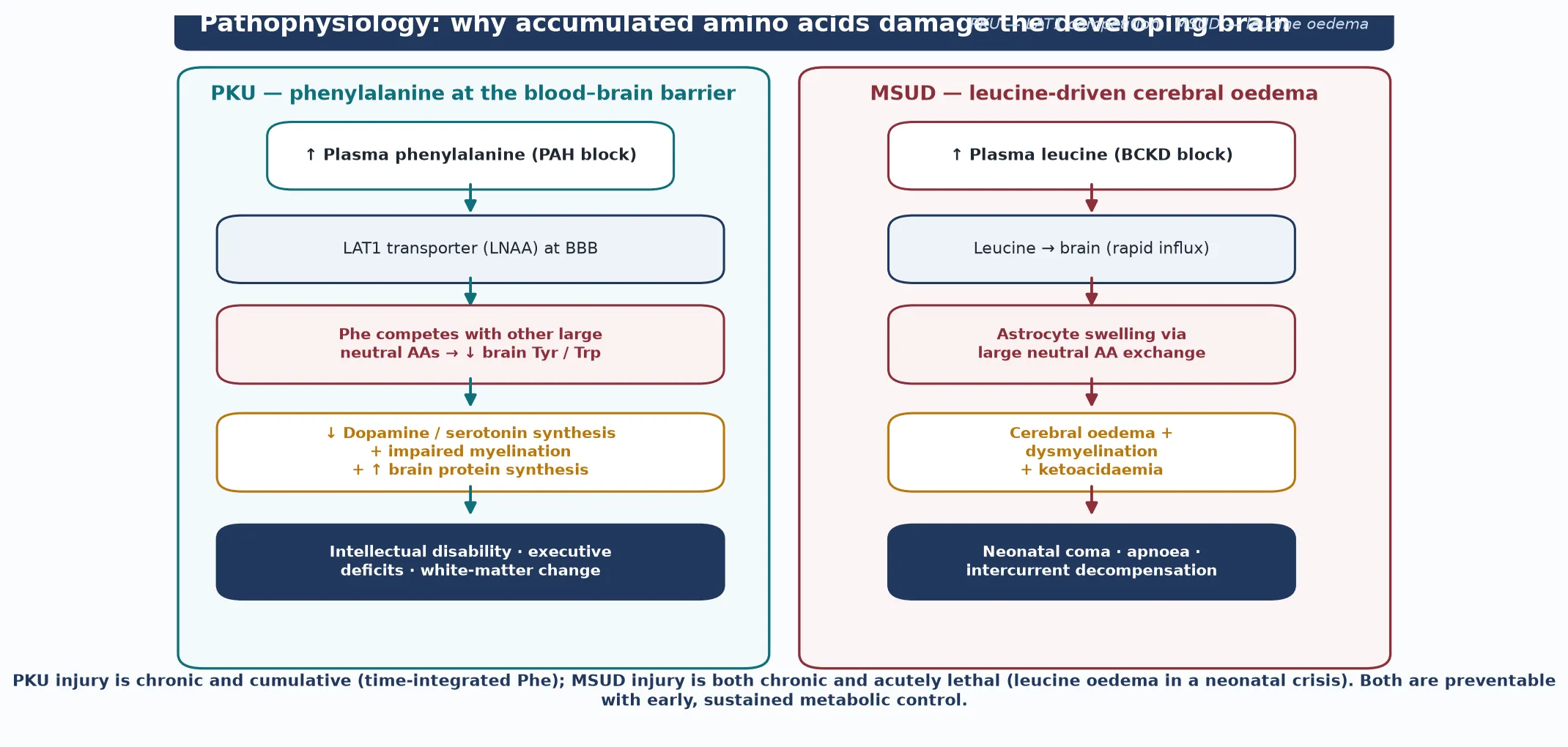
Maple syrup urine disease injures the brain through a different but parallel mechanism. The branched-chain α-ketoacid dehydrogenase complex normally oxidises leucine, isoleucine and valine; when it is blocked, all three accumulate, but leucine is the most neurotoxic. Leucine crosses rapidly into the brain, drives astrocyte swelling through large neutral amino acid exchange, and produces cerebral oedema, dysmyelination and, in the acute crisis, coma and apnoea. The blocked pathway also generates the branched-chain α-ketoacids, which cause the characteristic ketoacidosis and the maple-syrup odour. Tyrosinaemia type I is different again: the accumulating fumarylacetoacetate and its derivative succinylacetone are directly hepatotoxic and nephrotoxic, and succinylacetone also inhibits porphobilinogen synthase, producing the painful neurovisceral crises that mimic acute porphyria. [7] [10]
Homocystinuria sits at the vascular end of the spectrum. Cystathionine β-synthase normally condenses homocysteine and serine into cystathionine; when it is blocked, homocysteine accumulates and methionine rises. Free homocysteine is toxic to the lens zonules, bone and the vascular endothelium, which is why the disease expresses as downward ectopia lentis, a marfanoid habitus, osteoporosis and a life-long risk of arterial and venous thrombosis. Understanding the mechanism of each disorder is what lets you predict its complications and defend the therapy at viva. [11]
Clinical Presentation
The presentation is shaped by whether newborn screening caught the disorder first. Phenylketonuria is now usually detected on the bloodspot before any symptom, and an early-treated child is often clinically normal. The untreated or late-diagnosed child, however, shows the full phenotype: progressive intellectual disability, fair skin and hair (from tyrosine and melanin depletion), eczema, a mousy or musty body odour from phenylacetate, seizures, and behavioural and psychiatric disturbance including autism features and executive dysfunction. A child with unexplained developmental delay and these features warrants a plasma phenylalanine. [1] [12]
Maple syrup urine disease presents most dramatically in its classic neonatal form: a term baby, well at birth, who deteriorates 48 to 120 hours into protein-containing feeds with poor feeding, vomiting, lethargy, and a characteristic neurological syndrome of alternating hypertonia and hypotonia, dystonia, opisthotonic posturing and boxing movements, progressing to seizures, coma and apnoea. The odour — maple syrup, caramel, burnt sugar or earwax — in the urine, cerumen or skin is pathognomonic and should be sought deliberately. The intermittent and intermediate forms present later, in infancy or childhood, as episodes of ataxia, lethargy, ketoacidosis or encephalopathy provoked by illness or a high-protein meal, and they are the form most often mislabelled as a gastroenteritis or a toxin. [7] [8]
Tyrosinaemia type I presents in early infancy, usually before six months in the acute form, with liver failure — a bleeding tendency, jaundice, hypoglycaemia and a coagulopathy out of proportion to the transaminitis — alongside a renal Fanconi syndrome causing rickets and growth failure, and painful neurovisceral crises that mimic acute intermittent porphyria. The chronic form presents later with a hepatitic picture and a high risk of hepatocellular carcinoma. The boiled-cabbage or rancid-butter odour is a clue. Classical homocystinuria presents more slowly and is the great Marfan mimic: a tall, thin child with a marfanoid habitus, downward dislocation of the lens, myopia, osteoporosis, scoliosis, intellectual disability and a thromboembolic event that can be fatal in childhood. [10] [11]
Differential Diagnosis
The differential depends on the presentation, but the discipline is the same: ask what is accumulating, and order the metabolite that distinguishes the mimics. Persistent hyperphenylalaninaemia on the newborn screen must first be separated from transient neonatal tyrosinaemia, common in preterm infants on high-protein feeds, which resolves as feeding matures; from liver dysfunction, which impairs tyrosine and phenylalanine handling; and from a BH4 cofactor defect, which needs pterin analysis and a sapropterin response test. A normal or transient rise should not be dismissed as benign until a cofactor defect is excluded, because a missed DHPR deficiency presents with progressive dystonia. [1] [12]
A neonate with encephalopathy and ketoacidosis raises maple syrup urine disease against the organic acidaemias (propionic, methylmalonic, isovaleric), sepsis, and a urea cycle disorder. The discriminating pattern is the metabolite: MSUD shows raised leucine, isoleucine and valine with the pathognomonic allo-isoleucine and no hyperammonaemia; the organic acidaemias show a high anion-gap metabolic acidosis with an abnormal acylcarnitine and organic-acid profile and often a raised ammonia; a urea cycle disorder shows hyperammonaemia with NO acidosis and NO ketosis. Succinylacetone is the single test that confirms tyrosinaemia type I and distinguishes it from galactosaemia, hereditary fructosaemia, neonatal haemochromatosis and mitochondrial hepatopathies as causes of neonatal liver failure. [7] [10]
The marfanoid child with a lens dislocation raises classical homocystinuria against Marfan syndrome and the other lens-ectopia syndromes. The direction of the lens, the presence of thrombosis, the intellectual disability, and a markedly elevated total homocysteine with a high methionine set the diagnosis; a LOW methionine with a high homocysteine redirects the work-up to the remethylation defects (MTHFR deficiency, cobalamin C or D disease), which behave and are treated differently. Keeping the differential metabolite-driven — not label-driven — is what guarantees the right investigation at the first visit. [11]
Clinical & Bedside Assessment
The bedside assessment runs at two speeds. The acute stabilisation asks one question first: what is the level, and is this child about to lose their brain or their liver. You take a focused history of onset relative to feeds and the presence of a catabolic trigger, you note any odour, and you examine for the signs of cerebral oedema, hepatic failure or a coagulopathy — while simultaneously drawing the blood gas, glucose, ammonia, quantitative amino acids, and the targeted metabolites (succinylacetone, total homocysteine, acylcarnitines). Resuscitation is never delayed for a complete history. [7] [8]
The diagnostic characterisation follows once the child is safe. Build a three-generation pedigree asking explicitly about consanguinity, neonatal or unexplained deaths, developmental delay, and — for phenylketonuria — the mother's own diet and phenylalanine control in pregnancy, because an untreated mother with PKU can devastate a genetically unaffected fetus. Examine for the stigmata: fair skin and hair, eczema and a mousy odour in PKU; dystonia, opisthotonus and an odour in MSUD; hepatosplenomegaly, bruising and rickets in tyrosinaemia; ectopia lentis, marfanoid habitus and livedo in homocystinuria. These findings direct the investigation, but they never close a search on their own. [1] [11]
The bedside assessment converts directly into a parallel investigation order rather than a sequential work-up. Quantitative plasma amino acids confirm and follow PKU, MSUD and tyrosinaemia; succinylacetone confirms tyrosinaemia type I; total homocysteine confirms classical homocystinuria; acylcarnitines and organic acids screen the organic acidaemias; and pterin analysis with a sapropterin response test separates a BH4 cofactor defect from classical PKU. Assess the child's practical context — formula access, schooling, distance from a metabolic centre — because a location-specific emergency regimen is the intervention that changes outcome at home. [12] [3]
Investigations
Newborn bloodspot tandem mass spectrometry is the first-tier screen for the three disorders it detects well. Phenylketonuria is flagged by a raised phenylalanine and an elevated phenylalanine-to-tyrosine ratio; maple syrup urine disease by a raised leucine (with isoleucine and allo-isoleucine); and tyrosinaemia type I by a raised tyrosine together with succinylacetone. The limitation that examiners test is that homocystinuria may be missed, because methionine is variable and several newborn-screen programmes measure only amino acids and acylcarnitines, not homocysteine directly — so a normal newborn screen does not exclude it. [1] [11]
Quantitative plasma amino acids are the confirmatory test and the treatment-monitoring tool for PKU and MSUD. In PKU, phenylalanine is followed to the treatment target (the European guidelines recommend a target of 120 to 360 micromoles per litre across the age range, with strict control maintained before and during pregnancy). In MSUD, leucine, isoleucine and valine are followed, and allo-isoleucine is the pathognomonic metabolite that confirms the diagnosis and separates it from other causes of neonatal ketoacidosis. In tyrosinaemia type I, succinylacetone in blood or urine is both diagnostic and a marker of control, and alpha-fetoprotein with liver imaging forms the surveillance programme for hepatocellular carcinoma. [2] [10]
Why a BH4 (sapropterin) response test is part of the work-up
A rise in phenylalanine on the newborn screen could be classical PKU or a BH4 cofactor defect, and they are managed differently. A sapropterin (synthetic BH4) loading test, supported by urinary and plasma pterin analysis and molecular testing of GCH1, PTS, QDPR and PCBD1, separates them. A responsive fall in phenylalanine means the child may tolerate diet loosening on sapropterin; a cofactor defect additionally needs neurotransmitter replacement with levodopa/carbidopa and 5-hydroxytryptophan, because the same BH4 feeds the tyrosine and tryptophan hydroxylases. Missing a cofactor defect at this point is a serious and avoidable error. [1] [12]
Classical homocystinuria is confirmed by a markedly elevated total homocysteine with a raised methionine, which separates it from the remethylation defects (MTHFR deficiency, cobalamin C/D disease) where methionine is LOW. Molecular confirmation of the responsible gene (PAH, BCKDHA/BCKDHB/DBT/DLD, FAH, CBS, or the BH4-pathway genes) then defines the exact diagnosis, predicts cofactor responsiveness where relevant, and enables cascade carrier testing, prenatal diagnosis and preimplantation genetic testing for the family. [11] [12]
Management — Resuscitation
Resuscitation for an acute metabolic decompensation — most often an MSUD crisis — follows one principle: switch off catabolism and clear the toxin, and begin before the molecular diagnosis returns. The first moves are to stop all natural protein, establish intravenous access, and begin aggressive calorie provision with glucose plus insulin and intralipid, because switching the child from catabolism to anabolism is the single most important physiological intervention. Insulin is used not for hyperglycaemia but as an anabolic signal that drives amino acids into protein synthesis. [7] [8]
The disease-specific medical food is introduced early and in parallel. In MSUD, a branched-chain-amino-acid-free formula is given with added isoleucine and valine — the surprising but critical point that leucine falls faster when the other two branched-chain amino acids are repleted, because they re-enter the cycle and promote protein synthesis. The ketoacids are cleared by the kidney and, in severe cases, by haemofiltration or haemodialysis. A leucine level above roughly 1000 micromoles per litre with encephalopathy, or a clearly rising trend despite medical therapy, prompts extracorporeal removal; the objective is to halve leucine within hours, because every hour of cerebral oedema adds to permanent injury. [7] [9]
For a suspected or known phenylketonuria neonate, the resuscitation question is gentler but no less important: start a phenylalanine-restricted, not phenylalanine-free, diet, because some phenylalanine is an essential amino acid needed for growth, and over-restriction causes catabolism and a falling phenylalanine that itself impairs growth. The phenylalanine is titrated to the target range with frequent monitoring in the first weeks. Tyrosinaemia type I in acute liver crisis is treated with nitisinone (NTBC) and a tyrosine-restricted diet, and supportive management of the coagulopathy and Fanconi syndrome. [1] [10]
Management — Definitive & Stepwise
Once the acute crisis is controlled, definitive management is a lifelong, multidisciplinary framework built around diet, cofactor or enzyme therapy, prevention of catabolism, and — for selected severe disease — transplantation. For phenylketonuria, the ladder is a phenylalanine-restricted diet with a phenylalanine-free amino-acid formula, a trial of sapropterin (synthetic BH4) for responsive patients that can loosen the diet, and — for adults whose phenylalanine remains uncontrolled — pegvaliase, a pegylated phenylalanine ammonia lyase that lowers phenylalanine through an alternate degradation pathway. The pegvaliase dosing regimen of induction, titration and maintenance established in phase 2 studies made adult metabolic control achievable for the first time. [1] [6]
For maple syrup urine disease, the ladder is a leucine-restricted diet with a branched-chain-amino-acid-free formula, deliberate supplementation with isoleucine and valine to their target ranges, a thiamine trial for the thiamine-responsive form, and a written emergency regimen that the family can execute at the first sign of illness. Liver transplantation is an effective definitive therapy for severe MSUD, because the graft supplies enough enzyme activity to normalise branched-chain amino acids and abolish the risk of decompensation; the case series after transplantation show preserved or improved cognitive and adaptive functioning. [8] [9]
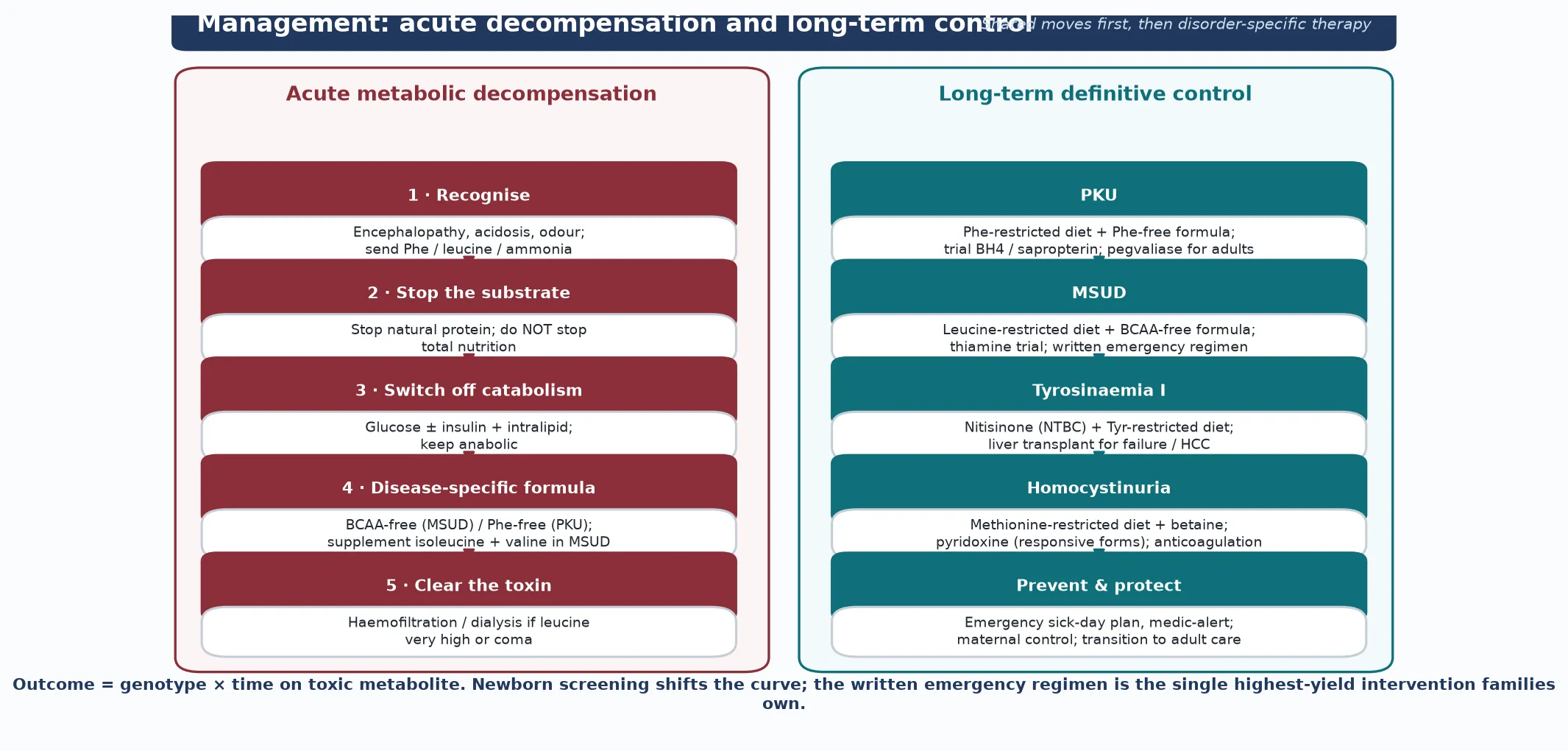
Tyrosinaemia type I is transformed by nitisinone (NTBC), which blocks 4-hydroxyphenylpyruvate dioxygenase upstream of the fumarylacetoacetate hydrolase block, preventing the formation of the toxic fumarylacetoacetate and succinylacetone. Nitisinone must be paired with a tyrosine-restricted diet, because blocking upstream raises tyrosine itself, and it converts a disease that was once near-uniformly fatal from liver failure into a medically manageable condition. Liver transplantation is reserved for acute liver failure unresponsive to nitisinone, suspected or confirmed hepatocellular carcinoma, and the rare non-responder. Corneal crystals and a high tyrosine are watched for on therapy. [10]
Classical homocystinuria is managed with a methionine-restricted diet, betaine (which donates a methyl group to remethylate homocysteine back to methionine, lowering the toxic homocysteine), pyridoxine (vitamin B6) in the responsive forms, plus folate and vitamin B12 to support remethylation. The life-saving intervention is thromboprophylaxis vigilance: dehydration, surgery, the peri-partum period and immobilisation all raise the thrombotic risk, and they must be managed with aggressive hydration, anticoagulation and metabolic optimisation. Every transition point — school, adolescence, pregnancy, surgery — is a moment to re-plan. [11]
C.O.F.A.C.T.O.R. — the therapy hooks
Specific Subtypes & Scenarios
Classical severe phenylketonuria — an untreated phenylalanine above about 1200 micromoles per litre — differs from mild hyperphenylalaninaemia and variant PKU, in which phenylalanine is only modestly raised and the child is often clinically well. The distinction matters because mild hyperphenylalaninaemia may not need a strict diet, but it does need confirmation that it is not a BH4 cofactor defect and, in girls and women, lifelong pre-conception counselling. The BH4 defects themselves are a distinct subtype: GTP cyclohydrolase deficiency (Segawa disease) presents as a dopa-responsive dystonia that often fluctuates diurnally, and dihydropteridine reductase deficiency is the most severe, needing lifelong neurotransmitter replacement alongside the diet. [1] [12]
Maple syrup urine disease has several subtypes that share the same enzyme block but differ in residual activity and severity. The classic form has essentially no residual activity and presents neonatally; the intermediate form presents in infancy with developmental delay and intermittent decompensation; the intermittent form is well between episodes provoked by catabolism; the thiamine-responsive form improves on pharmacological thiamine doses; and the rare E3 (dihydrolipoamide dehydrogenase) deficiency affects all three α-ketoacid dehydrogenase complexes and produces a combined lactic acidosis. Each subtype carries the same lifelong risk of cerebral oedema during illness. [7] [8]
Tyrosinaemia type I must be distinguished from the milder type II (Richner–Hanhart syndrome, corneal and palmar-plantar keratosis from tyrosine aminotransferase deficiency) and type III, in which nitisinone is NOT indicated because the toxic succinylacetone is not generated. Classical homocystinuria (CBS, with a HIGH methionine) must be distinguished from the remethylation defects — MTHFR deficiency and the cobalamin C and D defects — where methionine is LOW and the therapy is hydroxocobalamin, folate and betaine rather than methionine restriction. The direction of the methionine is the discriminator that sets the treatment. [10] [11]
The pregnant woman with phenylketonuria is a high-stakes scenario. An untreated or poorly controlled mother carries a phenylalanine that crosses the placenta and causes the maternal PKU syndrome — microcephaly, congenital heart disease, intrauterine growth restriction and intellectual disability in a fetus who may not even inherit the disease. The only prevention is strict phenylalanine control (target 120 to 360 micromoles per litre) achieved BEFORE conception and through the first trimester, which is why every girl and woman with PKU needs pre-conception counselling. The pregnant woman with homocystinuria is managed with hydration, betaine, folate and anticoagulation through the thrombosis-prone puerperium. [1] [11]
Complications & Pitfalls
The complications divide into the acute injuries, the chronic burden, and the cognitive traps that cost marks. The acute injuries are cerebral oedema and coma in MSUD, hepatic failure and neurovisceral crises in tyrosinaemia, and thromboembolism in homocystinuria — each of which can be fatal or permanently disabling if the diagnosis is delayed. The chronic burden is the cognitive, psychiatric and physical cost of time on the toxic metabolite: intellectual disability, executive dysfunction, white-matter change, anxiety and depression in PKU; osteoporosis and a neurocognitive burden in MSUD; hepatocellular carcinoma and renal disease in tyrosinaemia; and lens dislocation, osteoporosis and recurrent thrombosis in homocystinuria. [1] [9]
The most dangerous pitfall is over-restriction of an essential amino acid. Phenylalanine, leucine and methionine are all essential, and a diet that drives them too low causes catabolism, growth failure, skin rashes and, in MSUD, an actual worsening of leucine through muscle breakdown. The second pitfall is treating a BH4 cofactor defect as 'just PKU', because the child then develops a preventable, progressive dystonia from untreated dopamine deficiency. The third is forgetting maternal PKU, because a fetus with no genetic disease can still be devastated by the mother's phenylalanine. The fourth is mislabelling homocystinuria as Marfan and missing a treatable thrombotic risk. [1] [11]
Prognosis & Disposition
Prognosis is governed by three factors: the specific disorder, the age at diagnosis and treatment onset, and the lifetime, time-integrated exposure to the toxic metabolite — which in practice means adherence. Early-treated phenylketonuria allows near-normal intellect and a normal life; untreated or poorly controlled PKU carries a heavy cognitive, educational and psychiatric burden, and maternal PKU carries its own fetal prognosis. The European guideline data show that sustained phenylalanine control to the treatment target is the single strongest modifier of outcome, which is why adherence — not biology — is usually what limits a child's trajectory. [1] [2]
Maple syrup urine disease has a good intellectual outcome when acute crises are prevented and leucine is kept in range, but it carries a real lifelong risk of decompensation-related brain injury, so the emergency regimen is the load-bearing intervention. Tyrosinaemia type I, with nitisinone, has transformed from a near-uniformly fatal disease into one in which survival into adulthood is expected, though surveillance for hepatocellular carcinoma continues. Homocystinuria's prognosis is dominated by the thrombotic and ophthalmic complications, which rigorous metabolic control and thromboprophylaxis can reduce but never abolish. [9] [10]
Disposition is shared, lifelong, multidisciplinary care. The specialist metabolic service owns the diet, the cofactor and enzyme therapy, and the transplantation pathway; the general paediatrician or family doctor owns coordination, immunisation, growth and developmental surveillance, and the emergency sick-day plan; and the family owns the day-to-day vigilance that prevents catabolism. Every transition — into school, into adolescence, into pregnancy, and into adult metabolic services — is a high-risk point at which the emergency plan must be re-taught, re-written and reconciled. [3] [4]
Special Populations
The same amino-acid disorder behaves differently across populations because access, recognition and service models are unevenly distributed. In remote and Indigenous communities, later presentation, distance from a metabolic centre, the cost of medical formula, and the need for aeromedical retrieval during a neonatal MSUD crisis all lengthen the window between onset and treatment, so a written, location-specific emergency plan and a low threshold to send quantitative amino acids in any encephalopathic neonate are disproportionately important. In migrant, refugee and consanguineous families, the autosomal-recessive pre-test probability is higher, language barriers complicate the emergency teaching, and a trained interpreter must own the conversation. [7] [2]
The woman with phenylketonuria is a special population in her own right, because her pre-conception phenylalanine control dictates her fetus's outcome regardless of the fetus's own genotype. Every girl with PKU is counselled from adolescence that pregnancy must be planned, with phenylalanine brought to the target range before conception and through the first trimester. The woman with homocystinuria is managed through the thrombosis-prone pregnancy and puerperium with hydration, betaine, folate and anticoagulation. The adolescent transitioning to adult metabolic care is the highest-risk group for adherence slip, a lost emergency plan and an unplanned pregnancy, so the transition is planned, documented and rehearsed. [1] [11]
Socioeconomic disadvantage and the high cost of medical formula, sapropterin, pegvaliase and nitisinone create real inequity in outcome, and the general paediatrician is often the advocate who secures funding, arranges supply, and coordinates the school and welfare supports that make adherence possible. In families managing complex chronic metabolic disease, fragmentation of care is the chief threat, and a written, shared, reconciled care plan that travels with the child is the intervention that matters most. [4] [2]
Evidence, Guidelines & Regional Differences
The evidence base rests on consensus guidelines, longitudinal cohort and registry data, and a series of drug-development programmes that have become paradigms for metabolic therapy. The European guidelines on the diagnosis and treatment of phenylketonuria, updated in the first revision of 2025, set the current international standard for phenylalanine targets, BH4 responsiveness testing and the place of pegvaliase. The web-based nutrition management guideline for PKU (2016, updated for pegvaliase in 2023) and the parallel maple syrup urine disease nutrition guideline (2014) codify the medical-food approach, while the PKU Scientific Review Conference (2014) defined the evidence gaps that still drive research. [2] [3]
The drug-development story is itself high-yield viva material. Sapropterin (synthetic BH4) established the principle of pharmacological chaperone and cofactor therapy in PKU. Pegvaliase — a pegylated phenylalanine ammonia lyase that degrades phenylalanine through an alternate pathway, with an induction, titration and maintenance dosing regimen — made adult metabolic control achievable for patients in whom diet and sapropterin had failed, and it is a template for enzyme-substitution therapy across metabolic disease. Nitisinone transformed tyrosinaemia type I from a near-uniformly fatal liver disease into a medically manageable condition, and the MSUD liver-transplantation case series demonstrate that a graft can correct the enzyme deficit and abolish the risk of decompensation. [6] [9]
In Australia and New Zealand, the aminoacidopathies are managed through the state-based metabolic services (the major children's hospitals in each state and Starship in New Zealand), with the newborn screening programmes detecting PKU, MSUD and tyrosinaemia type I on the bloodspot tandem mass spectrometry panel. Phenylalanine targets follow the European guidelines, and access to sapropterin and pegvaliase is funded through specialist metabolic programmes with defined criteria, so the general paediatrician usually refers early and co-manages growth, immunisation and the emergency sick-day plan. Aeromedical retrieval to a tertiary metabolic and intensive-care centre is the expected pathway for an MSUD crisis, and continuous kidney replacement therapy is available at the paediatric intensive care units that receive these patients. Genetic counselling, carrier testing and prenatal or preimplantation diagnosis are coordinated through the clinical genetics services.
Exam Pearls
A fellowship candidate answering on the amino-acid disorders should land six anchor points and avoid four classic traps. The anchors are the shared toxic-buildup mechanism, the newborn-screen and metabolite signatures of PKU, MSUD, tyrosinaemia type I and homocystinuria, the acute 'switch off catabolism, clear the toxin' protocol for an MSUD crisis, the cofactor and enzyme-substitution therapies (sapropterin and pegvaliase in PKU, thiamine in MSUD, nitisinone in tyrosinaemia, betaine and pyridoxine in homocystinuria), the lens-direction and thrombosis points for homocystinuria, and maternal PKU. The traps are missing late-onset MSUD behind a gastroenteritis, treating a BH4 defect as 'just PKU', forgetting maternal PKU, and mislabelling homocystinuria as Marfan. The candidate who can name allo-isoleucine as the pathognomonic MSUD metabolite and succinylacetone as both the diagnostic marker and the tissue toxin of tyrosinaemia will defend the topic well. [1] [7]
References
- [1]Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet, 2010.PMID 20971365
- [2]van Spronsen FJ, Blau N, Hardt S, et al. European guidelines on diagnosis and treatment of phenylketonuria: First revision. Mol Genet Metab, 2025.PMID 40378670
- [3]Camp KM, Parisi MA, Acosta PB, Berry GT, Bilder DA, Blau N, et al. Updated, web-based nutrition management guideline for PKU: An evidence and consensus based approach. Mol Genet Metab, 2016.PMID 27211276
- [4]Singh RH, Stainback-Cerquoz N, Hougaard Drettvold A, et al. Nutrition management of PKU with pegvaliase therapy: update of the web-based PKU nutrition management guideline recommendations. Orphanet J Rare Dis, 2023.PMID 37349772
- [5]Camp KM, Lloyd-Puryear MA, Yao L, Groft SC, Parisi MA, Berry GT, et al. Phenylketonuria Scientific Review Conference: state of the science and future research needs. Mol Genet Metab, 2014.PMID 24667081
- [6]Thomas J, Levy H, Amoroso C, Longo N, Jones M, Hustvedt B, et al. Induction, titration, and maintenance dosing regimen in a phase 2 study of pegvaliase for control of blood phenylalanine in adults with phenylketonuria. Mol Genet Metab, 2018.PMID 30146451
- [7]Strauss KA, Puffenberger EG, Morton DH. Maple Syrup Urine Disease. GeneReviews, 1993.PMID 20301495
- [8]Frazier DM, Allgeier C, Homer C, Marriage BJ, Ogata B, Rohr F, et al. Nutrition management guideline for maple syrup urine disease: an evidence- and consensus-based approach. Mol Genet Metab, 2014.PMID 24881969
- [9]Mazariegos GV, Morton DH, Sindhi R, Soltys K, Nayyar N, Bond G, et al. Cognitive and adaptive functioning after liver transplantation for maple syrup urine disease: a case series. Pediatr Transplant, 2011.PMID 20946191
- [10]Chinsky JM, Singh R, Ficicioglu C, van Karnebeek CDM, Nyhan WL, Scott CR, et al. Tyrosinemia Type I. GeneReviews, 1993.PMID 20301688
- [11]Morris AAM, Kožich V, Santra S, Andria AM, Ben-Omran T, Chakrapani AB, et al. Homocystinuria due to Cystathionine Beta-Synthase Deficiency. GeneReviews, 1993.PMID 20301697
- [12]Mitchell JJ, Trakadis YJ, Scriver CR. Phenylalanine Hydroxylase Deficiency. GeneReviews, 1993.PMID 20301677