Paeds · genetics-dysmorphology-and-metabolism
Fatty-acid oxidation disorders
Also known as FAOD · Fatty acid beta-oxidation disorders · Medium-chain acyl-CoA dehydrogenase deficiency · MCAD deficiency · MCADD · Long-chain fatty acid oxidation disorders · LC-FAOD · VLCAD deficiency · LCHAD deficiency
A fellowship approach to the fatty-acid oxidation disorders: recognise the child who cannot switch to fat-burning during fasting and so presents with hypoketotic hypoglycaemia, cardiomyopathy, rhabdomyolysis or sudden death; treat the acute crisis with intravenous glucose to shut off fatty-acid mobilisation; confirm with plasma acylcarnitines and molecular testing; and prevent recurrence with avoidance of fasting, an emergency sick-day plan, and for long-chain defects triheptanoin and carnitine.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who thinks in three layers simultaneously. The first is the child in front of you: a hypoglycaemic, encephalopathic infant after a viral illness, or an adolescent with exercise-induced muscle pain and dark urine. The immediate question is not which enzyme is defective but whether the child can make ketones. The second is the physiology: fasting triggers lipolysis, fatty acids enter the mitochondrion via the carnitine shuttle, and beta-oxidation trims two carbons at a time to generate acetyl-CoA, ketone bodies and ATP. A block anywhere in this chain produces an energy deficit and, in the long-chain defects, toxic accumulation. The third is the family: these are autosomal recessive conditions, newborn screening detects most of them, and a well-constructed sick-day plan is what keeps the child safe between episodes. [1] [4]
Overview & Definition
The fatty-acid oxidation disorders are a group of inherited metabolic diseases caused by defects in the transport of fatty acids into the mitochondrion or in their subsequent beta-oxidation to generate energy. The pathway begins with the carnitine shuttle — carnitine palmitoyltransferase I (CPT I), the carnitine-acylcarnitine translocase (CACT), and carnitine palmitoyltransferase II (CPT II) — which ferries long-chain fatty acids across the impermeable inner mitochondrial membrane. Once inside, the beta-oxidation spiral progressively shortens the fatty-acyl chain through a repeating cycle of dehydrogenation, hydration, dehydrogenation and thiolysis, with each enzyme class acting on a different chain-length range: very long-chain acyl-CoA dehydrogenase (VLCAD), long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD, part of the mitochondrial trifunctional protein), medium-chain acyl-CoA dehydrogenase (MCAD), and short-chain acyl-CoA dehydrogenase (SCAD). [1] [4]
Clinically, the fatty-acid oxidation disorders sit within the family of fasting-induced energy-failure inborn errors: conditions in which a blocked catabolic pathway prevents the body from using its largest energy reserve — stored fat — during periods of caloric deficit. This framing matters because it dictates both the presentation (crisis during fasting, infection or exercise) and the management (prevent catabolism, provide alternative fuel). Medium-chain acyl-CoA dehydrogenase deficiency is by far the most common, accounting for the majority of diagnosed cases, and it was the first condition for which newborn screening by tandem mass spectrometry demonstrated a clear outcome benefit. The long-chain defects (VLCAD, LCHAD, CPT I, CPT II, CACT) are individually rare but collectively important because they cause cardiomyopathy, liver dysfunction and rhabdomyolysis in addition to hypoglycaemia. [1] [7]
Classification
The clinically useful classification is by chain-length specificity, because the chain length of the fatty acid the enzyme handles determines both the organs affected and the severity of energy failure. The medium-chain enzyme — MCAD — processes the bulk of dietary and stored fatty acids, and its deficiency produces the classic isolated hypoketotic hypoglycaemia without cardiomyopathy or chronic muscle disease. The long-chain enzymes handle the energy-hungry tissues — heart and skeletal muscle — and their defects cause a more severe multisystem phenotype with hypertrophic or dilated cardiomyopathy, hepatic dysfunction, and exercise-induced rhabdomyolysis. [1] [2]
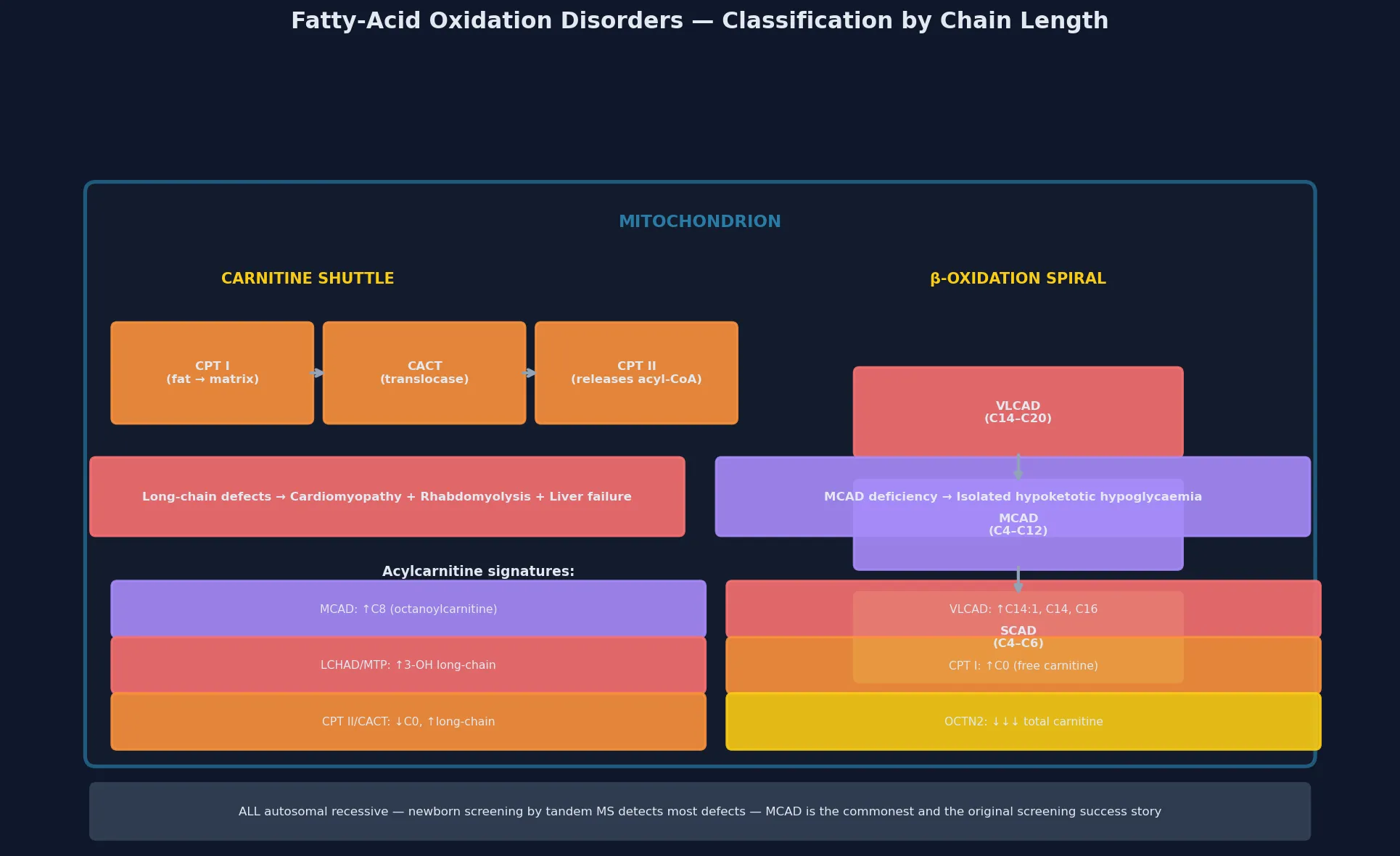
A defect at the carnitine shuttle (CPT I, CACT, CPT II) blocks entry of long-chain fats into the mitochondrion, producing a severe neonatal or childhood energy failure with cardiomyopathy, liver dysfunction, and — in CPT II — recurrent exercise-induced rhabdomyolysis and myoglobinuria. The primary carnitine transporter defect (OCTN2, SLC22A5) causes systemic carnitine depletion and a dilated cardiomyopathy that responds dramatically to oral carnitine supplementation. A defect at VLCAD or MTP/LCHAD blocks long-chain beta-oxidation inside the mitochondrion, producing the full multisystem phenotype of cardiomyopathy, retinal pigmentary changes, peripheral neuropathy, recurrent rhabdomyolysis, and — in LCHAD — a maternal history of acute fatty liver of pregnancy or HELLP syndrome. A defect at MCAD blocks the central portion of the spiral and produces isolated hypoketotic hypoglycaemia. Glutaric acidemia type II (multiple acyl-CoA dehydrogenase deficiency, MADD) is caused by defects in electron transfer from the acyl-CoA dehydrogenases to the respiratory chain via ETF and ETF-dehydrogenase, and it produces a severe neonatal-onset phenotype with hypotonia, cardiomyopathy, hepatomegaly and metabolic acidosis. [2] [11]
Epidemiology & Risk Factors
Medium-chain acyl-CoA dehydrogenase deficiency has an estimated incidence of roughly one in 9,000 to 15,000 live births across most screened populations, making it the most common fatty-acid oxidation disorder and one of the most common inborn errors of metabolism detected by newborn screening. The Dutch natural-history study by Derks and colleagues, drawn from one of the best-characterised populations, demonstrated that in the pre-screening era a first metabolic crisis carried a mortality of 20 to 25 percent and a high rate of residual neurological injury among survivors — a stark figure that underpins the case for universal newborn screening. [5] [6]
The Australian study by Wilcken and colleagues is one of the landmark outcome comparisons: children identified by expanded newborn screening were compared with unscreened historical controls at age six, and the screened cohort showed a dramatic reduction in developmental disability and death. This study cemented tandem mass spectrometry as the standard of care for detecting fatty-acid oxidation disorders in the newborn period. The cost-effectiveness evidence from Pandor and colleagues corroborated this from a health-systems perspective. [7] [12]
The major risk factor for a metabolic crisis is a catabolic stressor that increases fatty-acid mobilisation — intercurrent infection (especially gastroenteritis with poor oral intake), prolonged fasting, surgery, exercise, or the overnight period in an infant who is night-weaned too early. The infant who sleeps through the night and skips a feed, or the toddler with a viral illness who does not eat for 12 to 18 hours, is the prototypical presentation of MCAD deficiency. The long-chain defects are additionally provoked by sustained exercise, cold exposure, and the physiological fasting of the neonatal period. A family history of sudden infant death, unexplained developmental disability, or consanguinity raises the pre-test probability, as does a maternal history of acute fatty liver of pregnancy — which is associated with fetal LCHAD deficiency. [5] [11]
Pathophysiology
The molecular story begins with the fasting state. When dietary glucose is exhausted, the body turns to stored triglyceride: hormone-sensitive lipase releases free fatty acids from adipose tissue, they travel bound to albumin, and they are taken up by liver, heart and skeletal muscle for energy production. In the liver, beta-oxidation generates acetyl-CoA that is converted into ketone bodies (acetoacetate and beta-hydroxybutyrate) for export to the brain, which cannot metabolise fatty acids directly. In heart and skeletal muscle, beta-oxidation generates ATP directly, and fatty acids are the preferred substrate at rest. [4] [11]
The carnitine shuttle transports long-chain fatty acyl-CoA esters across the inner mitochondrial membrane. CPT I on the outer membrane conjugates the acyl group to carnitine, the translocase (CACT) exchanges acylcarnitine in for free carnitine out, and CPT II on the matrix side reconverts acylcarnitine to acyl-CoA, freeing carnitine to recycle. Medium- and short-chain fatty acids cross the membrane without this shuttle, which is why MCAD deficiency does not cause a carnitine transport block. Once inside the matrix, each cycle of beta-oxidation removes two carbons from the chain, generates one FADH2 and one NADH for the electron transport chain, and releases one acetyl-CoA — so a single molecule of palmitate (C16) yields 106 ATP. A block at any step therefore produces a direct energy deficit proportional to the chain length handled and the tissue's reliance on that fuel. [4] [2]
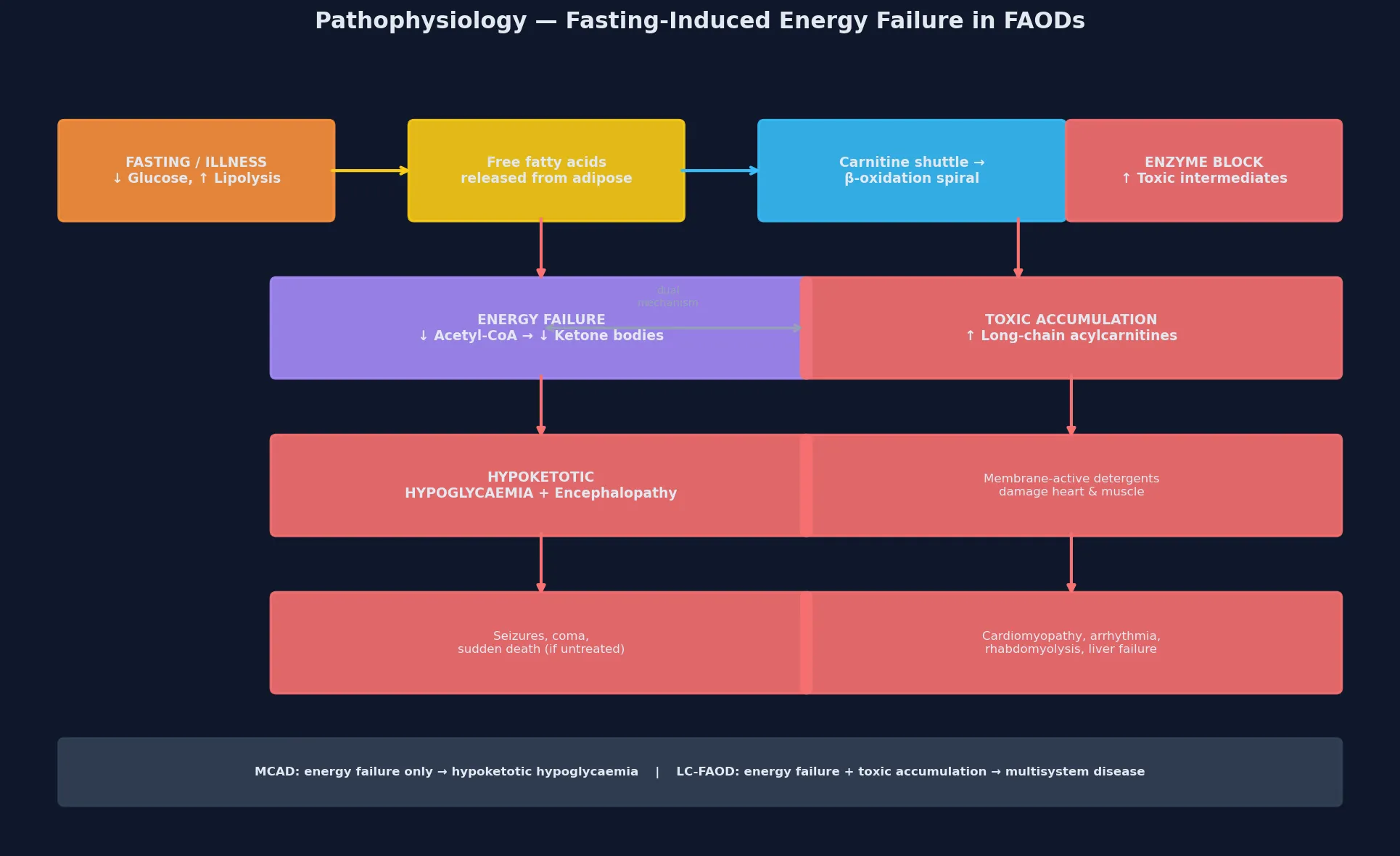
In the long-chain defects, the accumulation of partially oxidised intermediates is itself toxic. Long-chain acylcarnitines are detergent-like molecules that destabilise the cardiac and skeletal-muscle cell membrane, contributing to the cardiomyopathy, arrhythmia and rhabdomyolysis that characterise VLCAD, LCHAD and CPT II deficiency. This dual mechanism — energy failure plus membrane-active toxin accumulation — explains why the long-chain defects are more severe than MCAD deficiency and why carnitine supplementation must be used cautiously in some of them: increasing acylcarnitine export could theoretically worsen arrhythmogenic potential. The odd-chain fatty acid triheptanoin works by bypassing the block and providing anaplerotic substrate (the five-carbon intermediates replenish the citric acid cycle), which is the rationale for its use in LC-FAOD management. [2] [8]
Clinical Presentation
The presentation of MCAD deficiency is the textbook metabolic emergency: a previously well infant or young child, typically between three months and three years of age, who develops vomiting and lethargy during a viral illness or after a period of reduced oral intake, and is found obtunded or seizing with a profoundly low blood glucose. The age window reflects the loss of maternal metabolic support and the typical night-weaning interval: before three months the baby feeds frequently enough to maintain glucose, and after three years the child has larger glycogen reserves and better fasting tolerance. The laboratory signature is the inappropriately low or absent ketones in the face of hypoglycaemia — the single most important diagnostic clue at the bedside. [5] [10]
The long-chain defects present in two patterns. The severe neonatal form of VLCAD, LCHAD, CACT or CPT II deficiency manifests in the first days of life with hypotonia, poor feeding, hypertrophic cardiomyopathy, hepatomegaly with liver dysfunction, and metabolic acidosis — a picture of multisystem energy failure that may be fatal without rapid recognition. The late-onset or episodic form presents in older infants, children or adults with exercise-induced muscle pain, recurrent rhabdomyolysis with markedly elevated creatine kinase and myoglobinuria, or intermittent cardiomyopathy triggered by illness or exertion. An adolescent or adult who presents with recurrent rhabdomyolysis after exercise, fasting or illness, with or without a history of childhood complications, should be evaluated for a long-chain fatty-acid oxidation defect. [2] [11]
The primary carnitine transporter defect (OCTN2) is worth separate mention because it is the one fatty-acid oxidation disorder with a dramatic response to therapy. It presents with progressive dilated cardiomyopathy, hypotonia and failure to thrive in infancy or early childhood, often with a history of sibling sudden death. Plasma carnitine is profoundly low (often below five micromoles per litre), and high-dose oral carnitine supplementation produces a rapid and often complete recovery of cardiac function — one of the most satisfying treatments in metabolic medicine. Missing this diagnosis by failing to check a plasma carnitine in a child with unexplained cardiomyopathy is a classic and avoidable error. [1] [11]
Differential Diagnosis
The differential diagnosis of hypoketotic hypoglycaemia is broader than the fatty-acid oxidation disorders alone, and naming the full list is a fellowship expectation. The hyperinsulinaemic hypoglycaemias (including congenital hyperinsulinism, sulfonylurea ingestion, and insulinoma) produce hypoketotic hypoglycaemia because insulin suppresses both ketogenesis and lipolysis — but they show a detectable insulin and C-peptide at the time of hypoglycaemia, and a characteristic response to glucagon. The glycogen storage diseases produce fasting hypoglycaemia but with ketosis (except GSD I, which shows lactic acidosis and hyperlipidaemia) and hepatomegaly. The organic acidaemias and the mitochondrial hepatopathies produce hypoglycaemia with metabolic acidosis and a high lactate, and they are distinguished by urinary organic acids and lactate/pyruvate ratios. [1]
The endocrine causes — growth hormone deficiency, cortisol deficiency (primary adrenal insufficiency or hypopituitarism), and hypothyroidism — produce fasting hypoglycaemia through impaired gluconeogenesis or counter-regulatory failure, and they are excluded by the critical sample taken at the time of hypoglycaemia. The toxins and drugs — ethanol, salicylates, beta-blockers, and oral hypoglycaemic agents — enter the differential in any unexplained hypoglycaemia. The key principle is that the critical sample (glucose, insulin, C-peptide, ketones, lactate, free fatty acids, growth hormone, cortisol, ammonia and acylcarnitines drawn at the time of the low glucose) converts the undifferentiated presentation into a pathway-specific answer. [1] [11]
Clinical & Bedside Assessment
The bedside assessment runs at two speeds. In the acute crisis, the priority is resuscitation and the critical sample — airway, breathing, circulation, then a rapid glucose check, intravenous access, and the immediate administration of a dextrose bolus followed by a maintenance infusion. While the dextrose is running, draw the critical sample: glucose, insulin, C-peptide, blood gas, lactate, beta-hydroxybutyrate, free fatty acids, cortisol, growth hormone, ammonia, and the all-important plasma acylcarnitine profile. Do not delay dextrose while waiting for laboratory results — the hypoglycaemic brain is being injured minute by minute. [1] [10]
In the stable or recovering child, build a structured picture with a focused history. Ask explicitly about the circumstances of the episode: how long since the last meal, whether there was an intercurrent illness, whether the child was fasting overnight or had been exercised, and whether this is the first episode or a pattern. A three-generation pedigree asks about sudden infant or childhood deaths, unexplained developmental disability, recurrent hospitalisations for hypoglycaemia or dehydration, maternal acute fatty liver of pregnancy or HELLP syndrome, and consanguinity. The examination looks for hepatomegaly (suggesting a storage or long-chain defect), cardiomyopathy (a gallop, murmur or signs of cardiac failure pointing to VLCAD, LCHAD, CACT, CPT II or carnitine transporter defect), hypotonia, and the pigmentary retinopathy and peripheral neuropathy of LCHAD deficiency. [1] [11]
The bedside assessment converts directly into the investigation plan: the plasma acylcarnitine profile is the first-tier diagnostic test and localises the defect by the chain-length signature of the accumulating species, while molecular genetic testing of the candidate gene (or a metabolic gene panel) provides confirmation and enables cascade family testing and reproductive counselling. In a child already identified by newborn screening, the assessment at the first clinic visit confirms the screen result, characterises any residual metabolic instability, and builds the emergency sick-day plan and family education that prevent the first crisis from ever occurring. [7] [1]
Investigations
The first-tier investigation is the plasma acylcarnitine profile, measured by tandem mass spectrometry on a dried blood spot or plasma. Each defect produces a characteristic acylcarnitine signature that reflects the accumulating intermediate: MCAD deficiency shows a marked elevation of medium-chain species, dominated by octanoylcarnitine (C8) alongside C6 and C10; VLCAD deficiency shows elevated long-chain species (C14:1, C14, C16, C18:1); LCHAD and MTP deficiency show elevated 3-hydroxy-long-chain species; CPT I deficiency shows a high free carnitine (C0) with low long-chain acylcarnitines; CPT II and CACT deficiency show low free carnitine with high long-chain acylcarnitines. The ratio of C8 to C10 and C8 to C12 enhances the sensitivity for MCAD deficiency. [1] [2]
The supporting tests define the metabolic context and exclude the mimics. Plasma total and free carnitine identifies the carnitine transporter defect (profoundly low total carnitine) and guides carnitine supplementation decisions. Urinary organic acids characterise the organic acidaemias and glutaric acidemia type II. Blood gas and lactate assess for metabolic acidosis. The critical sample taken at the time of documented hypoglycaemia — glucose, insulin, C-peptide, beta-hydroxybutyrate, free fatty acids, cortisol, growth hormone and ammonia — separates the fatty-acid oxidation disorder (low insulin, low ketones, high free fatty acids) from hyperinsulinism (inappropriately high insulin, low ketones, low free fatty acids) and endocrine failure (low counter-regulatory hormones). [1] [11]
Why a normal acylcarnitine profile between crises does not always exclude a FAOD
In some long-chain defects, especially the milder or late-onset VLCAD and CPT II variants, the acylcarnitine profile may be normal when the child is well and fed. If the clinical suspicion is strong — exercise-induced rhabdomyolysis, a family history, or an at-risk sibling — repeat the acylcarnitine profile during an acute episode, or proceed directly to molecular testing with a metabolic gene panel or exome sequencing. The acylcarnitine profile is most diagnostic when the pathway is under stress, which is precisely when the child is most vulnerable. [2] [11]
Molecular confirmation is then undertaken by sequencing the candidate gene suggested by the acylcarnitine pattern (ACADM for MCAD, ACADVL for VLCAD, HADHA/HADHB for MTP/LCHAD, CPT1A for CPT I, CPT2 for CPT II, SLC22A5 for the carnitine transporter), or by a metabolic gene panel or exome when the pattern is ambiguous. The common ACADM pathogenic variant c.985A>G (K304E) accounts for the majority of disease alleles in Northern European populations, and homozygosity or compound heterozygosity for pathogenic variants confirms the diagnosis. For the LCHAD/MTP common variant c.1528G>C (E474Q), there is a well-established genotype-phenotype correlation with isolated LCHAD versus full trifunctional protein deficiency, which carries prognostic implications. Confirming the molecular defect enables cascade carrier testing, prenatal and preimplantation genetic diagnosis, and — for the emerging therapies — eligibility assessment. [1] [10]
Management — Resuscitation
The cardinal principle of resuscitation is to switch off lipolysis immediately by providing exogenous glucose, because the metabolic crisis is driven by the mobilisation of fatty acids that the blocked pathway cannot process. The moment hypoglycaemia is identified in a child with a suspected or known fatty-acid oxidation disorder, give an intravenous bolus of 2 millilitres per kilogram of 10 percent dextrose (200 milligrams per kilogram), followed by a continuous infusion of 10 percent dextrose at a rate sufficient to maintain the blood glucose above five millimoles per litre — typically 6 to 8 milligrams per kilogram per minute, titrated upward as needed. If intravenous access is delayed, glucagon is ineffective in these disorders (there is no insulin excess) and intraosseous access should be used. [1] [2]
In the long-chain defects, the acute crisis may also involve cardiogenic shock from the energy-starved myocardium, rhabdomyolysis with acute kidney injury from myoglobinuria, and liver dysfunction with coagulopathy. These require parallel supportive management: inotropes for cardiogenic shock, aggressive intravenous hydration and monitoring of renal function and creatine kinase for rhabdomyolysis, and correction of coagulopathy. Carnitine supplementation (100 to 200 milligrams per kilogram per day intravenously or orally) is given to enhance the excretion of accumulating toxic acyl-CoA intermediates as acylcarnitines, though its role in the long-chain defects is debated and should be guided by the metabolic team. In the carnitine transporter defect, high-dose carnitine is both acute and chronic therapy and is life-saving. [2] [3]
Once the child is stabilised, the dextrose infusion is weaned gradually as oral intake is re-established with a low-fat, high-carbohydrate diet, and the family is re-educated on the emergency sick-day plan. The duration of intravenous therapy depends on the underlying defect and the severity of the precipitating illness — MCAD deficiency may stabilise within 24 to 48 hours, while a severe long-chain defect with cardiomyopathy may require a prolonged PICU admission. The Spiekerkoetter workshop cohort of 75 individuals with long-chain defects demonstrated that early and aggressive intravenous dextrose during illness is the single most important determinant of outcome, and that delayed or inadequate treatment is the chief cause of morbidity and mortality. [3] [2]
Management — Definitive & Stepwise
Once the acute crisis is controlled, definitive management is built around three pillars: avoidance of fasting, an emergency sick-day plan, and disease-specific pharmacological therapy. Avoidance of fasting is the cornerstone: MCAD-deficient children should not fast longer than the age-appropriate safe interval — typically 8 to 10 hours for infants under six months, 10 to 12 hours for older infants and toddlers, and 12 hours for school-age children. Night-time feeds, late-evening snacks of complex carbohydrate (such as cornstarch), and careful management of the night-weaning transition are practical applications of this principle. Uncooked cornstarch (1 to 2 grams per kilogram) provides slow-release glucose and extends fasting tolerance overnight. [1] [10]
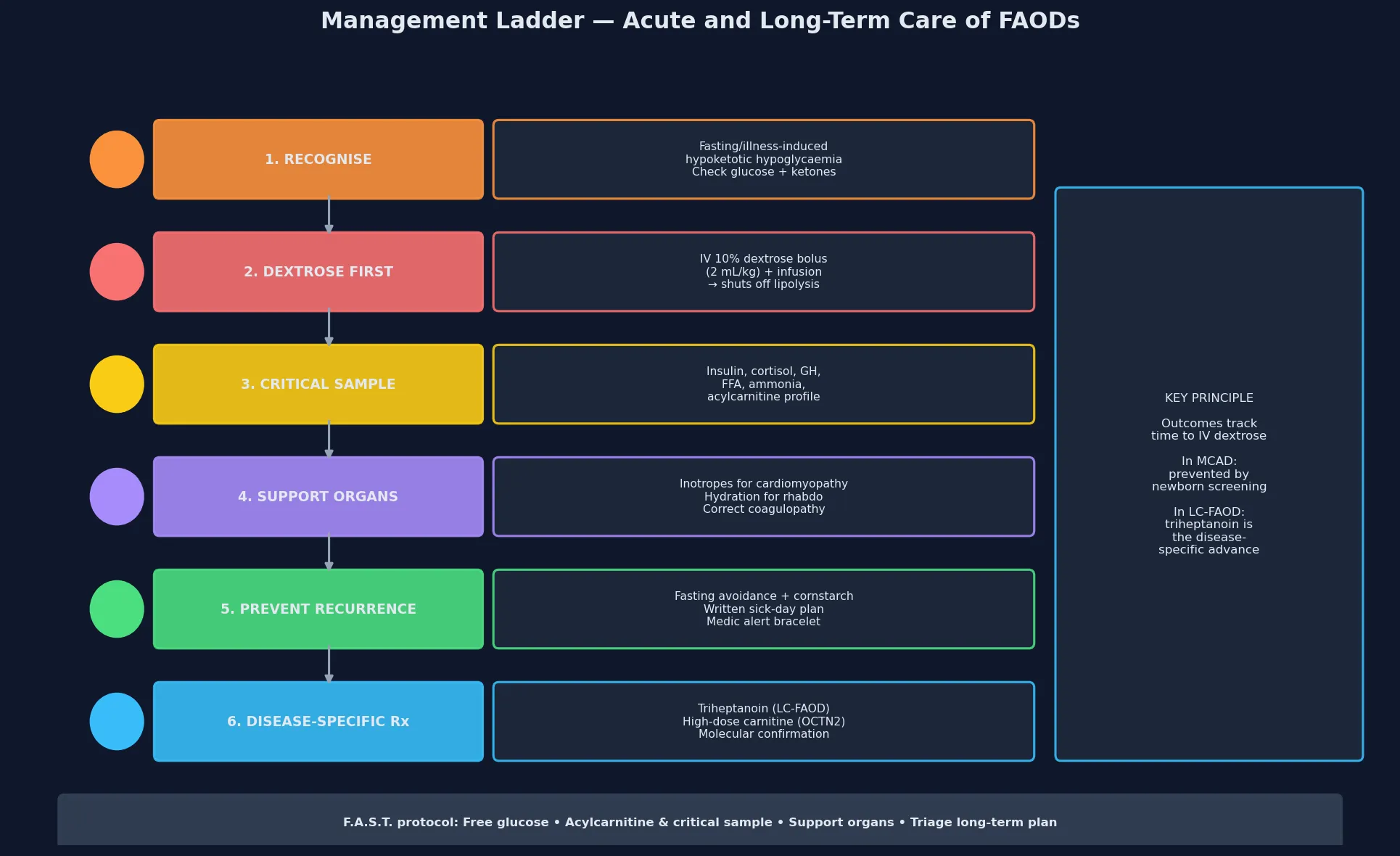
The emergency sick-day plan is the single most important safety net, and it should be written, specific, and carried with the child at all times. At the first sign of illness — a fever, vomiting, poor feeding, or lethargy — the family stops the fasting interval, gives frequent carbohydrate-rich fluids or oral rehydration with glucose, and presents early for intravenous management if oral intake fails or the glucose is borderline. A medic alert bracelet or card, a written emergency letter, and direct contact with the metabolic service are essential. Prevention of catabolism is also achieved by ensuring up-to-date immunisation (to reduce the frequency and severity of intercurrent illness) and by perioperative fasting protocols that use intravenous dextrose from the moment oral intake stops. [1] [5]
Disease-specific pharmacological therapy is evolving rapidly for the long-chain defects. Triheptanoin (a seven-carbon odd-chain triglyceride) was developed to bypass the long-chain beta-oxidation block and provide anaplerotic substrate — the five-carbon propionyl-CoA it generates replenishes the citric acid cycle and restores energy production. The long-term extension study by Vockley and colleagues demonstrated clinically meaningful reductions in the frequency and severity of rhabdomyolysis, cardiomyopathy and hospitalisation in patients with VLCAD, LCHAD and MTP deficiency treated with triheptanoin. For the carnitine transporter defect, high-dose oral carnitine (100 to 400 milligrams per kilogram per day) restores plasma and tissue carnitine, reverses cardiomyopathy and is essentially curative. For MCAD deficiency, no specific pharmacological therapy is needed beyond fasting avoidance — the key intervention is prevention, and the prognosis is excellent. [8] [9]
F.A.S.T. \u2014 the acute FAOD protocol
Specific Subtypes & Scenarios
Medium-chain acyl-CoA dehydrogenase deficiency is the prototype and the most common fatty-acid oxidation disorder. It presents classically between three months and three years of age with hypoketotic hypoglycaemia after a viral illness or fasting, and the acylcarnitine profile shows a dominant elevation of octanoylcarnitine (C8). The prognosis is excellent when the diagnosis is made by newborn screening and the family is educated on fasting avoidance and the emergency sick-day plan — the Wilcken study demonstrated near-complete elimination of crisis-related death and disability in screened populations. The common pathogenic variant c.985A>G accounts for the majority of alleles in Northern European populations, and carrier frequency is roughly one in 50 to 70. [5] [7]
VLCAD deficiency is the most common long-chain defect and presents across a spectrum from severe neonatal cardiomyopathy and liver failure to mild, late-onset exercise-induced rhabdomyolysis. The severe infantile form carries the highest mortality and may present with ventricular arrhythmia from long-chain acylcarnitine accumulation. Management combines fasting avoidance, a low-long-chain-fat diet with medium-chain triglyceride (MCT) supplementation (which bypasses the carnitine shuttle), triheptanoin, and aggressive intravenous dextrose during illness. [2] [8]
LCHAD and mitochondrial trifunctional protein (MTP) deficiency share the c.1528G>C (E474Q) common variant and are distinguished by the extent of the enzyme deficiency. Both produce the full multisystem phenotype — cardiomyopathy, liver dysfunction, retinal pigmentary degeneration, peripheral sensorimotor neuropathy, and recurrent rhabdomyolysis. A unique and high-yield association is that mothers carrying a fetus with LCHAD deficiency are at significantly elevated risk of acute fatty liver of pregnancy (AFLP) and HELLP syndrome — the metabolic products of the affected fetus overwhelm the heterozygous mother's hepatic beta-oxidation capacity. Every infant born to a mother with a history of AFLP or HELLP should be evaluated for LCHAD deficiency. [2] [11]
CPT II deficiency has a classic adult-onset form that presents with recurrent exercise- or fasting-induced rhabdomyolysis and myoglobinuria in an otherwise healthy adolescent or young adult, often with a history of recurrent childhood muscle pain that was attributed to growing pains or viral myositis. The severe infantile form presents with hypoketotic hypoglycaemia, cardiomyopathy and liver dysfunction. The distinction matters because the adult form has a good prognosis with exercise modification and avoidance of fasting, while the infantile form requires full metabolic management. [11]
Complications & Pitfalls
The complications divide into the acute injuries, the chronic multisystem burden, and the cognitive traps. The acute injuries during a metabolic crisis are hypoglycaemic encephalopathy, cerebral oedema, seizures, coma and death — and in the long-chain defects, cardiogenic shock, ventricular arrhythmia from acylcarnitine accumulation, rhabdomyolysis with acute kidney injury, and hepatic failure with coagulopathy. The Derks natural-history study showed that the first unrecognised MCAD crisis carried a 20 to 25 percent mortality and a high rate of residual neurological injury, which is why universal newborn screening is now the standard of care. [5] [6]
The chronic complications of the long-chain defects are driven by cumulative toxic injury and episodic energy failure: progressive hypertrophic or dilated cardiomyopathy, pigmentary retinopathy (in LCHAD), progressive peripheral neuropathy (in LCHAD and MTP), recurrent exercise-induced rhabdomyolysis with cumulative muscle damage, and intellectual disability from recurrent hypoglycaemic insults. The retinopathy and neuropathy of LCHAD deficiency may progress despite good metabolic control, reflecting ongoing low-level toxicity from accumulated intermediates. [2] [11]
The chief cognitive trap is delayed dextrose — either failing to check a glucose in a vomiting, lethargic infant because the picture seems like gastroenteritis, or collecting the critical sample before giving dextrose and thereby prolonging the hypoglycaemic brain injury. The second trap is missing the carnitine transporter defect by not checking a plasma carnitine in a child with unexplained cardiomyopathy, when the treatment is simple and curative. The third is failing to counsel the family on the emergency plan, particularly in the transition from hospital to home, when the child is well and the urgency seems to have passed. [1] [5]
Prognosis & Disposition
Prognosis is determined by three factors: whether the diagnosis is made before or after the first crisis, the specific enzyme defect, and the quality and consistency of long-term management. MCAD deficiency identified by newborn screening and managed with fasting avoidance and a sick-day plan carries an excellent prognosis — near-normal life expectancy and neurodevelopmental outcome, with the main residual risk being the possibility of a first crisis if the plan is not followed during a severe illness. MCAD deficiency diagnosed only after a first crisis carries the 20 to 25 percent mortality and significant neurological morbidity of that presentation, which is the entire justification for newborn screening. [5] [7]
The long-chain defects carry a more guarded prognosis because of the cumulative cardiomyopathy, retinopathy, neuropathy and episodic rhabdomyolysis. The Vockley triheptanoin extension study showed meaningful improvement in hospitalisation rates, rhabdomyolysis frequency and cardiac function, representing a genuine advance in management but not a cure. The severe neonatal-onset forms of VLCAD, LCHAD, CACT and CPT II deficiency carry significant mortality in infancy even with optimal management. The carnitine transporter defect, by contrast, has an excellent prognosis with carnitine supplementation — one of the most dramatic treatment responses in inherited metabolic disease. [8] [9]
Disposition is shared, lifelong, multidisciplinary care. A specialist metabolic service owns the diagnostic confirmation, the dietary plan, the pharmacological therapy and the molecular counselling. The general paediatrician or GP owns coordination, immunisation, growth monitoring and the front-line implementation of the emergency sick-day plan. The family owns the day-to-day vigilance — recognising the early signs of decompensation, giving the carbohydrate-rich emergency fluids, and presenting early. Every transition — from breast to bottle feeding, from home to childcare, from primary to secondary school, and from paediatric to adult metabolic services — is a high-risk point, and the emergency plan must be re-communicated at each stage. [1] [2]
Special Populations
The same fatty-acid oxidation disorder behaves differently across populations because access, recognition and service models are unevenly distributed. In remote and Indigenous communities, later presentation, longer retrieval times, and higher rates of intercurrent infection mean that the window between onset and treatment is longer and outcomes are worse — so a location-specific emergency plan, telehealth access to the metabolic service, and aeromedical retrieval thresholds are disproportionately important. In migrant, refugee and asylum-seeking families, consanguinity raises the pre-test probability of autosomal recessive defects, language barriers complicate the emergency teaching, and an interpreter must be used for every counselling session. [1] [7]
In adolescents transitioning to adult metabolic care, the move is a high-risk point: dietary adherence may slip, the emergency plan may not transfer, and exercise and alcohol introduce new triggers for long-chain defects. Adolescents with CPT II deficiency need specific counselling about alcohol as a precipitant of rhabdomyolysis. In families managing complex chronic metabolic disease — especially the multisystem long-chain defects with cardiomyopathy, retinopathy and neuropathy — fragmentation of care across multiple subspecialties (cardiology, ophthalmology, neurology, metabolic) is the chief threat, and a written, shared, reconciled care plan with a named coordinator is the intervention that matters most. [2] [11]
In the neonatal period, the baby who is identified by newborn screening but has not yet had confirmatory testing should be managed presumptively: avoid prolonged fasting, ensure regular feeds (every three to four hours), and educate the family on the emergency sick-day plan while the acylcarnitine profile and molecular testing are pending. A normal newborn screen does not entirely exclude a fatty-acid oxidation disorder — some long-chain defects and milder variants may be missed — so any infant with a suggestive clinical picture should have a repeat acylcarnitine profile and molecular testing regardless of the screen result. [7] [1]
Evidence, Guidelines & Regional Differences
The evidence base rests on three pillars: the natural-history and outcome data from the pre-screening era, the newborn screening outcome studies that justified universal implementation, and the emerging therapeutic trials for the long-chain defects. The Derks natural-history study of MCAD deficiency in the Netherlands (2006) documented the mortality and morbidity of the pre-screening era, providing the benchmark against which the benefit of screening was measured. The Wilcken Australian outcome study (2009) compared screened and unscreened cohorts at age six and demonstrated a dramatic reduction in death and disability — this is the single most influential study in the field and is the foundation of expanded newborn screening programmes worldwide. [6] [7]
The cost-effectiveness evidence from the Pandor systematic review (2004) corroborated the case from a health-economics perspective, and it underpinned the adoption of tandem mass spectrometry screening in the United Kingdom, Australia, the United States and most developed health systems. The Spiekerkoetter workshop cohort (2009) of 75 individuals with long-chain defects provided the first structured management framework for the LC-FAODs, establishing the principles of fasting avoidance, aggressive intravenous dextrose during illness, and disease-specific dietary manipulation that remain the standard of care. [3] [12]
The therapeutic evidence is moving fast. The triheptanoin long-term extension study by Vockley and colleagues (2023) showed clinically meaningful reductions in hospitalisation, rhabdomyolysis and cardiomyopathy across the major LC-FAOD subtypes, and triheptanoin is now an approved therapy in several jurisdictions. The biochemistry reviews by Houten and colleagues and by Tein provide the mechanistic foundation that explains both the energy failure and the rationale for the anaplerotic approach. [9] [4]
In Australia and New Zealand, fatty-acid oxidation disorders are included in the universal newborn screening programme, which uses tandem mass spectrometry on the heel-prick bloodspot collected at 48 to 72 hours of life. MCAD deficiency, VLCAD deficiency and several other acylcarnitine-targeted conditions are routinely detected. Confirmatory testing — repeat acylcarnitine profile, plasma carnitine, urinary organic acids and molecular sequencing — is coordinated through the state-based metabolic services at the major children's hospitals in each state and Starship in New Zealand. Triheptanoin is available through the metabolic services for confirmed LC-FAOD cases, and aeromedical retrieval to a tertiary metabolic and intensive-care centre is the expected pathway for a severe acute crisis. Genetic counselling, carrier testing and prenatal or preimplantation diagnosis are provided through the clinical genetics services linked to each metabolic centre. [7] [1]
Exam Pearls
A fellowship candidate answering on the fatty-acid oxidation disorders should land six anchor points and avoid three classic traps. The anchors are the hypoketotic hypoglycaemia signature, the fasting-and-illness trigger pattern, the acylcarnitine chain-length signature that localises the defect, the intravenous-dextrose-first emergency protocol, the role of triheptanoin for LC-FAODs and carnitine for the transporter defect, and the AFLP/HELLP maternal clue for LCHAD deficiency. The traps are delayed dextrose (waiting for the critical sample while the brain is being injured), missing the carnitine transporter defect in an unexplained cardiomyopathy, and failing to provide a written emergency sick-day plan. The candidate who can name the C8 elevation of MCAD, the C14:1 of VLCAD, and the profound carnitine deficiency of OCTN2 deficiency, and who can explain why glucagon is ineffective in a fatty-acid oxidation disorder, demonstrates the depth that earns distinction marks. [1] [2] [5]
References
- [1]Merritt JL 2nd, Norris M, Kanungo S. Fatty acid oxidation disorders. Ann Transl Med, 2018.PMID 30740404
- [2]Vockley J, Burton B, Berry GT, Longo N, Phillips J, Sanchez-Valle A, et al. Long-chain fatty acid oxidation disorders and current management strategies. Am J Manag Care, 2020.PMID 32840329
- [3]Spiekerkoetter U, Lindner M, Reitzer R, Kreisel I, Schulze A, Haussinger D, et al. Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis, 2009.PMID 19399638
- [4]Houten SM, Violante S, Ventura FV, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis, 2010.PMID 20195903
- [5]Wilcken B. Fatty acid oxidation disorders: outcome and long-term prognosis. J Inherit Metab Dis, 2010.PMID 20049534
- [6]Derks TG, Duran M, Waterham HR, Reijngoud DJ, Ten Brink HJ, IJlst L, et al. The natural history of medium-chain acyl CoA dehydrogenase deficiency in the Netherlands: clinical presentation and outcome. J Pediatr, 2006.PMID 16737882
- [7]Wilcken B, Haas M, Joy P, Wiley V, Bowling FG, Carpenter KH, et al. Expanded newborn screening: outcome in screened and unscreened patients at age 6 years. Pediatrics, 2009.PMID 19620191
- [8]Vockley J, Marsden D, Dewey MA, Hsu C, Nagan N, Wynn M, et al. Long-term major clinical outcomes in patients with long chain fatty acid oxidation disorders before and after transition to triheptanoin treatment. Mol Genet Metab, 2015.PMID 26116311
- [9]Vockley J, Burton B, Berry GT, Longo N, Phillips J, Sanchez-Valle A, Tanasi I, et al. Triheptanoin for the treatment of long-chain fatty acid oxidation disorders: Final results of an open-label, long-term extension study. J Inherit Metab Dis, 2023.PMID 37276053
- [10]Mason E, Hindmarch CCT, Dunbar M, Hartley J, Hartley R, Rajakulendran S, et al. Medium-chain acyl-CoA dehydrogenase deficiency: pathogenesis, diagnosis, and treatment. Endocrinol Diabetes Metab, 2023.PMID 36300606
- [11]Tein I. Disorders of fatty acid oxidation. Handb Clin Neurol, 2013.PMID 23622388
- [12]Pandor A, Eastham J, Beverley C, Chilcott J, Paisley S. Clinical effectiveness and cost-effectiveness of neonatal screening for inborn errors of metabolism using tandem mass spectrometry: a systematic review. Health Technol Assess, 2004.PMID 14982654