Paeds · genetics-dysmorphology-and-metabolism
Disorders of metal metabolism: Wilson and Menkes disease
Also known as Wilson disease · Hepatolenticular degeneration · ATP7B deficiency · Menkes disease · Menkes kinky hair disease · ATP7A deficiency · Copper transport disorders
A fellowship approach to the two inherited copper-transporting ATPase disorders: Wilson disease, in which a defective ATP7B gene causes copper to accumulate and poison the liver and basal ganglia, and Menkes disease, in which a defective ATP7A gene prevents copper from ever leaving the gut and reaching the brain so that copper-dependent enzymes fail. Recognise Wilson by its hepatic and neuropsychiatric faces and the Kayser-Fleischer ring, confirm with low ceruloplasmin and high urinary copper, and treat with chelation or zinc, reserving transplant for acute failure. Recognise Menkes in the hypotonic, failing infant with kinky hair and seizures, confirm with low serum copper, and treat with subcutaneous copper histidinate as early as possible.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who holds the single unifying idea while answering each disease in depth. That idea is the copper-transporting ATPase: a membrane pump that moves copper across a barrier. Two sister genes encode two such pumps. ATP7B sits in the hepatocyte and loads copper into ceruloplasmin and into bile, so when it fails, copper stays in the body and accumulates. ATP7A sits in the gut enterocyte and the blood-brain barrier and ferries dietary copper into the circulation and the brain, so when it fails, copper is trapped in the enterocyte and the body is starved. The same ion, the same protein family, the opposite clinical problem - and this single contrast explains every clinical, biochemical and therapeutic fact a fellowship examiner will probe. [1] [5]
Overview & Definition
Wilson disease and Menkes disease are inherited disorders of copper transport caused by defects in two homologous copper-transporting ATPases. The first, Wilson disease (hepatolenticular degeneration), follows autosomal recessive loss of the ATP7B copper transporter on chromosome 13. Because ATP7B normally loads copper into the plasma protein ceruloplasmin and excretes excess copper into bile, its failure leaves copper trapped in the hepatocyte; copper then accumulates, poisons the liver, and eventually spills into the blood to deposit in the brain, cornea and other organs. It is a disorder of copper toxicity. [1] [3]
The second, Menkes disease (kinky hair disease), follows X-linked recessive loss of the ATP7A copper transporter. ATP7A normally shuttles dietary copper out of the gut enterocyte into the bloodstream and across the blood-brain barrier, so its failure traps copper in the enterocyte and denies it to the body and brain. The result is a functional systemic copper deficiency in which copper-dependent enzymes - lysyl oxidase, dopamine-beta-hydroxylase, tyrosinase and cytochrome c oxidase among them - fail to mature, producing the characteristic hair, vascular, pigmentation and neurodegenerative features. It is a disorder of copper deficiency. [5] [6]
These two conditions are paired deliberately because the contrast is the teaching. Both are diseases of copper, both are caused by a pump that cannot move copper, and both produce devastating brain injury - but the direction of the error is opposite, and so is the treatment. Remove the copper in Wilson, replace the copper in Menkes. The candidate who internalises this contrast will never confuse the biochemistry, the lab values, or the management. [1] [5]
Classification
The classification that matters clinically is molecular and directional, because the gene and the direction of the copper error determine everything downstream. There are two genes, each with a classic severe disease and a milder allelic variant, and the inheritance pattern differs between them. [1] [5]
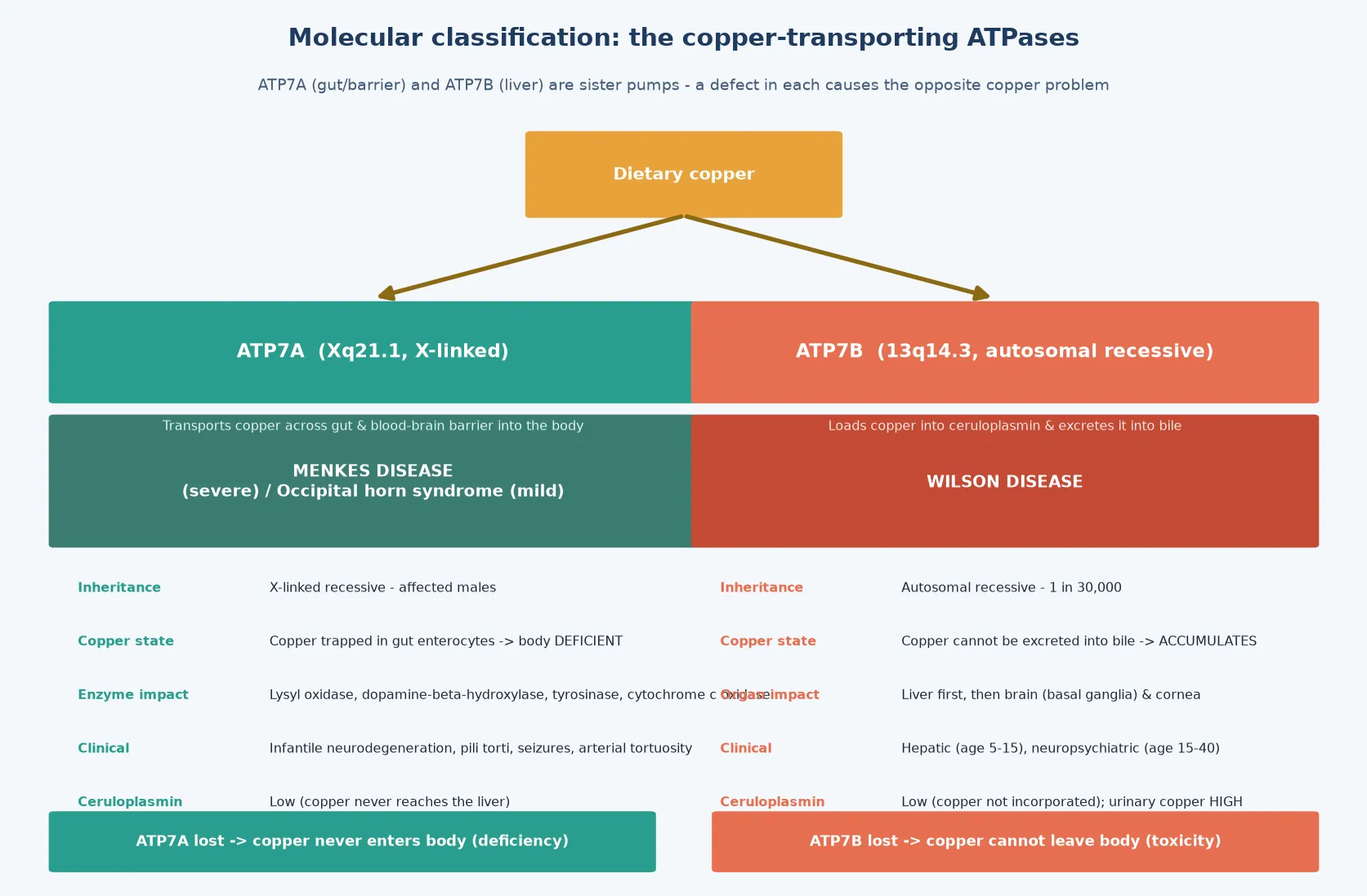
Wilson disease is caused by biallelic pathogenic variants in ATP7B. Hundreds of variants are known, and the genotype does not predict the phenotype reliably because of modifier genes and environmental copper intake, though loss-of-function variants are increasingly associated with worse hepatic survival, as Nayagam and colleagues demonstrated in their genotype-outcome cohort. A milder ATP7B-related phenotype exists in which presentation is delayed into adulthood. The inheritance is autosomal recessive, so siblings carry a one-in-four risk and screening of siblings is mandatory at diagnosis. [10] [1]
Menkes disease is the severe infantile phenotype of ATP7A loss. The same gene produces a milder allelic disorder, occipital horn syndrome, in which partial ATP7A function survives and the phenotype is dominated by connective-tissue features (cutis laxa, bladder diverticula, occipital exostoses) with little or no neurodegeneration. Because ATP7A is X-linked, the severe disease affects males; female carriers are usually asymptomatic, though a minority show mild connective-tissue or skin changes from skewed X-inactivation. This inheritance difference is the first counselling fork: Wilson disease counsels siblings, Menkes disease counsels the mother and her future sons. [5] [6]
Epidemiology & Risk Factors
Wilson disease has a prevalence of roughly one in 30,000 live births, with a carrier frequency of about one in 90. It occurs worldwide but is slightly more common in populations with consanguinity and in certain isolates, and it is probably underdiagnosed: a presentation in middle age or beyond, or a purely psychiatric presentation, is easily missed. The mean age at hepatic presentation is around 10 to 13 years and at neurological presentation around 19 years, but the range extends from early childhood into the forties, which is why Wilson disease belongs on the differential of any unexplained liver disease or movement disorder in a young person. [1] [3]
Menkes disease is rarer, with an estimated incidence of about one in 100,000 live births, and because it is X-linked it almost exclusively affects males. The ATP7A gene is large and de novo variants occur, so a negative family history does not exclude the diagnosis - roughly one-third of cases arise from new variants. The risk factor that matters most for Menkes disease is timing: the treatment window closes in the first weeks of life, so any family history of a male infant who regressed and died in infancy on the maternal side should trigger urgent consideration. [5] [4]
For both conditions, consanguinity raises the probability of autosomal recessive Wilson disease in migrant, refugee and certain Indigenous communities, and a family history of unexplained liver disease, movement disorder, psychiatric illness, or infant male deaths is the single highest-yield epidemiological clue. Siblings of a proband with Wilson disease must be screened biochemically and molecularly, and the mother of a boy with Menkes disease must be offered carrier testing because her recurrence risk in future male pregnancies is substantial if she is a carrier. [1] [6]
Pathophysiology
Copper is an essential trace metal that serves as a cofactor for a family of enzymes - among them cytochrome c oxidase (energy), lysyl oxidase (cross-linking collagen and elastin), dopamine-beta-hydroxylase (catecholamine synthesis), tyrosinase (melanin) and superoxide dismutase (antioxidant defence). The body obtains copper from the diet, absorbs it in the duodenum, distributes it in the blood largely bound to ceruloplasmin, and excretes the surplus into bile. Two copper-transporting ATPases, ATP7A and ATP7B, move copper across the relevant membranes. When either pump fails, the enzymes that depend on copper malfunction, and the clinical disease follows. [6] [8]
In Wilson disease, the hepatocyte cannot excrete copper into bile because ATP7B is absent. Copper accumulates in the liver progressively from birth, initially producing no symptoms, then a transaminitis or chronic hepatitis, then cirrhosis. Once hepatic storage capacity is exceeded, non-ceruloplasmin-bound (free) copper spills into the circulation and deposits in the brain - preferentially in the basal ganglia, where it produces the movement disorder - in the cornea at the limbus (the Kayser-Fleischer ring), and in the kidney, heart and other organs. Free copper is redox-active: it generates reactive oxygen species, damages mitochondria, and causes the hepatocyte necrosis that underlies the acute Wilsonian crisis with its characteristic Coombs-negative haemolytic anaemia. [3] [11]

In Menkes disease, the gut enterocyte absorbs dietary copper normally, but because ATP7A is absent the copper cannot exit the enterocyte into the portal blood. It is trapped and then lost when the enterocyte sloughs, so the body - and critically the brain - receives almost no copper. The consequence is the failure of every copper-dependent enzyme: lysyl oxidase failure weakens elastin and collagen, producing arterial tortuosity, bladder diverticula and loose skin; tyrosinase failure depigments the hair and skin; dopamine-beta-hydroxylase failure disrupts catecholamine synthesis, producing the characteristic autonomic instability and hypothermia; and cytochrome c oxidase failure cripples oxidative phosphorylation in the brain, driving the seizures and neurodegeneration that dominate the clinical course. [5] [6]
Clinical Presentation
Wilson disease presents across a spectrum whose three faces - hepatic, neurological and psychiatric - can appear alone or together. The hepatic presentation is the commonest in children and often the first: anything from an asymptomatic transaminitis picked up on routine bloods, through chronic hepatitis mimicking autoimmune liver disease, to decompensated cirrhosis or the dramatic acute liver failure of the Wilsonian crisis with its Coombs-negative haemolytic anaemia and acute kidney injury. A child with chronic hepatitis of unclear cause, or with autoimmune serology that does not quite fit, should have Wilson disease excluded. [1] [3]
The neurological presentation of Wilson disease typically emerges later, in adolescence or young adulthood, and reflects copper deposition in the basal ganglia. The movement disorder is the signature: a tremor (often a wing-beating tremor), dystonia, parkinsonism with bradykinesia and rigidity, dysarthria, dysphagia and gait disturbance. The psychiatric presentation is equally important and often precedes the movement disorder by months or years: depression, personality change, irritability, cognitive decline, and frank psychosis. A deteriorating adolescent with new behavioural change or deteriorating school performance, with or without a movement disorder and liver signs, should prompt a slit-lamp examination and a copper work-up. [11] [7]
Menkes disease presents in a very different tempo. The affected infant is typically normal at birth and for the first six to eight weeks, because residual maternal and placental copper has sustained enzyme function. Then, from two to three months, the infant loses milestones, becomes profoundly hypotonic and lethargic, feeds poorly, develops intractable seizures (often myoclonic or infantile spasms), and fails to thrive. The hair is the external signature: sparse, brittle, twisted (pili torti) and often lightly pigmented. The skin is pallid, the facies characteristic with a sagging jowled appearance, and there may be hypothermia from autonomic failure. Without treatment, the course is one of relentless neurodegeneration, and most untreated children die by two to three years of age. [5] [4]
Differential Diagnosis
For Wilson disease, the hepatic differential is the differential of chronic liver disease in a child or young adult: autoimmune hepatitis (which can coexist with and mimic Wilson disease - check autoantibodies and immunoglobulins), chronic viral hepatitis, non-alcoholic fatty liver disease, alpha-1 antitrypsin deficiency, and drug-induced liver injury. The discriminator is the integrated copper work-up: a low ceruloplasmin with a high urinary copper and high hepatic copper, supported by Kayser-Fleischer rings and molecular confirmation. When the presentation is acute liver failure, the key discriminator from other causes is the Coombs-negative haemolytic anaemia alongside the liver failure and renal injury - the non-ceruloplasmin copper that floods the circulation lyses red cells directly. [3] [1]
The neurological and psychiatric differential of Wilson disease is equally broad: a tremor or dystonia in a young person raises juvenile Parkinson disease, essential tremor, Huntington disease, drug effects, and the expanding genetic dystonias. The psychiatric presentation mimics primary depression, psychosis or a personality disorder. The lesson is that Wilson disease is one of the treatable causes of both young-onset movement disorder and adolescent psychiatric disturbance, which is why it must not be missed: a slit-lamp examination and a copper panel are cheap, and the disease is entirely treatable if caught. [11]
For Menkes disease, the infantile differential is the differential of the floppy, failing, seizing infant with developmental regression: this includes other neurodegenerative disorders (neuronal ceroid lipofuscinosis, Tay-Sachs, the mitochondrial encephalopathies), the congenital myopathies, spinal muscular atrophy, severe cerebral palsy of unknown cause, and non-accidental injury when the subdural collections of the tortuous Menkes vasculature are misread. The hair changes are the discriminator: pili torti is unusual outside Menkes and occipital horn syndrome, and low serum copper and ceruloplasmin with the characteristic arterial tortuosity on imaging completes the picture. [5] [6]
Clinical & Bedside Assessment
The bedside assessment differs for each disease because the tempo and the question differ. For a child with suspected Wilson disease, the examination looks for three things. First, the hepatic stigmata: hepatomegaly, splenomegaly, jaundice, signs of chronic liver disease and portal hypertension, and the ascites and encephalopathy of decompensation. Second, the neurological signs: a tremor (ask the child to hold the arms forward and to flap), rigidity, bradykinesia, dysarthria, dystonia and gait disturbance. Third, the Kayser-Fleischer ring - a golden-brown copper deposit at the periphery of the cornea in Descemet membrane, best seen at the slit lamp, which is present in nearly all neurological Wilson disease and in the majority of hepatic presentations, and which resolves with treatment. [3] [2]
The history asks explicitly about the onset and tempo of liver, neurological and psychiatric symptoms, about a family history of liver disease or movement disorder, about consanguinity, and about any prior unexplained psychiatric diagnosis. Take a careful medication history - immunosuppressants for presumed autoimmune hepatitis that did not work are a common clue. The examination then converts directly into the investigation plan: ceruloplasmin, 24-hour urinary copper, slit-lamp examination, liver function and synthetic function, and molecular testing of ATP7B. [1]
For suspected Menkes disease, the infant is examined for the characteristic facies (sagging cheeks, full lips), the hair (sparse, brittle, lightly pigmented, twisted on microscopy), the skin (pallid, loose, cutis laxa-like), the neurological state (hypotonia, lethargy, seizures, absent developmental milestones) and the autonomic features (hypothermia, labile blood pressure). The history asks about the normal early weeks followed by regression, and about a maternal family history of male infant deaths or developmental disorders. Imaging often shows the tortuous cerebral arteries, and the examination converts into the plan to measure serum copper, ceruloplasmin, and plasma catecholamines (the characteristic neurochemical pattern of Menkes disease), followed by molecular testing of ATP7A. [5] [4]
Investigations
The investigation of Wilson disease rests on an integrated panel interpreted together, because no single test is sufficient. Serum ceruloplasmin is low (under 0.2 grams per litre) in the majority, but it is an acute-phase reactant and can be normal in up to 15 percent of Wilson disease and in pregnancy or active inflammation, so a normal value does not exclude it. The 24-hour urinary copper is the most useful single test: it is elevated above 0.6 to 1 micromole per 24 hours (approximately 40 micrograms) in symptomatic Wilson disease, and markedly elevated above 4 micromoles per 24 hours in acute Wilsonian failure. A slit-lamp examination for Kayser-Fleischer rings and sunflower cataracts is both diagnostic and non-invasive. [1] [3]
When these are equivocal, the hepatic copper concentration on a liver biopsy is the gold standard, with a value above 250 micrograms per gram dry weight being diagnostic, though overlapping values occur in cholestatic liver disease. The biopsy also characterises the stage of liver disease. The final step is molecular testing of ATP7B: identifying biallelic pathogenic variants confirms the diagnosis, enables sibling screening, and supports family counselling, and it is increasingly the first-tier confirmatory test alongside the biochemical panel. [2] [10]
The Leipzig score, developed by Ferenci and colleagues, integrates these data into a validated diagnostic instrument: it assigns points for ceruloplasmin, hepatic copper, urinary copper, neurological features, Kayser-Fleischer rings and ATP7B variants, and a high score confirms the diagnosis without need for liver biopsy in typical cases. This score is the structure the fellowship candidate should name and reproduce. The Nayagam genotype-outcome data add a modern refinement: patients with loss-of-function ATP7B variants on both alleles show worse hepatic survival, which may inform the intensity of monitoring and the timing of transplant assessment. [2] [10]
Why a normal ceruloplasmin does not exclude Wilson disease
Ceruloplasmin is synthesised in the liver, and in Wilson disease it is low because copper cannot be loaded into it. But ceruloplasmin is also an acute-phase reactant, so it rises with inflammation and in pregnancy - and some ATP7B variants preserve enough function to maintain a near-normal ceruloplasmin. Roughly 10 to 15 percent of patients with Wilson disease have a ceruloplasmin in the normal range, particularly those presenting with liver disease. This is why the Leipzig score integrates ceruloplasmin with the 24-hour urinary copper, the slit-lamp examination and the ATP7B genotype rather than relying on any single value. [1] [2]
For Menkes disease, the biochemistry is the mirror image. Serum copper and ceruloplasmin are low, but because these are normally low in the neonate and rise over the first months, a single low value in a young infant is not diagnostic, and the trend and the neurochemical pattern matter. The characteristic plasma catecholamine profile - a high ratio of dihydroxyphenylacetic acid to dihydroxyphenylglycol, reflecting dopamine-beta-hydroxylase failure - is a highly sensitive and specific biochemical marker, because dopamine-beta-hydroxylase is a copper-dependent enzyme. Skull and skeletal imaging may show wormian bones and metaphyseal spurring, and cerebral angiography or magnetic resonance angiography demonstrates the pathognomonic tortuosity of the cerebral and visceral arteries. The diagnosis is confirmed by molecular testing of ATP7A, and placental copper analysis or neonatal molecular testing enables early or prenatal diagnosis in at-risk families. [5] [6]
Management — Resuscitation
The resuscitation question differs sharply between the two diseases. For Wilson disease, the true emergency is the acute Wilsonian crisis: a child or young adult in acute liver failure with a Coombs-negative haemolytic anaemia, jaundice, coagulopathy and acute kidney injury. This is a transplant emergency, because the massive release of free copper causes hepatic necrosis and intravascular haemolysis that medical therapy alone often cannot reverse. The immediate moves are to stabilise the patient, to begin copper removal with chelation and - as a bridge - plasma exchange, haemofiltration or albumin dialysis to lower the circulating free copper, and to refer urgently to a liver transplant centre for listing, because without transplantation the mortality of acute Wilsonian failure approaches 100 percent. [9] [1]
For a child with chronic decompensated Wilson disease - worsening liver function, ascites, encephalopathy - the resuscitation is the standard management of decompensated cirrhosis (fluid, electrolytes, nutrition, treatment of infection and encephalopathy) alongside the initiation of chelation therapy under specialist guidance, with a low threshold for transplant assessment. Chelation must be introduced carefully in advanced liver disease because penicillamine can transiently worsen the neurological and hepatic picture by mobilising copper from tissue stores into the circulation, which is why trientine or zinc is often preferred for initial treatment in the decompensated or neurologically presenting patient. [7] [11]
For Menkes disease, there is no acute resuscitation equivalent in the usual sense, because the disease declares itself as a progressive encephalopathy rather than an acute collapse. The urgent intervention is symptomatic: control seizures (which are often refractory and may require multiple anticonvulsants), support nutrition (many infants need a gastrostomy), and manage the intercurrent infections and aspiration that complicate severe neurodisability. The most urgent treatment decision, however, is not resuscitation but the initiation of subcutaneous copper histidinate at the earliest possible moment, because the window for benefit is narrow and closes within the first months of life. [4] [12]
Management — Definitive & Stepwise
The definitive management of Wilson disease is a lifelong commitment built around three pillars: copper removal, copper absorption blockade, and - for acute or refractory disease - liver transplantation. Chelation therapy is the foundation. Penicillamine has been the traditional first-line agent: it binds copper and promotes its urinary excretion, and it has decades of evidence behind it, but it has a significant side-effect profile (initial neurological worsening, skin changes, nephropathy, bone-marrow suppression) and requires pyridoxine supplementation. Trientine is an alternative chelator with fewer side effects, increasingly preferred for initial therapy, and it is the agent of choice when penicillamine is not tolerated. [7] [8]

Zinc acts by a different mechanism: it induces metallothionein in the enterocyte, which binds dietary copper and prevents its absorption, so that the copper is lost when the enterocyte sloughs. Zinc is well tolerated and is used for maintenance therapy, for pre-symptomatic siblings identified by screening, and in pregnancy. The modern approach, refined by the 2022 AASLD guidance and the treatment-perspective review of Litwin and colleagues, individualises the choice of agent to the clinical context: trientine or zinc for the patient presenting with liver disease in whom penicillamine might worsen things, a chelator plus zinc combination in selected patients, and lifelong adherence with serial monitoring of 24-hour urinary copper to confirm adequacy. [1] [7]
Liver transplantation is curative for Wilson disease because the donor liver carries a normal ATP7B gene, restoring biliary copper excretion. The indications are acute Wilsonian failure, decompensated cirrhosis unresponsive to medical therapy, and - more selectively - refractory neurological disease. The evidence assembled by Schilsky and by Ferrarese and colleagues shows excellent long-term survival after transplant and meaningful neurological recovery in selected patients, reinforcing that transplantation is both a rescue for acute failure and a definitive therapy for the severe hepatic genotype, though it does not always reverse established neurological injury, which is why medical therapy remains the backbone. [9] [1]
For Menkes disease, the definitive therapy is subcutaneous copper histidinate. By complexing copper with histidine and injecting it subcutaneously, the treatment bypasses the gut block and delivers bioavailable copper to the circulation, allowing partial metalation of the copper-dependent enzymes. The landmark study by Kaler and colleagues demonstrated that neonatal diagnosis and treatment with copper histidinate improved survival and neurological outcome in the classic severe disease, though the benefit is greatest when treatment begins in the first month of life and is limited in patients with null ATP7A variants who cannot utilise the delivered copper. The experimental gene therapy approach of Haddad and colleagues - cerebrospinal-fluid-directed AAV9-delivered ATP7A combined with copper histidinate - has shown survival benefit in a mouse model and represents the leading translational frontier. [4] [12]
C.O.P.P.E.R. \u2014 the management contrast
Specific Subtypes & Scenarios
The asymptomatic sibling is the scenario that tests screening discipline. When a proband is diagnosed with Wilson disease, all first-degree relatives must undergo biochemical screening (ceruloplasmin, liver function, 24-hour urinary copper) and molecular testing for the family's ATP7B variants. A sibling who is homozygous or compound heterozygous for pathogenic variants but is pre-symptomatic is started on zinc or chelation to prevent accumulation, and this pre-symptomatic treatment is one of the most effective interventions in all of inherited metabolic medicine, because it prevents the disease from ever declaring itself. [1] [3]
The acute Wilsonian crisis deserves its own scenario because it is the highest-stakes presentation. A child or young adult in acute liver failure with a Coombs-negative haemolytic anaemia, a low or normal ceruloplasmin, a markedly elevated urinary copper and acute kidney injury has Wilson disease until proven otherwise. The team must begin copper removal immediately (chelation plus plasma exchange or haemofiltration as a bridge), list the patient urgently for liver transplantation, and arrange the logistics of transfer to a transplant centre. The mortality without transplantation is near-total, and the window for a successful outcome is short. [9] [11]
The pregnant patient with Wilson disease is a scenario that tests adherence. Chelation must not be stopped in pregnancy, because withdrawal risks acute decompensation and hepatic failure, but penicillamine is teratogenic at high doses and zinc is the preferred agent or the chelator dose is reduced. The plan is shared with the obstetric and metabolic teams, and the neonate is assessed for copper deficiency from maternal chelation. For Menkes disease, the key scenario is the at-risk pregnancy: the mother of an affected boy who is confirmed as a carrier is offered prenatal or preimplantation genetic diagnosis for future male pregnancies, because the recurrence risk for a carrier mother is 50 percent of male offspring being affected. [1] [5]
Occipital horn syndrome is the milder ATP7A allelic disorder and is worth naming because it sits at the boundary. It is dominated by the connective-tissue consequences of lysyl oxidase failure - cutis laxa, bladder diverticula, inguinal herniae, skeletal features including the occipital exostoses that give it its name - with relatively spared cognition and a near-normal or only mildly reduced lifespan. It is treated with copper supplementation along the same principle as Menkes disease, and the candidate who can distinguish it from classic Menkes at viva demonstrates the depth the examiner rewards. [6]
Complications & Pitfalls
The complications of Wilson disease divide into the hepatic, neurological and treatment-related. The hepatic complications are those of chronic liver disease: portal hypertension, variceal bleeding, ascites, hepatocellular carcinoma (rare but documented) and the acute liver failure of the Wilsonian crisis. The neurological complications are the movement disorder, dysphagia with aspiration risk, and the dementia that accompanies advanced disease. The treatment-related complications are substantial: penicillamine can cause an initial worsening of neurological symptoms by mobilising tissue copper, as well as skin changes, nephropathy and bone-marrow suppression, which is why patients are monitored closely and many centres now prefer trientine or zinc for initial therapy. [7] [11]
The chief pitfall in Menkes disease is diagnostic delay, because the infant is normal for the first weeks and the initial regression is non-specific, so the diagnosis is often made after the treatment window has closed. The second is misreading the investigations: a single low serum copper and ceruloplasmin in a young infant is normal, and it is the trend, the neurochemical pattern and the molecular result that make the call. The third is using the wrong treatment route: oral copper does not bypass the enterocyte block, so the copper must be given subcutaneously as copper histidinate, and families must be taught the technique and supported with a reliable supply. [5] [4]
Prognosis & Disposition
For Wilson disease, the prognosis is excellent if the diagnosis is made early and the treatment is taken faithfully: pre-symptomatic siblings identified by screening and treated from childhood live essentially normal lives. For symptomatic disease, the hepatic and neurological outcomes depend on the extent of damage at diagnosis and on adherence, and the major threat to long-term survival is non-adherence to lifelong therapy, which precipitates decompensation and acute failure. After liver transplantation for acute failure or decompensated cirrhosis, long-term survival is excellent, and the neurological outcome is favourable when transplantation occurs before irreversible injury. The lesson is that Wilson disease is one of the few chronic liver diseases that is both preventable (by screening) and curable (by transplant), which is why the diagnosis matters so much. [9] [3]
For Menkes disease, the prognosis remains poor for the classic severe form: untreated, most children die by two to three years of age from neurodegeneration, infection or vascular complications. Early copper histidinate treatment improves survival and, in a subset of patients - particularly those with some residual ATP7A function - it preserves cognition and improves the neurological trajectory, as the Kaler neonatal treatment cohort demonstrated. The emerging gene therapy approaches offer hope for the future but are not yet standard care. The disposition is a shared, multidisciplinary one: the metabolic and neurology services own the disease-specific therapy and the seizure management, the general paediatrician owns nutrition, infection prevention and family support, and palliative care is engaged early because the disease trajectory, even with treatment, often involves significant neurodisability. [4] [12]
In both diseases, transition from paediatric to adult metabolic or hepatology services is a high-risk point: adherence slips, the emergency plan may not transfer, and the young adult may disengage. A written, shared care plan, an engaged adult service, and proactive transition counselling are the interventions that preserve the gains of paediatric care. [1] [11]
Special Populations
The same copper disorder behaves differently across populations because access, recognition and service models are unevenly distributed. In remote and Indigenous communities, later presentation, distance from a metabolic or transplant service, and the need for aeromedical retrieval during a Wilsonian crisis mean that the window between onset and definitive treatment is longer and outcomes are worse - so a written, location-specific emergency plan and a low threshold to check ceruloplasmin and urinary copper in any young person with unexplained liver disease are disproportionately important. In migrant, refugee and asylum-seeking families, consanguinity raises the pre-test probability of autosomal recessive Wilson disease, and language barriers complicate the teaching of a lifelong chelation regimen and a sick-day plan. [1]
In adolescents transitioning to adult care, adherence to chelation is the single greatest threat to survival in Wilson disease, because the disease is silent on treatment and the patient feels well, so the motivation to take an unpleasant medication daily is low. Structured transition, mental-health support, and a medication regimen that is as simple as possible (often zinc monotherapy for the stable patient) are the interventions that matter. For Menkes disease, the special population is the carrier mother and her future pregnancies: genetic counselling, carrier testing of at-risk female relatives, and access to prenatal or preimplantation genetic diagnosis are obligations of the index diagnosis, because the recurrence risk for a carrier mother's male offspring is 50 percent. [5] [6]
In families managing complex chronic metabolic disease, the chief threat is fragmentation of care: the child who sees a hepatologist, a neurologist, a metabolic physician and a general practitioner, with no single owner of the integrated plan. A written, shared, reconciled care plan and a named case coordinator are the interventions that prevent the gaps through which these patients fall. [1] [11]
Evidence, Guidelines & Regional Differences
The evidence base for Wilson disease rests on three pillars. The first is the consensus clinical guidelines, of which the 2022 AASLD Practice Guidance (Schilsky and colleagues, executive summary PMID 36152019) is the current international standard, building on decades of clinical experience. It sets the diagnostic algorithm, the chelation and zinc regimens, the transplant indications, and the family-screening obligations that most national programmes adopt. The second is the diagnostic instrument: the Leipzig score of Ferenci and colleagues (PMID 12955875) remains the validated, widely used tool that integrates ceruloplasmin, urinary copper, hepatic copper, the Kayser-Fleischer ring and the ATP7B genotype into a diagnostic probability. [1] [2]
The third pillar is the genotype-outcome and treatment-perspective evidence: the Nayagam cohort (PMID 36096368) demonstrating worse hepatic survival with loss-of-function ATP7B variants, and the Litwin treatment-perspectives review (PMID 31179305) consolidating the modern, individualised approach to chelation, zinc and combination therapy. The liver-transplantation evidence is assembled in the Schilsky and Ferrarese reviews (PMIDs 24820352 and 38899966), which confirm excellent long-term survival and selected neurological recovery. The classic Brewer monograph (PMID 1635439) remains the foundation for the zinc-therapy mechanism, and the Mulligan and Bronstein neurological overview (PMID 32279718) grounds the movement-disorder management. [9] [10]
For Menkes disease, the evidence rests on the Kaler neonatal copper histidinate trial (PMID 18256395), which established that early subcutaneous copper histidinate improves survival and neurological outcome; the Tümer and Møller review (PMID 19888294) and the Horn and Wittung-Stafshede metalation paper (PMID 33917579) ground the molecular biology of ATP7A and the copper-dependent enzyme cascade; and the Haddad gene-therapy study (PMID 30090842) represents the leading translational advance. The evidence is necessarily limited by the rarity of the disease, and much of the treatment guidance is expert consensus rather than randomised trial, which the fellowship candidate should acknowledge honestly. [4] [5]
In Australia and New Zealand, Wilson disease is managed through the state-based paediatric hepatology and metabolic services (most coordinated through the major children's hospitals in each state and Starship in New Zealand), with liver transplantation centralised at the quaternary paediatric transplant units. Newborn screening does not detect Wilson disease or Menkes disease, so a normal newborn screen does not exclude either diagnosis, and Wilson disease is identified by clinical suspicion and the integrated copper panel. For Menkes disease, plasma catecholamine analysis and ATP7A molecular testing are accessed through the specialist metabolic services, and subcutaneous copper histidinate is supplied through the national pharmacy arrangements on a named-patient basis, with the metabolic service coordinating the family training and the supply chain. Genetic counselling, carrier testing, and prenatal or preimplantation genetic diagnosis are available through the state genetic services, and aeromedical retrieval to a tertiary centre is the expected pathway for an acute Wilsonian crisis. [1] [4]
Exam Pearls
A fellowship candidate answering on the inherited copper disorders should land six anchor points and avoid three classic traps. The anchors are the unifying concept (a defect in a copper-transporting ATPase causes either accumulation or deficiency), the three faces of Wilson disease (hepatic, neurological, psychiatric) with the Kayser-Fleischer ring, the integrated copper panel and the Leipzig score, the treatment principle (chelation or zinc for Wilson, copper histidinate for Menkes, transplant for acute Wilsonian failure), the X-linked counselling of Menkes versus the autosomal recessive sibling screening of Wilson, and the honest statement that Menkes disease remains largely palliative while Wilson disease is treatable and curable. [1] [5]
The traps are three. The first is relying on a single ceruloplasmin to exclude Wilson disease - integrate the panel. The second is starting penicillamine carelessly in a neurologically presenting patient without warning of the initial deterioration. The third is confusing the direction of treatment - removing copper in Menkes or replacing it in Wilson is a catastrophic error that the contrast (remove in Wilson, replace in Menkes) should prevent. The candidate who can name the Kayser-Fleischer ring at the corneal limbus, the pili torti of Menkes hair, the Coombs-negative haemolysis of the Wilsonian crisis, and the neonatal treatment window for copper histidinate will hold the topic firmly at viva. [3] [4]
References
- [1]Schilsky ML, Roberts EA, Bronstein JM, Dhawan A, Hamilton JP, Nolan NM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: Executive summary of the 2022 Practice Guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology, 2023.PMID 36152019
- [2]Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int, 2003.PMID 12955875
- [3]Schilsky ML. Wilson Disease: Diagnosis, Treatment, and Follow-up. Clin Liver Dis, 2017.PMID 28987261
- [4]Kaler SG, Holmes CS, Goldstein DS, Tang J, Godwin SC, Donsante A, et al. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med, 2008.PMID 18256395
- [5]Tümer Z, Møller LB. Menkes disease. Eur J Hum Genet, 2010.PMID 19888294
- [6]Horn N, Wittung-Stafshede P. ATP7A-Regulated Enzyme Metalation and Trafficking in the Menkes Disease Puzzle. Biomedicines, 2021.PMID 33917579
- [7]Litwin T, Dzieżyc K, Członkowska A. Wilson disease-treatment perspectives. Ann Transl Med, 2019.PMID 31179305
- [8]Brewer GJ, Yuzbasiyan-Gurkan V. Wilson disease. Medicine (Baltimore), 1992.PMID 1635439
- [9]Schilsky ML. Liver transplantation for Wilson's disease. Ann N Y Acad Sci, 2014.PMID 24820352
- [10]Nayagam JS, Jeyaraj R, Foskett P, Griffiths J, Mielke-Vega C, Heaton N, et al. ATP7B Genotype and Chronic Liver Disease Treatment Outcomes in Wilson Disease: Worse Survival With Loss-of-Function Variants. Clin Gastroenterol Hepatol, 2023.PMID 36096368
- [11]Mulligan C, Bronstein JM. Wilson Disease: An Overview and Approach to Management. Neurol Clin, 2020.PMID 32279718
- [12]Haddad MR, Choi EY, Zerfas PM, Yi L, Hofmann SL, Bhattacharya R, et al. Cerebrospinal Fluid-Directed rAAV9-rsATP7A Plus Subcutaneous Copper Histidinate Advance Survival and Outcomes in a Menkes Disease Mouse Model. Mol Ther Methods Clin Dev, 2018.PMID 30090842