Paeds · genetics-dysmorphology-and-metabolism
Fragile X syndrome
Also known as FMR1-related disorders · FRAXA syndrome · Martin-Bell syndrome · Fragile X-associated disorders · FXS
A fellowship approach to fragile X syndrome: recognise the X-linked FMR1 CGG-repeat expansion as the commonest inherited cause of intellectual disability, confirm the molecular diagnosis with PCR plus methylation analysis, build multidisciplinary developmental and behavioural support around the child, and run cascade testing of the wider family — because no curative therapy exists and every at-risk relative is a candidate for counselling.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who thinks in three layers at once. The first layer is the child in front of you: their intellectual level, their autism and ADHD comorbidity, their seizures and sensory profile, and the supports that change their trajectory. The second is the molecular mechanism: a CGG expansion that expands across generations, a silenced gene, and a missing synaptic brake. The third is the family: a premutation mother at risk of fragile X-associated tremor/ataxia syndrome (FXTAS) and ovarian insufficiency, siblings who may be carriers or affected, and relatives who deserve cascade testing and counselling. [1] [9]
Overview & Definition
Fragile X syndrome is an X-linked neurodevelopmental disorder caused by a full mutation (more than 200 CGG repeats) in the 5-prime untranslated region of the FMR1 gene. The expansion triggers hypermethylation of an adjacent CpG island, transcriptionally silencing the gene and stopping production of fragile X mental retardation protein (FMRP). Without FMRP, the developing brain loses a critical regulator of synaptic protein synthesis, and the result is intellectual disability, autism features, behavioural dysregulation, seizures, and a characteristic (but not universal) physical phenotype. [3] [5]
Clinically, fragile X syndrome sits within the broader family of FRAXopathies, the FMR1-related disorders that span the allele classes from normal repeats through premutation to full mutation. The syndrome itself is the full-mutation phenotype, but the same gene causes separate premutation-associated conditions in carriers — fragile X-associated tremor/ataxia syndrome and fragile X-associated primary ovarian insufficiency — which is why the family, not just the child, is the unit of care. [5]
Classification
The clinical classification that matters most is molecular, because the CGG-repeat class determines methylation, FMRP level, and phenotype. Four allele classes run from normal repeats to the full mutation, and the boundary between premutation and full mutation — at about 200 CGG repeats — is where the gene tips from being expressed into being silenced. [5] [3]
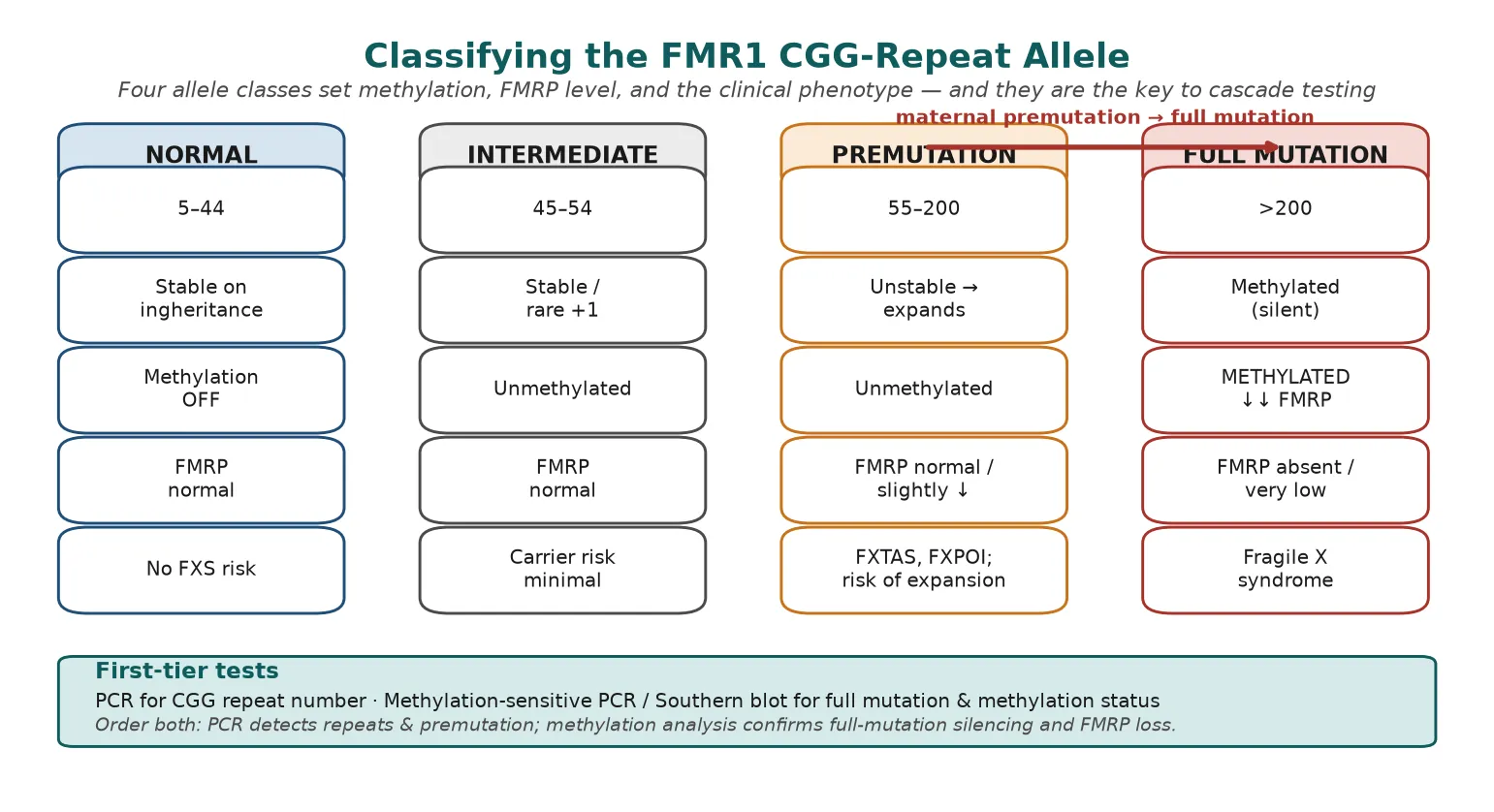
A normal allele carries 5 to 44 repeats, is stable on inheritance, and produces normal FMRP. An intermediate allele (45 to 54 repeats) is essentially stable and carries minimal carrier risk, though it can occasionally gain a few repeats. A premutation (55 to 200 repeats) is unstable and unmethylated; it produces normal or slightly reduced FMRP but carries the risk of expansion to a full mutation in the next generation and the separate adult-onset conditions FXTAS and FXPOI. A full mutation (over 200 repeats) is methylated and silenced, so FMRP is absent or severely reduced, producing fragile X syndrome. [3] [5]
The expansion risk rises sharply with premutation size, and it is governed mainly by the maternal transmission: a mother carrying a premutation of around 90 repeats or more has a very high chance of expanding to a full mutation in her offspring, whereas paternal premutations almost always pass to daughters as premutations. This maternal-expansion rule is the genetic engine behind anticipation in fragile X families and the reason cascade testing of female relatives matters. [9]
Epidemiology & Risk Factors
Fragile X syndrome is the leading inherited cause of intellectual disability and the most common known single-gene cause of autism. The systematic review by Hunter and colleagues pooled the prevalence at roughly one in 7,000 males and one in 11,000 females with a confirmed full mutation, with premutation carriers far more common at around one in 150 females and one in 450 males. [2]
The premutation is not rare, and that fact carries two practical consequences for a paediatric service. First, premutation carrier mothers are encountered in almost every general genetics clinic, and each is a conduit through which full-mutation children are born. Second, a large newborn study of 51,000 infants in China confirmed that FMR1 allele frequencies are broadly similar across populations, so ethnicity does not usefully lower pre-test probability in the way clinicians sometimes assume. [7]
The major risk factor is a family history of intellectual disability in males, particularly a maternal pattern consistent with X-linked inheritance. The next is a child already diagnosed with autism spectrum disorder, of whom a meaningful proportion carry an FMR1 full mutation. Advanced understanding of the maternal-expansion rule means that a mother with a large premutation is the highest-yield screening target in any affected family. [1] [2]
Pathophysiology
The molecular story begins with the CGG repeat in the 5-prime untranslated region of FMR1. When the repeat exceeds about 200 copies, the adjacent CpG island becomes hypermethylated, the promoter is transcriptionally silenced, and FMR1 mRNA is not produced. The downstream protein, FMRP, is therefore absent or present only in trace amounts, and it is the loss of FMRP that drives the entire phenotype. [3] [4]
FMRP is an RNA-binding protein that shuttles into dendrites and acts as a brake on the translation of target mRNAs at the synapse. In its absence, group 1 metabotropic glutamate receptor (mGluR)-linked long-term depression runs unchecked, AMPA receptor internalisation and local protein synthesis are excessive, and dendritic spines remain long, thin, and immature. This synaptic dysmaturation is the cellular signature of fragile X syndrome and the reason the disorder presents as altered plasticity, epilepsy, sensory hypersensitivity, and intellectual disability. [3] [4]
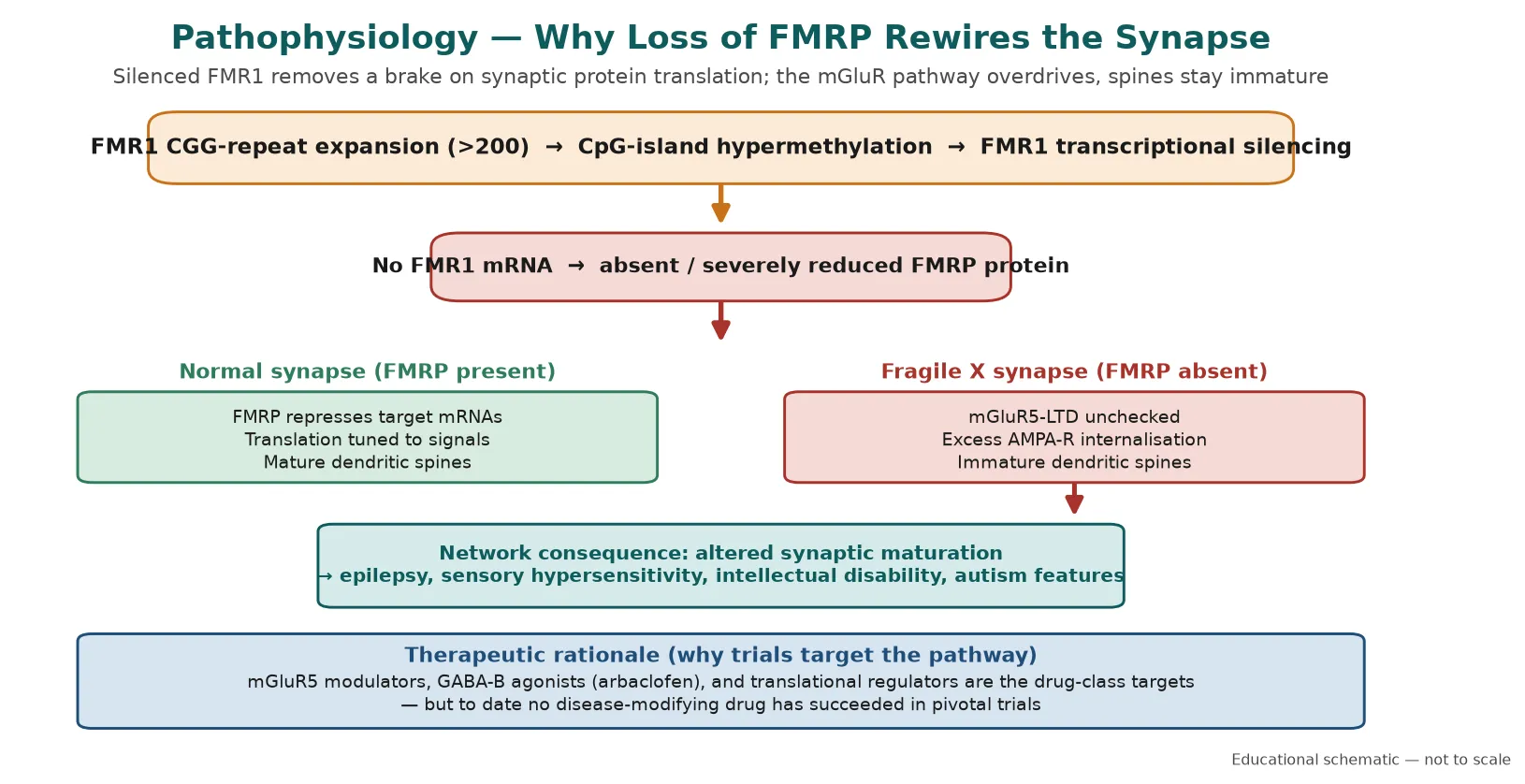
The mGluR theory of fragile X has shaped two decades of drug development, because it predicted that dampening mGluR5 signalling or restoring GABAergic inhibition might correct the core synaptic defect. Multiple targeted agents — mGluR5 negative modulators, the GABA-B agonist arbaclofen, and downstream translational regulators — showed promise in cellular and early human work, yet to date no disease-modifying drug has succeeded in pivotal trials. Management remains supportive and comorbidity-driven. [6] [8]
Clinical Presentation
The presentation is age-dependent, and the prepubertal child rarely shows the classic facial gestalt. In infancy and early childhood the picture is developmental: global delay, late walking, speech and language delay, hypotonia, and behavioural features such as gaze aversion, social anxiety, hand-flapping, and tactile defensiveness. Autism features are very common, as is ADHD with hyperactivity and short attention. Seizures occur in a sizeable minority and are usually responsive to standard anticonvulsants. [1] [4]
After puberty the physical phenotype becomes more recognisable: a long face, large or prominent ears, a high arched palate, macro-orchidism (enlarged testes), joint laxity, pes planus, and sometimes mitral valve prolapse. However, these features are variable and may be subtle, and a clinician who waits for the "textbook face" before testing will miss many affected children. Girls are usually more mildly affected than boys because of X-inactivation mosaicism, and they may present with milder intellectual disability, learning difficulty, shyness, or selective mutism rather than the full syndrome. [1] [10]
Differential Diagnosis
The differential is the differential of unexplained intellectual disability and autism, and fragile X is one highly testable node within it. The first task is to separate fragile X from the broader genetic and environmental causes of developmental delay — Down syndrome (recognised by phenotype and confirmed by karyotype or microarray), Rett syndrome (a regression in a girl with a MECP2 variant), tuberous sclerosis (skin signs, epilepsy, TSC1/2 variants), and Prader-Willi and Angelman syndromes (imprinting disorders of 15q11-q13). [5]
From the other causes of non-syndromic intellectual disability, fragile X is distinguished by the molecular test. Chromosomal microarray is the first-tier test for unexplained intellectual disability because it detects copy-number variants across the genome, but it does not detect the FMR1 repeat expansion, so fragile X PCR plus methylation analysis is ordered in parallel when the phenotype or family history fits. Exome sequencing is increasingly used as a first-tier test and likewise requires a separate fragile X assay. [1] [10]
Clinical & Bedside Assessment
The bedside assessment begins with a structured developmental history and examination, then converts the findings into an investigation plan. Quantify the developmental delay across domains using a standardised tool, screen for autism with the M-CHAT and refer for ADOS where indicated, and record the family history in a three-generation pedigree that explicitly asks about intellectual disability in males, autism, early ovarian failure, and adult tremor or ataxia. [1]
The examination looks for the physical features that support (but never exclude) the diagnosis: long face, large or prominent ears, high arched palate, macro-orchidism in the post-pubertal male, joint hypermobility, and soft skin. Check for features of comorbidity — seizures, hypotonia, strabismus, otitis media, and cardiac murmur — because these shape the management plan regardless of the genetic result. The examination is also the moment to screen for the premutation-associated conditions in accompanying adults, by asking about tremor, ataxia, and ovarian function. [1] [9]
Investigations
The molecular investigation is a two-part test. FMR1 PCR measures the CGG repeat number precisely and detects normal, intermediate, and premutation alleles, including large premutations in carrier females that Southern blot alone can miss. Methylation-sensitive PCR or Southern blot determines whether a full mutation is methylated and silenced, confirms the repeat size of full mutations, and detects mosaicism. Ordering only one of the two is a common error: PCR alone may not size a very large full mutation, and methylation analysis alone may miss a premutation. [1] [10]
Chromosomal microarray should be ordered in parallel when the intellectual disability is unexplained, because copy-number variants are the commonest single finding in this group and may coexist with or mimic fragile X. Exome sequencing, where used as first-tier, still needs a dedicated fragile X assay because sequencing does not reliably size the CGG repeat. Baseline assessments that complete the diagnostic work-up include audiology and vision, baseline EEG when seizures are suspected, echocardiography for mitral valve prolapse, and a developmental or cognitive assessment to anchor educational planning. [1] [5]
Why karyotype and microarray are not enough
A standard karyotype does not detect the FMR1 repeat expansion, and neither does chromosomal microarray. If you send only a microarray for unexplained intellectual disability and it returns normal, fragile X has not been excluded — it was never tested. The reflex is to add FMR1 PCR plus methylation analysis whenever the microarray is non-diagnostic and the clinical picture fits. [1] [10]
Management — Resuscitation
Resuscitation in fragile X syndrome is rarely about an acute physiological collapse, but two scenarios demand urgent action. The first is status epilepticus or poorly controlled seizures, which occur in a sizeable minority and are managed with standard paediatric protocols — benzodiazepines first, then phenytoin, levetiracetam, or valproate, with the same dosing and airway priorities as any other child. The second is an acute behavioural disturbance in a child with severe intellectual disability and sensory overload, where the priority is environmental de-escalation, removal of triggers, and behavioural support before any pharmacological adjunct. [1]
In both, the underlying principle is that fragile X does not change the resuscitation algorithm — it changes the threshold to act. A child with limited communication may present seizures as behavioural change, sleep disruption, or regression, so a low threshold for EEG and for treating an identifiable seizure burden is warranted. Behavioural crises are managed first with safety, sensory regulation, and a calm environment, reserving short-acting medication for genuine risk. [1] [4]
Management — Definitive & Stepwise
Definitive management is a five-step framework that a fellowship candidate can recite and a general practitioner can deliver: confirm the molecular diagnosis, assemble a multidisciplinary baseline assessment, build developmental and educational support, treat comorbidities with behavioural and pharmacological tools, and run cascade testing with genetic counselling. No step is curative, but each changes the trajectory. [1] [10]

Developmental and educational support is the backbone: early intervention from diagnosis, individualised education plans, speech and language therapy, occupational therapy for sensory and motor needs, and augmentative and alternative communication where speech is limited. Behavioural management uses structured routines, visual supports, and applied behaviour analysis principles, delivered in a neurodiversity-affirming frame that builds on the child's strengths rather than framing difference as deficit. [1]
Pharmacological treatment is strictly comorbidity-led and never aimed at the core defect. Stimulants or alpha-2 agonists help ADHD symptoms, SSRIs help anxiety, and anticonvulsants control epilepsy, each titrated carefully because children with fragile X can be medication-sensitive. Targeted disease-modifying agents remain investigational, and no trial to date has produced an approved curative therapy, so families should be counselled honestly and protected from unsupported treatments. [6] [8]
M.A.C.R.O. \u2014 the fragile X examination set
Specific Subtypes & Scenarios
The affected girl is a scenario where under-diagnosis is the chief pitfall. Because of X-inactivation, girls with a full mutation are usually more mildly affected than boys, presenting with learning difficulty, shyness, selective mutism, or anxiety rather than overt intellectual disability. The reflex to test only boys in a fragile X family misses these girls, who still need educational support, reproductive counselling, and surveillance. [1] [5]
The mosaic child carries a mixture of methylated full-mutation and unmethylated premutation or normal-size cells, and the phenotype is correspondingly variable — sometimes milder than a classic full mutation because some FMRP is produced. Molecular testing must include methylation analysis to detect mosaicism, because PCR repeat sizing alone may misclassify the allele. The mosaic result still carries the same cascade-testing implications for the family. [3] [5]
The premutation carrier is a child or adolescent who tests positive for 55 to 200 repeats without intellectual disability. Most premutation children develop normally, but a subset show subtle neurodevelopmental features, anxiety, or ADHD, and a smaller group develop fragile X-associated tremor/ataxia syndrome in adulthood. Female premutation carriers risk fragile X-associated primary ovarian insufficiency, so the premutation result is an entry into lifelong surveillance, not a reassuring negative. [9]
Complications & Pitfalls
The complications divide into the clinical comorbidities that accumulate over a lifetime and the cognitive traps that cost marks. The clinical comorbidities are epilepsy, autism, ADHD, anxiety, sensory processing differences, sleep disturbance, mitral valve prolapse, recurrent otitis media, and the orthopaedic consequences of joint laxity. Each is treatable in its own right, and proactive surveillance prevents the secondary disability that neglect of comorbidity produces. [1] [4]
The chief cognitive trap is failure to test. Clinicians ration fragile X testing on the grounds that the result "would not change management", but this is wrong on three counts: the molecular diagnosis unlocks educational funding and support, it enables reproductive counselling, and it identifies at-risk relatives through cascade testing. The second trap is failing to test the family once an index case is confirmed, leaving carriers and affected relatives undiagnosed. The third is overpromising a cure, steering families toward unsupported treatments when honest counselling about supportive, evidence-based care is the better service. [1] [10]
Prognosis & Disposition
Prognosis is determined by the severity of intellectual disability, the burden of comorbidity, and — most powerfully — the quality and timing of support. A child diagnosed early, enrolled in early intervention, and supported through individualised education, behavioural therapy, and comorbidity treatment can achieve substantial functional independence, though most affected males will need lifelong support to varying degrees. Girls are typically more mildly affected and more likely to live and work independently. [1]
Life expectancy is near-normal in the absence of severe comorbidity, and the leading modifiers of outcome are seizure control, behavioural and mental health support, and educational inclusion. Disposition is shared, lifelong, medical-home care: the general paediatrician or GP owns the coordination and preventive care, the genetics service owns the counselling and cascade testing, and allied health and education own the developmental and behavioural support. Every transition — into school, into adolescence, and into adult services — is a point at which support can be lost, so the plan must travel with the child. [1] [10]
Special Populations
The same fragile X diagnosis behaves differently across populations because access, recognition, and service models are unevenly distributed. In remote and Indigenous communities, later presentation, limited genetic service access, and lower rates of cascade testing mean that affected children and their relatives are diagnosed late, if at all, and culturally safe genetic counselling is essential. In migrant, refugee, and asylum-seeking families, language barriers, incomplete family histories, and different prior test records complicate the pedigree and the counselling, and an interpreter must be used at every key consultation. [1]
In families managing complex disability, fragmentation of care is the chief threat to support; a written, shared care plan reconciled at every visit is the intervention that matters. In adolescents transitioning to adult care, the move is a high-risk point for loss of behavioural, educational, and mental-health support, so the transition plan must explicitly hand over the care plan and the genetic record. Affected girls and women are a special population in their own right, at risk of under-diagnosis and, if premutation carriers, of fragile X-associated primary ovarian insufficiency. [9]
Evidence, Guidelines & Regional Differences
The evidence base rests on three pillars: consensus clinical guidelines, molecular epidemiology, and the mechanistic and therapeutic literature. The AAP health supervision guideline (Hersh and Saul, 2011) sets the structure of surveillance, comorbidity screening, and family counselling that most national programmes adopt, and the Indian Academy of Pediatrics consensus demonstrates how the same framework is adapted in a different resource setting. [1] [10]
The molecular epidemiology rests on the systematic review by Hunter and colleagues, which pooled the global prevalence of full-mutation and premutation alleles, and on the large newborn screening studies, including the 51,000-infant Chinese cohort, that have confirmed carrier frequencies and informed screening policy. The mechanistic and therapeutic literature — the mGluR theory, the synaptic-pathology reviews, and the targeted-treatment trials — explains the biology but has not yet delivered a curative drug, which is the honest framing for families. [2] [3] [6] [7]
In Australia and New Zealand, fragile X testing is publicly funded when clinical criteria are met, and genetic counselling is delivered through state clinical genetics services with access in metropolitan, regional, and (by outreach or telehealth) remote settings. Cascade testing of at-risk relatives is coordinated through the genetics service once an index case is confirmed, and the Australian Immunisation Handbook and disability support frameworks (the NDIS in Australia, and the equivalent in New Zealand) fund the allied health and educational supports that anchor long-term care. Always confirm the current local eligibility criteria for genetic testing and disability funding, as these change.
[1][10]Exam Pearls
A fellowship candidate answering on fragile X syndrome should land five anchor points and avoid three classic traps. The anchors are the molecular definition (CGG expansion over 200, methylation, silenced FMR1, absent FMRP), the two-part investigation (PCR plus methylation analysis, not karyotype or microarray alone), the premutation-to-full-mutation maternal-expansion rule, the supportive comorbidity-driven management with no curative drug, and the cascade-testing obligation to the family. The traps are failing to test, waiting for the face, and overpromising a cure. [1] [3]
References
- [1]Hersh JH, Saul RA, Committee on Genetics. Health supervision for children with fragile X syndrome. Pediatrics, 2011.PMID 21518720
- [2]Hunter J, Rivero-Arias O, Angelov A, Kim E, Gethin Lynch, Noakes K, et al. Epidemiology of fragile X syndrome: a systematic review and meta-analysis. Am J Med Genet A, 2014.PMID 24700618
- [3]Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol, 2012.PMID 22017584
- [4]Maurin T, Zongaro S, Bardoni B. Fragile X Syndrome: from molecular pathology to therapy. Neurosci Biobehav Rev, 2014.PMID 24462888
- [5]Pirozzi F, Tabolacci E, Neri G. The FRAXopathies: definition, overview, and update. Am J Med Genet A, 2011.PMID 21739597
- [6]Protic D, Salcedo-Arellano MJ, Dy JB, Potter LA, Hagerman RJ. New targeted treatments for fragile X syndrome. Curr Pediatr Rev, 2019.PMID 31241016
- [7]Zhang JY, Wu DW, Yang RL, Liu HY, Du YS, Ma GX, et al. FMR1 allele frequencies in 51,000 newborns: a large-scale population study in China. World J Pediatr, 2021.PMID 34738199
- [8]Castagnola S, Bardoni B, Maurin T. The search for an effective therapy to treat fragile X syndrome: dream or reality? Front Synaptic Neurosci, 2017.PMID 29163124
- [9]Hagerman R, Hagerman P, Chudley AE, Tassone F, Huls HM, Wang JY, et al. Insight and recommendations for fragile X-premutation-associated conditions from the Fifth International Conference. Cells, 2023.PMID 37759552
- [10]Sachdeva A, Jain P, Gunasekaran V, Mahay H, Aneja S, Mathur N, et al. Consensus statement of the Indian Academy of Pediatrics on diagnosis and management of fragile X syndrome in India. Indian Pediatr, 2019.PMID 30954995