Paeds · genetics-dysmorphology-and-metabolism
Down syndrome
Also known as Trisomy 21 · Down syndrome · Down's syndrome · 21 trisomy · Mosaic Down syndrome
A fellowship approach to Down syndrome: recognise the three genetic mechanisms (free trisomy 21, Robertsonian translocation, mosaicism) and why a karyotype changes genetic counselling, map the comorbidities by organ system (cardiac, gastrointestinal, endocrine, airway and sleep, haematology, neurodevelopment), and apply an age-stratified health-supervision schedule that is a checklist rather than a single visit.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A newborn is noted on the postnatal ward to be floppier than expected, with a flat facial profile, upslanting palpebral fissures, and a single transverse palmar crease. The same baby may also have a heart murmur that is hard to hear, a duodenum that never opened, or a blood count crowded with blasts. The fellowship task is not to name the syndrome — the face and the hypotonia do that — but to anticipate, system by system, what trisomy 21 is about to do to this child, and to lay a surveillance net that catches each complication before it causes irreversible harm. [1] [5]
3 \u00b7 21 \u00b7 T.R.I.S.O.M.Y.
Overview & Definition
Down syndrome is the recognisable clinical pattern that follows when cells carry a third copy of the long arm of chromosome 21. It is the most common autosomal trisomy compatible with survival beyond infancy, and almost every general paediatrician will care for affected children across their working life. The face, the hypotonia, and the developmental trajectory make the diagnosis suspected at the bedside, but the confirmation is genetic and the confirmation matters — the mechanism determines recurrence risk for the parents and for siblings. [1] [2]
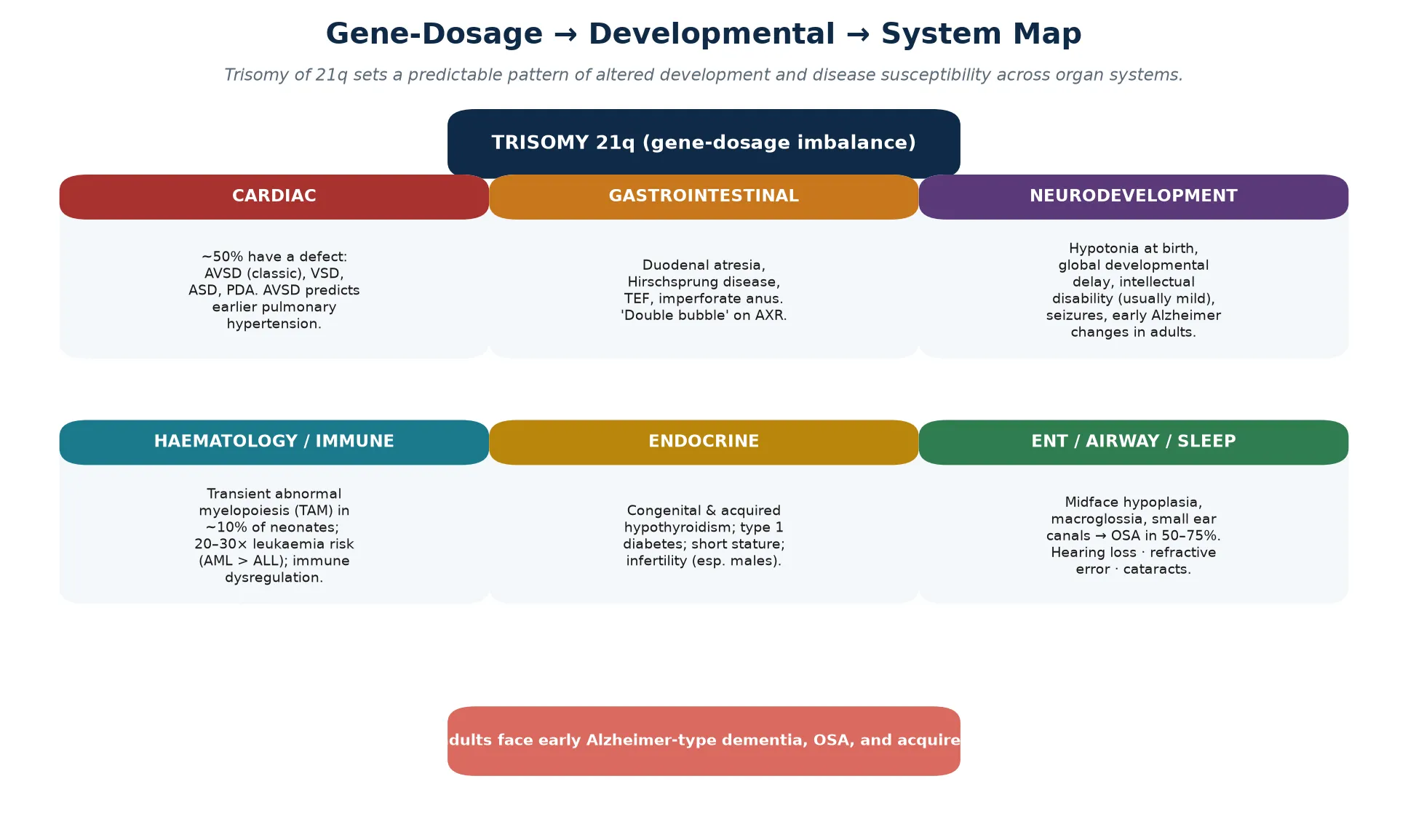
The phenotype is produced by gene-dosage imbalance: the extra copy of around 230 genes on chromosome 21 is overexpressed, and the overexpression of a defined subset drives the recognizable features, the congenital malformations, and the disease susceptibilities. Because the gene dosage is consistent, the comorbidities are predictable, and predictability is what makes a surveillance schedule possible. A fellowship answer that lists features without linking them to the underlying trisomy reads as a catalogue; the gene-dosage link is the teaching that holds the topic together. [1] [5]
The lifespan trajectory has transformed. A child born with Down syndrome in the mid-twentieth century had a life expectancy measured in single figures, dominated by untreated congenital heart disease and infection. Cardiac surgery, antibiotics, inclusion, and structured surveillance have moved life expectancy into the seventh decade, and the population prevalence is now rising because affected children survive into adulthood and old age. The clinical task has shifted accordingly — from keeping the infant alive to guiding a person through seven decades of predictable, preventable, and treatable complications. [4] [6]
Classification
Down syndrome has three genetic mechanisms, and each changes the genetic counselling. Free trisomy 21 accounts for around ninety-five per cent of cases and is caused by meiotic nondisjunction, usually during maternal meiosis I; the recurrence risk is close to one per cent and tracks with maternal age. Robertsonian translocation accounts for three to four per cent: the extra 21q material is fused onto another acrocentric chromosome (most often 14 or 21), and roughly a quarter of these are inherited from a parent who carries a balanced translocation. Mosaicism accounts for one to two per cent, with two cell lines — one normal, one trisomic — produced by post-zygotic nondisjunction, and it often carries a milder and less predictable phenotype. [1] [2]
The reason a karyotype is the diagnostic test rather than a chromosomal microarray alone is that the mechanism matters for the family. A child with free trisomy needs a different conversation from a child whose translocation came from a balanced-carrier parent, because a carrier parent has a recurrence risk that can reach ten to fifteen per cent, and siblings may also be carriers. Microarray confirms the diagnosis and quantifies the extra 21q material, but it cannot distinguish a free trisomy from a Robertsonian translocation or identify a carrier parent — only a karyotype can. Asking for both, in that order, is the safe habit. [1]
The Down syndrome critical region is the segment of 21q — around 21q22 — whose triplication is sufficient to produce the core phenotype. All three mechanisms share trisomy of this region, which is why the clinical overlap between free trisomy, translocation, and mosaicism is large. Severity is set by gene dosage and by individual genetic modifiers rather than by the mechanism alone, so a mosaic child may look identical to a child with free trisomy, or may be so subtly affected that the diagnosis is made late. [1] [5]

Epidemiology & Risk Factors
Down syndrome occurs in roughly one in every seven hundred to one in a thousand live births, and it is the commonest chromosomal cause of intellectual disability worldwide. The single strongest risk factor is advancing maternal age, because nondisjunction during meiosis I becomes more likely as oocytes age. The age-related risk rises gradually through the thirties and more steeply after thirty-five, although most affected infants are born to younger women simply because younger women have more babies. [1] [9]
Population prevalence is rising even as prenatal screening has expanded, and the reason is survival. Improved cardiac surgery, antibiotics, structured health surveillance, and inclusion have driven a dramatic improvement in life expectancy, and large population studies across ten regions of the United States documented steadily improving survival across birth cohorts. Because affected children now survive into adolescence and adulthood, the point prevalence of Down syndrome in the population has grown, and clinical practice has shifted from neonatal and infant care towards adolescent transition and adult medicine. [6] [9]
The risk factors for individual complications, separate from the trisomy itself, are the risk factors the surveillance schedule is built to modify. A child with an unrepaired atrioventricular septal defect is at risk of irreversible pulmonary hypertension; a child with untreated obstructive sleep apnea is at risk of behavioural, cardiac, and developmental harm; a child with undiagnosed hypothyroidism is at risk of further developmental compromise. Identifying these risks early is the epidemiology that matters at the bedside, and it is why a checklist beats memory. [3] [8]
Pathophysiology
The pathophysiology of Down syndrome is a gene-dosage problem. The extra copy of chromosome 21 means that its roughly 230 genes are overexpressed, and the overexpression of a defined subset — clustered in and around the Down syndrome critical region on 21q22 — drives the malformations, the developmental differences, and the disease susceptibilities. Several named genes carry specific effects that a fellowship candidate should be able to link to a phenotype. [1] [5]
DYRK1A, a kinase encoded on chromosome 21, contributes to the intellectual disability and to the early onset of Alzheimer-type changes when overexpressed, because it alters neurogenesis, dendritic development, and tau phosphorylation. APP, the amyloid precursor protein gene also on chromosome 21, is the molecular basis of the near-universal development of Alzheimer-type neuropathology by middle age. Overexpression of ETS2 and ERG contributes to the haematological phenotype and the leukaemia predisposition; COL6A1 and COL6A2 to the joint laxity and the characteristic facial and soft-tissue features; and SOD1 to oxidative-stress susceptibility. None of these acts alone — the phenotype is a summation — but each anchors a teaching point. [1] [5]
The haematological pathophysiology is distinctive and high-yield. Transient abnormal myelopoiesis affects around one in ten neonates with Down syndrome and is driven by acquired GATA1 mutations in the trisomic fetal liver, producing a self-limited proliferation of immature myeloid cells that resolves spontaneously in most infants. A subset, however, progress to acute megakaryoblastic leukaemia within the first few years of life. Beyond the neonatal period, children with Down syndrome carry a twenty- to thirty-fold increased risk of acute leukaemia, with acute myeloid leukaemia — particularly acute megakaryoblastic leukaemia — proportionally more common than in children without trisomy 21. [5]
The immune dysregulation in Down syndrome is real and contributes to the higher burden of respiratory infection, the autoimmune endocrinopathy (hypothyroidism, coeliac disease, type 1 diabetes), and the altered leukaemia risk. The mechanism is multifactorial, involving thymic and T-cell differences and chronic low-grade inflammation, and it is the biological link between the trisomy and the clinical observation that affected children have more frequent and more severe infections than their peers. [2] [5]
Clinical Presentation
The clinical presentation depends on the age at which the child is seen, and a fellowship answer earns depth by handling each life stage separately rather than collapsing the syndrome into a single newborn snapshot. The diagnosis is most often suspected in the newborn period, but mosaicism may declare itself only when a developmental delay brings the child to attention later in infancy or toddlerhood. [1] [2]
In the newborn, hypotonia is often the first sign — a floppy baby on the postnatal ward — accompanied by the characteristic facial features of a flat facial profile, upslanting palpebral fissures, a small nose with a flat bridge, small and low-set ears, and a protruding tongue. The hands show a single transverse palmar crease, short fingers, and clinodactyly of the fifth finger; the feet show a wide sandal gap between the first and second toes. Brushfield spots on the iris complete the classical picture. None of these signs is individually diagnostic, but their pattern is. [1] [3]
System-specific presentations dominate the early weeks. A heart murmur, or sometimes no murmur at all, may signal an atrioventricular septal defect or a ventricular septal defect. Polyhydramnios in pregnancy followed by bilious vomiting in the first day or two of life, with a 'double bubble' on abdominal radiograph, signals duodenal atresia. Delayed passage of meconium with abdominal distension raises Hirschsprung disease. A combination of congenital heart disease and gastrointestinal atresia in the same infant makes trisomy 21 a leading working diagnosis. [1] [7]
Across infancy and childhood, the presentation shifts to development and growth. Motor and language milestones are delayed, tone remains low, and growth tracks below the median on standard charts — which is why Down syndrome–specific growth charts are used. Behavioural change, snoring, restless sleep, daytime somnolence, or school deterioration may be the presenting feature of obstructive sleep apnea, and new motor regression or gait change raises atlantoaxial instability. Each presentation is a surveillance item wearing a clinical disguise. [3] [8]
Differential Diagnosis
The differential diagnosis of Down syndrome splits into two questions: what else produces neonatal hypotonia with dysmorphism, and what else explains the individual system findings. Most of the time the pattern is unmistakable, but the discriminating skill is to know when to keep the door open. [1] [2]
Neonatal hypotonia with dysmorphism raises Prader-Willi syndrome, which shares hypotonia and feeding difficulty in the newborn but lacks the facial gestalt and the chromosomal pattern; Zellweger syndrome, with its profound hypotonia, characteristic facies, and hepatic involvement; myotonic dystrophy, inherited from an affected or mildly affected mother; and a congenital myopathy or spinal muscular atrophy when the dysmorphism is subtle. The clinical context and the karyotype separate these, and a normal karyotype in a hypotonic dysmorphic infant should reopen the search for an alternative diagnosis rather than dismiss the phenotype. [1]
Some dysmorphic syndromes superficially resemble Down syndrome. Noonan syndrome shares short stature and a characteristic facies but is an autosomal dominant rasopathy with a normal karyotype and a different cardiac profile (hypertrophic cardiomyopathy rather than AVSD). A child with a ventricular septal defect and developmental delay may raise 22q11.2 deletion, Williams syndrome, or another chromosomal disorder, and the discriminating step is to match the phenotype to the expected cardiac lesion and the facial gestalt before committing to a label. [1] [2]
The trap that costs marks is relying on a chromosomal microarray to confirm Down syndrome without a karyotype. Microarray identifies the extra 21q material but cannot distinguish free trisomy from a Robertsonian translocation and cannot identify a carrier parent, so it can miss a family with a substantial recurrence risk. When the clinical diagnosis is Down syndrome, request a karyotype — and if it shows a translocation, karyotype both parents before the counselling conversation. [1] [3]
| Feature | Down syndrome | Prader-Willi | Noonan syndrome |
|---|---|---|---|
| Facial gestalt | Flat profile, upslant, Brushfield | Narrow bifrontal, almond eyes | Triangular, hypertelorism, downslant |
| Cardiac lesion | AVSD, VSD | Usually none | Hypertrophic cardiomyopathy, pulmonary stenosis |
| Karyotype / genetics | Trisomy 21 (karyotype) | 15q11 paternal deletion / UPD | RASopathy, normal karyotype |
| Newborn tone | Hypotonia | Severe hypotonia | Usually normal |
Clinical & Bedside Assessment
The bedside assessment of a child with Down syndrome is structured around the comorbidity map rather than a single complaint, because the point of the visit is to run the surveillance net as well as to address the presenting problem. Plot every child on Down syndrome–specific growth charts, because standard charts will mislabel normal growth as faltering. A complete assessment covers cardiac, thyroid, developmental, ENT and airway, ophthalmic, haematological, and musculoskeletal systems at every relevant visit. [2] [3]
The history explores feeding and growth in infancy, sleep and snoring through childhood, developmental milestones and behaviour, school progress, hearing and vision concerns, thyroid symptoms, bowel pattern, and any new neurological symptoms such as gait change or neck pain. Ask directly about sleep-disordered breathing, because parents under-report it and it is both common and treatable. A careful family history of chromosomal disorder and the parental carrier status, where a translocation is involved, frames the genetic counselling. [3] [8]
Examination begins with growth and tone and moves through the systems. Listen carefully to the heart, recognising that an AVSD may be silent. Examine the hips for dysplasia, the abdomen for organomegaly, the thyroid for a goitre, the eyes for strabismus and cataracts, and the ears for otitis media with effusion. A brief neurological assessment screens for new motor signs that would prompt cervical spine imaging. Skin inspection may reveal dry skin consistent with hypothyroidism or a coppery hue consistent with undiagnosed coeliac or hepatic disease. [1] [2]
Assess development with a standardised tool appropriate to the child's age, and interpret the result against Down syndrome–specific expectations rather than typical milestones. A fellowship answer makes clear that developmental delay in Down syndrome is expected and is not, by itself, a sign of a new problem — but regression, a plateau, or an asymmetric loss of skills is a red flag that demands investigation for a complication such as hypothyroidism, sleep apnea, atlantoaxial instability, or, in the adult, dementia. [3] [10]
Investigations
The investigation plan for Down syndrome is a surveillance schedule, not a one-off work-up. The first test is genetic confirmation — a karyotype to establish the mechanism — followed by an echocardiogram, thyroid testing, hearing and vision screening, a blood count, and targeted screening as the child grows. Every item on the schedule has a rationale tied to a preventable harm. [1] [3]
Genetic confirmation is by karyotype, and a chromosomal microarray may be added to exclude a concurrent microdeletion but never to replace the karyotype. The karyotype distinguishes free trisomy, translocation, and mosaicism, and when a translocation is found both parents should be karyotyped to identify a balanced carrier and to quantify the recurrence risk. Non-invasive prenatal testing and combined first-trimester screening belong to antenatal care and are not diagnostic in the postnatal child. [1]
Cardiac investigation is an echocardiogram for every infant, performed by six weeks of age and regardless of symptoms, because an AVSD can be silent. Thyroid function is tested in the newborn period through the blood spot screen, repeated at six and twelve months, and then annually for life, because both congenital and acquired hypothyroidism are common. Hearing is screened in the newborn and reassessed by auditory brainstem response or behavioural testing by six months, with annual review through childhood, because conductive and sensorineural loss are both over-represented. [3] [7]
Vision is assessed by six months and annually thereafter, looking for refractive error, strabismus, cataracts, and nystagmus. A full blood count in the newborn period screens for transient abnormal myelopoiesis, and a blood count with a blood film is repeated if there are blast cells, cytopenias, or hepatosplenomegaly, with haematology review if abnormalities persist. Coeliac screening begins around two to three years and is repeated if symptoms emerge. A sleep study is requested whenever there are symptoms of sleep-disordered breathing, and some centres offer routine screening, because the prevalence of obstructive sleep apnea exceeds fifty per cent. [5] [8]
Management — Resuscitation
Resuscitation in a newborn or infant with Down syndrome means managing the acute surgical and cardiac presentations that bring the child to attention in the first days of life. Two presentations dominate: duodenal atresia with bilious vomiting, and congenital heart disease with cardiovascular compromise. Both are emergencies that demand prompt recognition and a defined pathway. [1] [2]
A newborn with bilious vomiting and a 'double bubble' on abdominal radiograph has duodenal atresia until proven otherwise. Pass a nasogastric or orogastric tube to decompress the stomach, establish intravenous access, correct fluid and electrolyte losses, and involve the surgical team early. A concomitant cardiac assessment is essential, because a child with both duodenal atresia and a cardiac lesion is at substantially higher anaesthetic and surgical risk. Hirschsprung disease, with delayed meconium passage and abdominal distension, follows its own resuscitation pathway of rectal washout or decompression before definitive surgery. [1]
An infant with a large left-to-right shunt from an unrepaired AVSD or ventricular septal defect may present in the first weeks of life with respiratory distress, poor feeding, and failure to thrive as pulmonary vascular resistance falls. Manage with antifailure therapy — diuretics and caloric supplementation — and refer promptly for surgical repair, because delaying repair beyond the first months of life risks the development of irreversible pulmonary vascular disease, the Eisenmenger physiology that once defined the natural history. Cardiology and cardiothoracic involvement at the point of diagnosis, not at the point of decompensation, is the rule. [7]
Transient abnormal myelopoiesis in the neonate usually resolves without treatment, but it demands haematology review and monitoring, because a subset progress to acute megakaryoblastic leukaemia. High-risk features — a very high blast count, liver dysfunction, or adverse cytogenetics — may prompt low-dose cytarabine, but most infants are observed with serial blood counts. The principle is that a leukaemoid reaction in a Down syndrome neonate is never ignored, but neither is it treated as frank leukaemia without haematology guidance. [5]
Management — Definitive & Stepwise
Definitive management of Down syndrome is an age-stratified health-supervision schedule, and it is the core of fellowship-level competence. The schedule is a checklist that moves with the child from the newborn period through infancy, childhood, adolescence, and adulthood, and each band carries non-negotiable items that prevent specific, predictable harm. Holding the whole schedule in view — rather than reacting to the complaint of the day — is what distinguishes structured care from ad hoc care. [3] [2]
In the newborn period, the priorities are genetic confirmation by karyotype, an echocardiogram by six weeks, the newborn hearing screen, a thyroid function test, and screening for gastrointestinal atresia or Hirschsprung disease where the history or examination suggests it. Feeding support, the establishment of Down syndrome–specific growth monitoring, and a first ophthalmology review belong here too. The family is given honest, strengths-based information and connected to a Down syndrome support organisation. [1] [3]
Across infancy and childhood, the schedule layers thyroid testing at six and twelve months and then annually, hearing and vision review, developmental assessment and early intervention, routine immunisation, dental review, and an annual symptom review for sleep-disordered breathing with a low threshold for a sleep study. Coeliac screening begins around two to three years, and atlantoaxial counselling — advising the family to report new neurological symptoms — replaces routine cervical imaging. Growth, development, behaviour, and school progress are reviewed at every visit. [3] [8]
Adolescence brings transition planning, reproductive and menstrual health, mental health screening, and increasing attention to self-advocacy and guardianship. The transition to adult care should be planned, documented, and begun well before the eightteenth birthday, with a structured handover that preserves continuity of cardiac, endocrine, sleep, and cognitive surveillance. In adulthood, the schedule adds annual cardiac review, ongoing thyroid testing, sleep assessment, cognitive baseline and monitoring for Alzheimer-type decline, cancer surveillance, and bone health. The schedule does not end at twenty-one; it changes shape. [10] [4]

Specific Subtypes & Scenarios
Each major comorbidity of Down syndrome carries a distinctive management point, and a fellowship answer earns depth by handling them individually rather than as a single block. The surveillance items generalise, but the management is system-specific. [1] [3]
The cardiac scenario centres on the atrioventricular septal defect, which occurs in around a third of children with Down syndrome and a heart defect. Early surgical repair, usually in the first months of life, prevents the irreversible pulmonary hypertension that once defined the natural history. Children with simpler lesions — ventricular or atrial septal defects — follow standard paediatric cardiology pathways, and those without a cardiac lesion are discharged from cardiac follow-up after the initial echocardiogram, with lifelong awareness that acquired valve disease may emerge in adulthood. [7]
The gastrointestinal scenario includes duodenal atresia, Hirschsprung disease, tracheo-oesophageal fistula, and imperforate anus, each managed by the relevant surgical pathway. Feeding difficulty, gastro-oesophageal reflux, and constipation are chronic problems through infancy and childhood, and coeliac disease is over-represented. Growth monitoring on Down syndrome–specific charts prevents the mislabelling of normal growth as faltering and the over-investigation that follows. [1] [2]
The haematological scenario is dominated by transient abnormal myelopoiesis in the neonate and by leukaemia across childhood. Transient abnormal myelopoiesis resolves in most infants but mandates haematology follow-up, because a minority progress to acute megakaryoblastic leukaemia within the first few years. Any persistent cytopenia, hepatosplenomegaly, or blast excess beyond the neonatal period demands urgent haematology assessment, because the leukaemia risk is twenty- to thirty-fold higher than in the general population and the response to treatment is, paradoxically, often better than in children without trisomy 21. [5]
The endocrine scenario centres on hypothyroidism, both congenital and acquired, which together affect a substantial minority and may be autoimmune. Type 1 diabetes and growth and pubertal differences are also over-represented. Annual thyroid testing, lifelong, is the safeguard, because the symptoms of hypothyroidism — fatigue, constipation, school deterioration — overlap with features already attributed to the syndrome and are easily missed. [2] [10]
Why obstructive sleep apnea is the most under-diagnosed complication
More than half of children with Down syndrome have obstructive sleep apnea, driven by midface hypoplasia, macroglossia, adenotonsillar hypertrophy, and hypotonia. Parents often do not recognise snoring and restless sleep as abnormal, and the daytime consequences — behavioural change, school difficulty, and fatigue — are attributed to the syndrome rather than to a treatable cause. A low threshold for a sleep study, in any child with symptoms or before any decline is attributed to the underlying condition, is the single most rewarding habit in the schedule. [8] [2]
Complications & Pitfalls
The complications of Down syndrome are the comorbidities left undetected, and the pitfalls are the assumptions that lead clinicians to miss them. A fellowship answer handles both, because the harm in Down syndrome is rarely the trisomy itself — it is the preventable complication that the surveillance schedule was designed to catch. [1] [3]
Cardiac complications dominate the infant period. An unrepaired AVSD causes a large shunt, heart failure, faltering growth, and ultimately irreversible pulmonary vascular disease. The pitfall is assuming that a well-looking infant with no murmur does not need an echocardiogram; an AVSD can be silent, and the echocardiogram by six weeks is the safeguard that prevents the harm. [7]
Airway and sleep complications dominate childhood. Untreated obstructive sleep apnea drives behavioural disturbance, school difficulty, pulmonary hypertension, and developmental compromise. The pitfall is attributing the daytime consequences to the syndrome rather than to a treatable sleep disorder, and the safeguard is a low threshold for a sleep study in any symptomatic child. [8]
Neurological complications include atlantoaxial instability and, in the adult, early Alzheimer-type dementia. Atlantoaxial instability may declare itself with gait change, neck pain, or motor regression, and the pitfall is performing cervical manipulation — in sport, in anaesthesia, or in a radiology department — without imaging a symptomatic child first. Early dementia presents with cognitive and functional decline in the third to fifth decade and is under-recognised because the baseline cognitive difference masks the change without serial assessment. [3] [4]
Haematological and endocrine complications run through the lifespan. Leukaemia may present with cytopenias, blasts, or organomegaly at any age in childhood, and hypothyroidism may present at any age with non-specific decline. The pitfall common to both is attributing the symptoms to the syndrome and sending no tests, and the safeguard is the annual blood count and thyroid function test that the schedule prescribes. [5] [2]
Prognosis & Disposition
The prognosis of Down syndrome has transformed across a generation. Life expectancy has risen from single figures in the mid-twentieth century to around sixty years today, driven by cardiac surgery, structured surveillance, inclusion, and improved infection management. The population is ageing, and the clinical task has shifted accordingly — from keeping the infant alive to guiding a person through seven decades of predictable, preventable complications. [4] [6]
The determinants of prognosis are the comorbidities and how early they are found. A child whose AVSD is repaired in the first months of life, whose hypothyroidism is diagnosed and treated, whose sleep apnea is identified and managed, and whose development is supported with early intervention will follow a trajectory close to the population norms for the condition. A child whose comorbidities are missed will accumulate preventable harm at each step. The prognosis is therefore not fixed by the trisomy; it is shaped by the quality of the surveillance. [3] [7]
Quality of life is shaped as much by social factors as by medical ones. Inclusion in education and employment, family support, and a strengths-based clinical relationship measurably improve outcomes, and the fellowship answer frames prognosis in those terms rather than in deficit language alone. Adults with Down syndrome live, work, and form relationships, and the disposition plan should support that trajectory. [10] [1]
Disposition for a general paediatrician is shared, structured care. The paediatrician owns the surveillance schedule, the coordination of subspecialty input, the developmental and behavioural support, and the transition to adult care. Cardiology, ENT and audiology, ophthalmology, endocrinology, haematology, and child development each contribute at the relevant point, and a named coordinator prevents the fragmentation that is the enemy of a checklist-based schedule. Early referral to a structured Down syndrome clinic, where one exists, supports the family from the point of diagnosis. [2] [3]
Special Populations
Down syndrome interacts with the child's social, cultural, and developmental context, and the same comorbidity behaves differently across populations. Access, adherence, and late presentation all shape outcome, and a fellowship answer recognises that the surveillance schedule is only as good as the family's ability to engage with it. [3] [10]
Indigenous children, particularly in Australia and New Zealand, may face a higher background burden of respiratory infection, otitis media, and sleep-disordered breathing, alongside reduced access to specialist services in remote communities. These factors intensify the need for early, structured surveillance and for a low threshold to investigate symptoms, because the margin for delay is smaller when the baseline infection burden is higher. Telehealth and outreach services extend the surveillance net into communities that a clinic-based model would miss. [8] [2]
Migrant, refugee, and asylum-seeking families may have had no antenatal screening, may arrive with an unrecognised diagnosis, and may face language and trauma barriers to engaging with a complex schedule. A careful reconstruction of the history, confirmation of the diagnosis and the mechanism, an interpreter-mediated explanation, and a written schedule in the family's language are the foundations. Vaccination status should be confirmed and completed, because the immune dysregulation of Down syndrome adds to the risk of vaccine-preventable disease. [1] [2]
Socioeconomically disadvantaged families carry the burden that adherence to a multi-specialist schedule is harder, and the limiting step is often attendance rather than the medicine. Structuring the schedule around a single coordinated visit, providing written and visual materials, and linking the family to a support organisation and to transport and appointment support all improve engagement. The aim is to fit the surveillance to the family's reality rather than the reverse. [3] [10]
Adolescents and adults in transition are a population in their own right. The handover to adult care is a vulnerable point, because the structured paediatric surveillance ends and adult services may not inherit the schedule. A planned, documented transition that transfers the checklist into adult medicine, preserves the cardiac and endocrine and cognitive surveillance, and addresses reproductive health, mental health, and guardianship, is the safeguard. The schedule does not stop at the transition; it changes hands. [10] [4]
Evidence, Guidelines & Regional Differences
The evidence base for Down syndrome rests on landmark reviews, the American Academy of Pediatrics health-supervision clinical report, and large population studies of survival and comorbidity, supplemented by consensus guidelines for adolescent and adult care. The AAP clinical report is the operational document that most paediatricians build their schedule around, and it is updated periodically to reflect new evidence. [1] [3]
The Bull review in the New England Journal of Medicine remains the framing reference, organising the genetics, the comorbidity map, and the lifespan trajectory that a fellowship answer is built on. The Weijerman and de Winter clinical practice paper provides a European perspective on the care of children with Down syndrome, and the Van Cleve adolescent guidelines extend the schedule into the second and third decades. Together these documents define a structured, evidence-based approach that generalises across settings. [1] [2] [10]
Population evidence underpins the surveillance items. The survival trends across ten United States regions document the improvement in life expectancy that has reshaped the condition; the congenital heart defect trends quantify the cardiac burden and its management; and the prevalence studies track the growing population of children and adolescents living with the condition. The obstructive sleep apnea literature defines a complication that is both common and under-diagnosed, and the leukaemia reviews define the haematological risk and its paradoxically favourable treatment response. [6] [7] [8] [5]
Regional differences are practical rather than scientific. Australia and New Zealand apply the international surveillance frameworks with national newborn screening for hypothyroidism, structured Down syndrome clinics in the major centres, and telehealth and outreach to extend the schedule into rural and remote communities. Indigenous-health considerations intensify the need for early respiratory and audiological surveillance, and the funding and organisation of subspecialty access shape how the checklist is delivered. The principles are constant; the delivery adapts to the setting. [3] [8]
In Australia and New Zealand, every infant with Down syndrome receives an echocardiogram by six weeks, newborn hearing screening, and newborn blood spot screening for hypothyroidism, with structured Down syndrome clinics coordinating surveillance in the major centres. Telehealth and outreach extend the schedule into rural and remote communities, and Indigenous-health considerations prompt earlier and more intensive respiratory and audiological surveillance. Transition to adult care is increasingly structured, recognising the growing population of adults with Down syndrome who require lifelong cardiac, endocrine, sleep, and cognitive surveillance. [3] [4]
Exam Pearls
A fellowship candidate answering on Down syndrome should land five anchor points and avoid four classic traps. The anchors are the framework examiners listen for; the traps are where candidates lose easy marks. [1] [3]
Anchor one: the mechanism changes the counselling. Confirm the diagnosis with a karyotype, because a Robertsonian translocation — found in three to four per cent — identifies a carrier parent and a recurrence risk of up to ten to fifteen per cent, while free trisomy carries a one per cent recurrence. A microarray alone cannot make that distinction. [1]
Anchor two: the heart comes first. Around half of infants have a congenital heart defect, and the atrioventricular septal defect is the classic association. An echocardiogram by six weeks in every infant, regardless of symptoms, prevents the irreversible pulmonary hypertension that an undiagnosed AVSD can cause. [7] [1]
Anchor three: obstructive sleep apnea is common and under-diagnosed. More than half of children are affected, the daytime consequences are attributed to the syndrome, and a low threshold for a sleep study is the single most rewarding habit. Ask about sleep at every visit. [8]
Anchor four: the surveillance schedule is the management. Thyroid testing from the newborn period and annually, hearing and vision through childhood, a blood count in the newborn and when symptomatic, coeliac screening, and atlantoaxial counselling — each item prevents a specific, predictable harm. Hold the whole checklist, not just the complaint of the day. [3] [5]
Anchor five: speak in strengths-based language and plan the lifespan. Down syndrome is a lifespan condition, and the schedule extends from the newborn period through adolescence and transition into adult cognitive and cardiac surveillance. Inclusion, family support, and a structured transition to adult care measurably improve outcomes. [10] [4]
The four traps to avoid are confirming the diagnosis with a microarray but no karyotype, reassuring a well newborn without an echocardiogram, attributing snoring or school decline to the syndrome without a sleep study, and ordering routine cervical spine films in an asymptomatic child while failing to image a symptomatic one. Avoid these and the rest of the answer falls into place. [1] [3]
References
- [1]Bull MJ. Down Syndrome. N Engl J Med, 2020.PMID 32521135
- [2]Weijerman ME, de Winter JP. Clinical practice. The care of children with Down syndrome. Eur J Pediatr, 2010.PMID 20632187
- [3]Bull MJ, Committee on Genetics. Health supervision for children with Down syndrome. Pediatrics, 2011.PMID 21788214
- [4]Bittles AH, Bower C, Hussain R, Glasson EJ. The four ages of Down syndrome. Eur J Public Health, 2007.PMID 16857692
- [5]Hitzler JK, Zipursky A. Origins of leukaemia in children with Down syndrome. Nat Rev Cancer, 2005.PMID 15630411
- [6]Kucik JE, Shin M, Siffel C, Marengo L, Correa A. Trends in survival among children with Down syndrome in 10 regions of the United States. Pediatrics, 2013.PMID 23248222
- [7]Bergström S, Carr H, Petersson G, Stephansson O, Bonamy AK, Ludvigsson JF, Dahlström A, Palmér M, Johansson S. Trends in congenital heart defects in infants with Down syndrome. Pediatrics, 2016.PMID 27252035
- [8]Maris M, Verhulst S, Wojciechowski M, Van de Heyning P. Sleep problems and obstructive sleep apnea in children with down syndrome, an overwiew. Int J Pediatr Otorhinolaryngol, 2016.PMID 26857307
- [9]Shin M, Besser LM, Kucik JE, Lu C, Siffel C, Correa A, Congenital Anomaly Multistate Prevalence and Survival (CAMPS) Collaborative. Prevalence of Down syndrome among children and adolescents in 10 regions of the United States. Pediatrics, 2009.PMID 19948627
- [10]Van Cleve SN, Cannon S, Cohen WI. Part II: Clinical Practice Guidelines for adolescents and young adults with Down Syndrome: 12 to 21 Years. J Pediatr Health Care, 2006.PMID 16675381