Paeds · genetics-dysmorphology-and-metabolism
Chromosomal microarray, exome and genome sequencing
Also known as Chromosomal microarray (CMA) · Comparative genomic hybridisation (aCGH) · SNP array · Whole-exome sequencing (WES) · Whole-genome sequencing (WGS) · Rapid genomic sequencing · Next-generation sequencing
Fellowship approach to choosing between chromosomal microarray, whole-exome and whole-genome sequencing in a child with congenital anomalies, developmental delay or a suspected monogenic disorder: what each platform detects, the resolution ladder, diagnostic yields, variant interpretation, secondary findings, consent, and rapid sequencing in the critically ill infant.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
T.E.S.T. ladder
Overview & Definition
Genomic diagnostics is the laboratory arm of clinical genetics, and the choice of test is the single most important decision a clinician makes after completing the clinical assessment. The four platforms are not interchangeable: they interrogate the genome at different resolutions and answer different questions, and ordering the wrong test wastes time, money and the family's trust. The fellowship candidate must be able to defend each choice against the clinical phenotype and the published evidence. [1] [2]
A karyotype is the light-microscope view of whole chromosomes, resolving changes larger than roughly five to ten megabases; it remains the test of choice for a suspected balanced rearrangement such as a translocation or inversion, which the newer platforms cannot see. Chromosomal microarray (CMA) uses array comparative genomic hybridisation (aCGH) and single-nucleotide-polymorphism (SNP) arrays to detect copy-number variants — deletions and duplications — down to tens of kilobases, and it can flag regions of homozygosity suggestive of uniparental disomy or consanguinity. It cannot detect single-nucleotide variants or balanced rearrangements. [1]
Whole-exome sequencing (WES) captures and sequences the protein-coding exons, which make up about one and a half percent of the genome but harbour roughly eighty-five percent of disease-causing variants. It detects single-nucleotide variants and small insertions or deletions in coding regions and canonical splice sites, but it misses deep intronic, regulatory and most structural variants, and it does not reliably detect repeat expansions. Whole-genome sequencing (WGS) sequences the entire genome and therefore captures the coding variants, the non-coding and regulatory variants, and the structural and copy-number variants in a single assay — at the cost of generating vastly more data and more variants of uncertain significance. [2] [3]
The clinical decision is which platform matches the phenotype, the acuity, and the locally funded pathway. In the well child with unexplained developmental delay or congenital anomalies, testing proceeds in a tier. In the critically ill infant in the intensive care unit, the tier collapses and rapid whole-genome sequencing becomes the first and sometimes only test, because a molecular diagnosis within days can change acute management, withdraw or escalate life-sustaining treatment, or redirect care. [7] [8]
Classification
Classify the platforms along three axes — what they detect, the resolution at which they detect it, and where they sit in the diagnostic tier. The karyotype sits at the base of the resolution ladder and answers the question of large chromosomal abnormality: aneuploidy, large deletions or duplications, and balanced rearrangements. It is now a targeted rather than a screening test. [1]
Chromosomal microarray sits one rung up and answers the question of copy-number change. It is split into two technologies that are often combined on a single slide: aCGH, which compares the patient's DNA against a reference to find gains and losses, and the SNP array, which additionally genotypes thousands of markers to detect copy-neutral loss of heterozygosity — regions of homozygosity that flag uniparental disomy or consanguinity. A pure aCGH platform misses this, which is why the SNP-array component matters when Prader–Willi, Angelman or a recessive condition is in the differential. [1]

Sequencing sits at the top and answers the question of single-nucleotide and small-variant change. Whole-exome sequencing reads the coding fraction; whole-genome sequencing reads the whole genome and additionally calls structural variants and repeat expansions that the exome misses. Gene-panel sequencing is a focused, cheaper alternative that sequences a curated set of disease-associated genes and carries a smaller variant-interpretation burden, and it remains a reasonable first sequencing test when the phenotype points to a defined disease family such as a cardiomyopathy or a deafness panel. [2] [3]
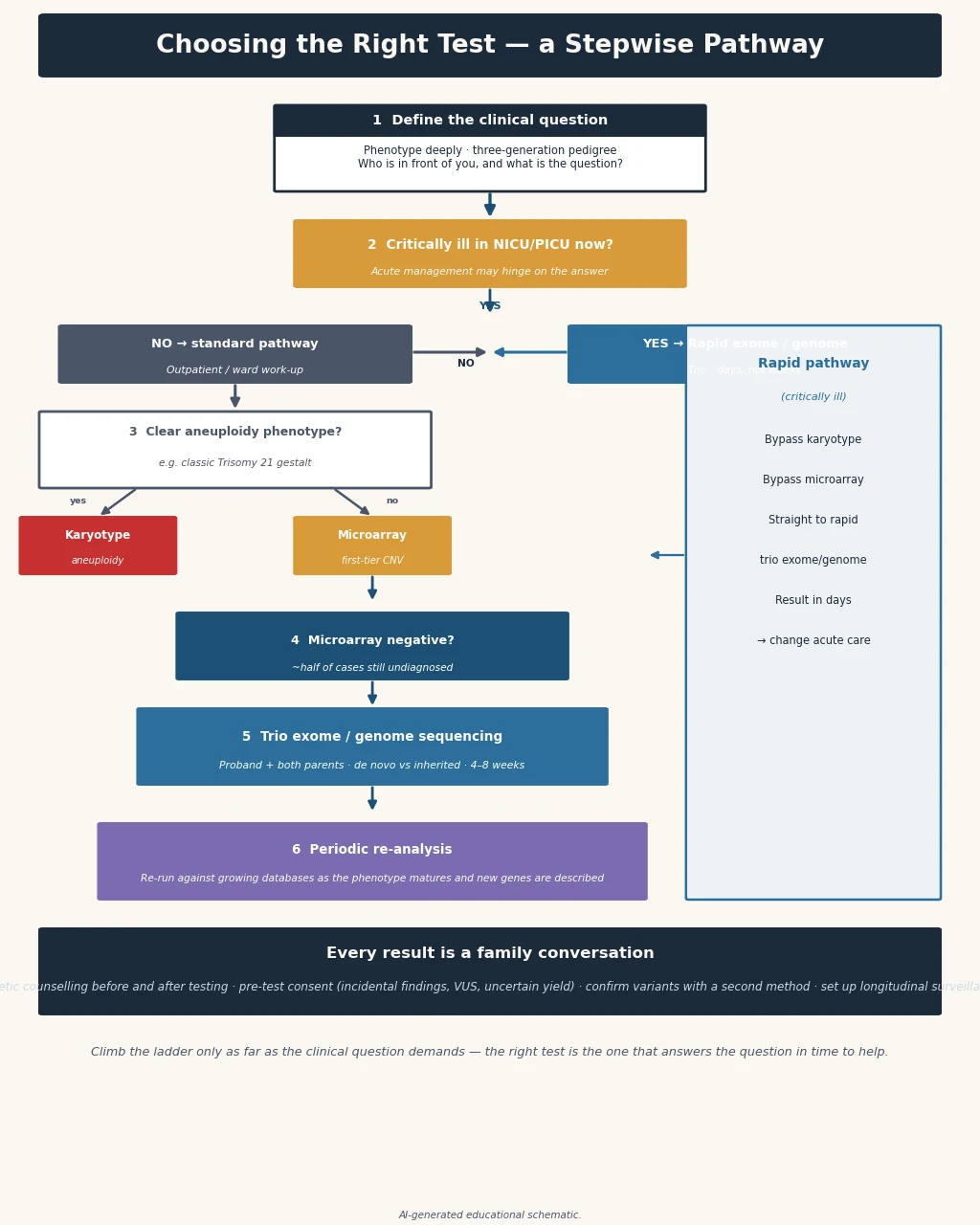
The tier follows the resolution. For the child with unexplained developmental delay, intellectual disability, autism with dysmorphism, or multiple congenital anomalies, chromosomal microarray is the established first-tier test and the karyotype is reserved for a suspected balanced rearrangement. Exome or genome sequencing is the second tier when the microarray is unrevealing, and — supported by the 2021 ACMG guideline and the 2019 meta-analytic consensus — is increasingly defensible as a first-tier test in its own right. Rapid whole-genome sequencing sits outside the tier: it is the first test for the critically ill infant with a suspected monogenic disorder. [1] [2] [3]
Epidemiology & Risk Factors
The population that drives genomic testing is large. Roughly two to four percent of liveborn infants have a major congenital anomaly, one to three percent have intellectual disability, and developmental delay affects a similar proportion of young children. Across this population, a molecular diagnosis was historically reached in only a minority, leaving most families without an answer and without recurrence-risk information. The newer platforms have shifted that curve. [1] [4]
The diagnostic yield rises as the platform ascends the resolution ladder. Chromosomal microarray returns a pathogenic copy-number variant in roughly fifteen to twenty percent of children with unexplained developmental delay, intellectual disability, autism or multiple congenital anomalies — a yield two to three times that of a karyotype, which is the evidence base that made microarray first-tier. Whole-exome sequencing adds a further diagnostic increment of roughly twenty-five to forty percent when applied after a normal microarray, and a trio exome (sequencing the child and both parents) lifts the yield further by resolving de novo status and inheritance. Whole-genome sequencing, particularly in critically ill infants, reports diagnostic yields of thirty to fifty percent. [1] [3] [7]
Risk factors that lower the threshold for, or alter the choice of, genomic testing include advanced parental age (which raises the rate of de novo point variants and of aneuploidy), consanguinity (which raises the prior probability of autosomal recessive disease and makes a SNP array or genome sequencing preferable to a pure aCGH), a family history of a known or suspected monogenic disorder, and a prior affected child. Critically, acuity is itself a risk factor: an infant in intensive care with a suspected monogenic disorder should not wait for tiered testing. [2] [7]
Pathophysiology
The resolution ladder exists because each platform interrogates the genome through a different physical principle, and understanding the principle explains both what each test finds and what it misses. The karyotype photographs metaphase chromosomes after culture and staining; it resolves gross number and structure but is blind to anything below the megabase range, including most pathogenic copy-number variants. It is the only routine test that detects a balanced translocation, because the total DNA content is unchanged and neither microarray nor sequencing reliably calls it. [1]
Chromosomal microarray hybridises fragmented, fluorescently labelled patient and reference DNA to thousands of probes tiling the genome, then reads the fluorescence ratio at each probe to call a gain or loss. The resolution is two orders of magnitude finer than a karyotype, which is why microarray finds the submicroscopic deletion behind 22q11.2 deletion syndrome or Williams syndrome that a karyotype cannot. The SNP-array component genotypes known polymorphic sites, and a long contiguous stretch of homozygous genotypes flags a region of homozygosity — the signature of uniparental disomy (relevant to Prader–Willi and Angelman) or of shared ancestry (relevant to recessive disease). Microarray cannot detect a single-base change, a balanced rearrangement, or a low-level mosaic variant below its detection threshold. [1]

Sequencing fragments genomic DNA, amplifies or captures the fragments, and reads them in massive parallel through next-generation sequencing. Exome sequencing first captures the coding exons with biotinylated probes, then sequences them; its strength is depth of coverage of the coding fraction, and its weakness is incomplete capture of some GC-rich or homologous exons, blindness to non-coding and regulatory variants, and an inability to call repeat expansions such as the FMR1 CGG repeat. Genome sequencing skips the capture step and sequences the entire molecule, so it recovers the coding variants and adds the intronic, regulatory, structural and repeat-expansion variants in one assay. Its blind spots are shorter-read limitations in highly repetitive regions and the data-interpretation burden of millions of variants. [2] [3]
The pathophysiology of interpretation is just as important as the sequencing itself. A sequencing run yields millions of variants, and the laboratory must filter them against the phenotype, the population frequency, the predicted effect on the protein, and the inheritance pattern, then classify each retained variant by the ACMG/AMP framework into pathogenic, likely pathogenic, uncertain significance, likely benign or benign. A trio strategy — sequencing the child and both parents — is powerful precisely because it resolves whether a variant is de novo, inherited from an unaffected carrier (which weakens pathogenicity), or inherited from an affected parent (which strengthens it). [6]
Clinical Presentation
The clinical presentations that trigger genomic testing fall into four cohorts, each with a different tempo. The first is the dysmorphic newborn or infant with multiple congenital anomalies — here the question is a syndromic copy-number or single-gene diagnosis, and the tiered pathway applies once the child is stable. The second is the older infant or child with unexplained global developmental delay, intellectual disability or autism, where the question is a neurodevelopmental genetic cause and the first-tier test is chromosomal microarray, escalating to exome or genome. [1] [4]
The third cohort is the child with a phenotype that points to a defined disease family — a progressive muscle weakness suggesting a muscular dystrophy, a cardiomyopathy, a deafness, or a syndromic obesity — where a targeted gene panel may be the most efficient first sequencing test, reserving exome or genome for an atypical or panel-negative presentation. The fourth, and most time-critical, is the critically ill infant in the neonatal or paediatric intensive care unit with a suspected monogenic disorder — here the question is whether a rapid molecular diagnosis will change acute management, and the test of choice is rapid whole-genome sequencing. [7] [8]
Across all cohorts, the clinical phenotype directs both the choice and the interpretation of testing. A sequencing result is only as good as the phenotypic information that accompanies it; a vague referral lowers the diagnostic yield because the laboratory cannot prioritise candidate genes. The clinician's job is to build a structured Human Phenotype Ontology description of the child, not merely to send a sample. [2] [4]
Differential Diagnosis
When choosing a test, the differential is a differential of variant classes, not of diseases. The first question is whether the suspected cause is a copy-number change, a single-nucleotide variant, a structural variant, a repeat expansion, or an epigenetic (methylation) abnormality — because each platform answers a different one of these questions. [1] [6]
A copy-number differential (a microdeletion or microduplication syndrome, aneuploidy, or a large deletion) points to chromosomal microarray. A single-nucleotide differential (a point mutation in a coding gene) points to exome or genome sequencing. A repeat-expansion differential — fragile X syndrome, myotonic dystrophy, Friedrich ataxia — points to a targeted repeat-primed PCR or Southern blot, because exome sequencing does not reliably call it. An imprinting or methylation differential — Prader–Willi, Angelman — points to methylation-specific testing or a SNP array that detects the resulting homozygosity, because a pure aCGH will miss it. [1] [4]
The can't-miss mimics are the results that look negative but are not. A normal microarray in a child with developmental delay does not exclude fragile X, a single-gene disorder, or an imprinting defect — it excludes only the common copy-number causes. A normal exome does not exclude a deep intronic variant, a structural variant, a repeat expansion, or a mosaic variant below the detection threshold. The clinician must hold both the result and its limitations in mind when counselling the family. [2] [3]
Clinical & Bedside Assessment
Define the clinical question before ordering
The highest-yield genomic test is the one ordered after a structured clinical assessment. Take a three-generation pedigree, ask explicitly about consanguinity, document the perinatal and developmental history, and perform a head-to-toe dysmorphology examination in standard terminology. Build a Human Phenotype Ontology term list, because the laboratory uses the phenotypic information to filter and prioritise candidate variants. A vague referral is the commonest cause of a missed diagnosis. [4] [5]
Choose the platform by the phenotype and acuity
In the stable child with unexplained developmental delay, intellectual disability, autism with dysmorphism, or multiple congenital anomalies, order chromosomal microarray as the first-tier test, and add a targeted test if the phenotype suggests a specific repeat-expansion or imprinting disorder. If the microarray is unrevealing and a monogenic syndrome remains likely, escalate to exome or genome sequencing, ideally as a trio. [1] [2]
Identify the critically ill infant who needs rapid sequencing
In the critically ill infant in the neonatal or paediatric intensive care unit with a suspected monogenic disorder, do not wait for the tier. The indication for rapid whole-genome sequencing is a sick infant in whom a timely molecular diagnosis could change acute management — escalating or withdrawing life-sustaining treatment, redirecting to palliative care, or instituting a specific therapy. The randomised NSIGHT1 trial and the Farnaes cohort showed that rapid sequencing delivers a diagnosis in days and changes management in a meaningful proportion of such infants. [7] [8] [9]
Consent before you test
Pre-test counselling is mandatory, not optional, because genomic sequencing can return findings the family did not expect. Explain what the test can and cannot find, the possibility of a variant of uncertain significance, the option to opt in or out of secondary findings in actionability genes, and the plan for data storage, re-analysis and family communication. Consent is a conversation, not a signature. [2] [10]
Investigations
The investigation strategy is the tier itself, and the candidate must be able to justify each rung. First tier — chromosomal microarray for unexplained developmental delay, intellectual disability, autism with dysmorphism, or multiple congenital anomalies, with a diagnostic yield of fifteen to twenty percent over karyotype. Request a platform that includes the SNP-array component if uniparental disomy or consanguinity is in the differential. [1]
Targeted testing alongside or instead of the tier. Send FMR1 repeat testing for any boy with unexplained developmental delay or autism regardless of the microarray, because microarray does not detect the fragile X expansion. Send methylation-specific testing for Prader–Willi or Angelman when the phenotype fits, because a pure aCGH microarray can miss it. Send a karyotype when a balanced translocation is suspected — recurrent miscarriages, a family history of a known translocation, or an abnormal prenatal karyotype — because microarray and sequencing do not reliably detect balanced rearrangements. [1] [4]
Second tier — exome or genome sequencing when the microarray is unrevealing and a monogenic cause remains likely. The 2021 ACMG guideline and the 2019 meta-analytic consensus support exome and genome sequencing as first- or second-tier tests for children with congenital anomalies, intellectual disability and neurodevelopmental disorders. A trio strategy (child plus both parents) increases the diagnostic yield and resolves inheritance, and is the preferred approach when parents are available. [2] [3]
Rapid whole-genome sequencing for the critically ill infant. In the neonatal or paediatric intensive care unit, the NSIGHT1 randomised trial demonstrated that rapid whole-genome sequencing shortens the time to an etiologic diagnosis and increases the proportion of infants receiving a diagnosis within a clinically useful timeframe. The Farnaes cohort and the PICU studies extended this to show decreased morbidity and cost of hospitalisation and clinical utility in the paediatric intensive care population. This is the one setting in which the tier collapses. [7] [8] [9]
Variant classification and reporting. Every reported variant is classified by the ACMG/AMP five-tier framework — pathogenic, likely pathogenic, uncertain significance, likely benign, benign — using population-frequency data, computational predictions, segregation in the family, and the phenotype–genotype match. Only pathogenic and likely pathogenic variants are reported as diagnostic. The variant of uncertain significance is a real finding whose meaning is not yet known; it must not be over-interpreted. [6]
Secondary findings. The ACMG secondary-findings list (currently version 3.2) recommends actively analysing and reporting pathogenic variants in a curated set of actionability genes — examples include the hereditary cancer genes such as BRCA1 and BRCA2 and the Lynch-syndrome mismatch-repair genes, and the potentially lethal cardiac genes such as those for long-QT and hypertrophic cardiomyopathy — regardless of the indication for testing, unless the family has opted out. These are real results with real surveillance and treatment implications, and consent must address them before testing. [10]
Management — Resuscitation
Genomic testing does not resuscitate a child, but in the critically ill infant the speed of a molecular diagnosis can redirect acute management. The immediate priority remains airway, breathing and circulation; the diagnostic question is addressed in parallel, not in place of, stabilisation. The role of rapid whole-genome sequencing in this setting is to deliver a diagnosis quickly enough to matter — to withdraw a harmful intervention, to institute a specific therapy such as a tailored metabolic or enzyme regimen, or to redirect from curative intent to palliative care. [7] [8]
Recognise the infants in whom rapid sequencing is indicated and refer early: the neonate with an unexplained encephalopathy, a suspected metabolic crisis, severe hypotonia, multi-organ failure of uncertain cause, or a dysmorphic pattern with decompensation. The diagnostic yield in this population is thirty to fifty percent, and a diagnosis changes management in a substantial proportion — the evidence base that justifies bypassing the tier. [7] [9]
Management — Definitive & Stepwise
- Complete the structured clinical assessment and build a Human Phenotype Ontology phenotypic description before ordering any test. [4]
- Consent the family on what the test will and will not find, the possibility of a variant of uncertain significance, and the secondary-findings opt-out. [2] [10]
- Choose the first-tier test by the phenotype: chromosomal microarray for unexplained developmental delay, intellectual disability, autism with dysmorphism, or multiple congenital anomalies; targeted testing (FMR1, methylation, karyotype) when the phenotype points to it. [1]
- Escalate to exome or genome sequencing, ideally as a trio, when the first tier is unrevealing and a monogenic cause remains likely. [2] [3]
- In the critically ill infant, request rapid whole-genome sequencing as the first test and refer through the local rapid-genomics pathway. [7] [8]
- Interpret the reported variant against the clinical phenotype and the ACMG/AMP classification; arrange segregation testing of parents when a variant of uncertain significance is found. [6]
- Communicate the result honestly and jargon-free, address recurrence risk, offer cascade testing of the family, and book a definite review. [2] [4]
- For a negative test, store the data and schedule periodic re-analysis as knowledge grows and the phenotype evolves. [2] [3]
Specific Subtypes & Scenarios
Stable child with unexplained developmental delay. The archetype of tiered testing. Order chromosomal microarray first; if unrevealing and a monogenic cause is likely, escalate to trio exome or genome. Add FMR1 testing in any boy with developmental delay or autism. Hold the family with a clear review point and a plan to re-analyse if the initial result is negative. [1] [4]
Child with multiple congenital anomalies. Microarray first-tier; escalate to exome or genome, because a substantial fraction of multi-organ malformation patterns are monogenic. Integrate the result into organ-targeted surveillance — echocardiography, renal ultrasound, hearing and vision screening — guided by the confirmed diagnosis. [1] [2]
Critically ill infant in intensive care. Rapid whole-genome sequencing as the first test, through the local rapid-genomics pathway. The NSIGHT1 trial and the Farnaes and PICU cohorts support its use when a diagnosis could change acute management — to escalate, to withdraw, or to redirect to palliation. Consent the family that a rapid result may change the goals of care. [7] [8] [9]
Consanguineous family. A SNP-array component or genome sequencing is preferable to a pure aCGH, because the long stretches of homozygosity flag the autosomal recessive disease that consanguinity makes likely. Weigh autosomal recessive conditions more heavily in the variant interpretation. [1] [6]
Known familial variant. When a pathogenic variant is already known in the family, offer targeted cascade testing rather than a full sequencing run — it is faster, cheaper, and answers the specific reproductive and surveillance question. [2] [4]
Complications & Pitfalls
The most dangerous pitfall is over-interpreting a variant of uncertain significance as pathogenic. A variant of uncertain significance is exactly that — uncertain — and treating it as a diagnosis can expose a child to unnecessary surveillance, anxiety and stigma while a re-classification later reverses it. Guard against this by interpreting every variant against the phenotype, by arranging segregation testing of the parents, and by refusing to build a management plan on a variant that does not match the clinical picture. [6]
The mirror pitfall is the false reassurance of a negative test. A normal microarray does not exclude fragile X, a single-gene disorder, or an imprinting defect. A normal exome does not exclude a deep intronic, regulatory, structural or mosaic variant. Counsel the family on what the test excluded and what it did not, and keep the door open for re-analysis. [2] [3]
Secondary findings are a pitfall of omission and of commission. Failing to offer the secondary-findings opt-out before testing deprives the family of choice; returning an actionable cancer-predisposition or cardiac variant without a counselling and surveillance plan abandons them at the moment of diagnosis. Address secondary findings in the consent and have a pathway to return them. [10]
A practical pitfall is sending a sample without phenotypic information. The laboratory filters millions of variants against the phenotype; a vague referral lowers the yield and delays the diagnosis. Invest the time in a structured Human Phenotype Ontology description before the sample leaves. [2] [4]
Prognosis & Disposition
The value of a molecular diagnosis extends beyond the label. A confirmed diagnosis unlocks guideline-based surveillance, secures access to therapy, funding and disability services, enables informed reproductive planning for the parents and the wider family, connects the family to peer support, and ends the diagnostic odyssey. Even when no diagnosis is reached, an explicit plan for periodic re-analysis protects the child, because a variant that was unclassifiable today may be re-interpretable within a year as the knowledge base grows. [2] [3]
Disposition is shared between the clinical genetics service, which owns the variant interpretation and the recurrence-risk counselling, and the general paediatric medical home, which owns the longitudinal care, the developmental and educational support, and the organ-targeted surveillance. Define who owns each piece and book the review. A negative result is not the end of the pathway — it is a checkpoint at which the data are stored and a re-analysis date is set. [4] [5]
Special Populations
Adapt the testing strategy to the child and the family in front of you. In a child from a consanguineous family, prefer a SNP-array component or genome sequencing and weigh autosomal recessive disease in the interpretation. In a child from an ethnic background under-represented in reference databases, recognise that a variant classified as rare or pathogenic in one population may be common and benign in another, and avoid over-calling a variant of uncertain significance against an inappropriate reference panel. [1] [6]
For rural and remote families, distance and cost can delay testing; use telehealth genetics and state-funded testing pathways to close the gap, and arrange interpreter and cultural-safety support so that language and distance do not delay diagnosis. For Māori and Pacific families in Aotearoa New Zealand and Aboriginal and Torres Strait Islander families in Australia, ensure the testing pathway is actively supported, because inequity in access to genomic medicine is well documented. [2]
The critically ill infant is a special population in its own right: the tempo and the test differ. In the intensive care unit, the tier collapses and rapid whole-genome sequencing is the first test, because the value of the diagnosis is measured in days, not weeks. [7] [9]
Evidence, Guidelines & Regional Differences
The evidence base for tiered genomic testing is strong and consistent. The 2010 consensus statement established chromosomal microarray as the first-tier test for unexplained developmental disability and congenital anomalies, on the strength of its fifteen to twenty percent diagnostic yield over karyotype. The 2019 meta-analysis and multidisciplinary consensus, and the 2021 ACMG evidence-based guideline, extended the tier upward, positioning exome and genome sequencing as first- or second-tier tests for selected children with congenital anomalies, intellectual disability and neurodevelopmental disorders, with yields of thirty to forty percent. [1] [2] [3]
For rapid sequencing in the critically ill infant, the randomised NSIGHT1 trial showed that rapid whole-genome sequencing increases the proportion of infants receiving an etiologic diagnosis within a clinically useful timeframe, and the Farnaes and PICU cohorts showed decreased morbidity and cost of hospitalisation and clinical utility in the intensive care population. The ACMG/AMP 2015 standards, and the current ACMG secondary-findings list, frame variant interpretation and reporting. [6] [7] [10]
Petrikin et al. 2018 — NSIGHT1 randomised controlled trial (NPJ Genomic Medicine)
Randomised controlled trial of rapid whole-genome sequencing versus standard testing in critically ill infants.
Key finding
Rapid whole-genome sequencing increased the proportion of infants receiving an etiologic diagnosis within a clinically useful timeframe, supporting its use as a first-line test when a timely diagnosis could change acute management.
Practice change
In the critically ill infant with a suspected monogenic disorder, rapid whole-genome sequencing is the test of choice — the tier collapses.
In Australia and Aotearoa New Zealand, genomic testing is state- and district-funded but access is uneven, with longer waits in rural and remote areas and for Māori, Pacific, Aboriginal and Torres Strait Islander families unless pathways are actively supported. Medicare-funded whole-genome and whole-exome items now exist for specified indications including suspected monogenic disorders in children, and rapid-genomics pathways for critically ill infants operate through tertiary networks. Confirm the local funded pathway, refer to the nearest clinical genetics service early, and provide cultural-safety and interpreter support so that distance and language do not delay diagnosis. [2]
Exam Pearls
- The resolution ladder is a perennial viva opener: karyotype (chromosome) → microarray (copy-number) → exome (coding base-pair) → genome (whole base-pair). Know the resolution and the blind spot of each. [1]
- Chromosomal microarray is first-tier for unexplained developmental delay, intellectual disability, autism with dysmorphism, or multiple congenital anomalies; the yield is fifteen to twenty percent. [1]
- Exome and genome sequencing are first- or second-tier; the yield is thirty to forty percent, and a trio lifts it further. [2] [3]
- A normal microarray does not exclude fragile X — send FMR1 testing for any boy with developmental delay or autism. [1] [4]
- Microarray misses balanced rearrangements — order a karyotype when a translocation is suspected. [1]
- A pure aCGH misses uniparental disomy — request the SNP-array component for Prader–Willi or Angelman, or send methylation testing. [1]
- The variant of uncertain significance is not a diagnosis — interpret it against the phenotype and arrange segregation testing. [6]
- Secondary findings in ACMG actionability genes are reported unless the family opts out — consent before testing. [10]
- Rapid whole-genome sequencing is the first test for the critically ill infant — the NSIGHT1 trial is the evidence. [7]
- Every negative test is re-interpretable — store the data and set a re-analysis date. [2]
References
- [1]Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. American Journal of Human Genetics, 2010.PMID 20466091
- [2]Manickam K, McClain MR, Demmer LA, Biswas S, Kearney HM, Malinowski J Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine, 2021.PMID 34211152
- [3]Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genetics in Medicine, 2019.PMID 31182824
- [4]Moeschler JB, Shevell M, Committee on Genetics Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics, 2014.PMID 25157020
- [5]Moeschler JB, Shevell M, American Academy of Pediatrics Committee on Genetics Clinical genetic evaluation of the child with mental retardation or developmental delays. Pediatrics, 2006.PMID 16740881
- [6]Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 2015.PMID 25741868
- [7]Petrikin JE, Cakici JA, Smith MJ, Kingsmore SF The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. NPJ Genomic Medicine, 2018.PMID 29449963
- [8]Farnaes L, Hildreth A, Sweeney NM, Clark MM, Chowdhury S, Nahas S Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genomic Medicine, 2018.PMID 29644095
- [9]Sanford EF, Mawn LA, Jordan E, Ackerman A, Champaigne J, Kingsmore SF Rapid whole genome sequencing has clinical utility in children in the PICU. Pediatric Critical Care Medicine, 2019.PMID 31246743
- [10]Miller DT, Lee K, Chung WK, Gordon AS, Hagstrom SA, Klein TE ACMG SF v3.2 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine, 2023.PMID 37347242