Paeds · genetics-dysmorphology-and-metabolism
Genetic deafness and blindness syndromes
Also known as Syndromic hearing loss · Usher syndrome · Pendred syndrome · Waardenburg syndrome · Jervell and Lange-Nielsen syndrome · Branchio-oto-renal syndrome · Leber congenital amaurosis · Norrie disease · Connexin 26 deafness · Dual sensory loss
A fellowship approach to genetic deafness and blindness syndromes: recognise the infant who fails newborn hearing screening as a candidate for GJB2 and Usher evaluation, build the diagnostic tier from audiology and ophthalmology through targeted gene panels, separate Usher as the dominant dual-sensory cause from single-sensory syndromes like Pendred, Waardenburg, Jervell and Lange-Nielsen, branchio-oto-renal, Leber congenital amaurosis and Norrie disease, and deliver early cochlear implantation, voretigene neparvovec for RPE65 retinal dystrophy, and cascade family testing.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The mark-winning candidate holds three layers together. The first is the child's sensory phenotype: what can they hear, what can they see, and at what age did the loss appear. The second is the molecular mechanism: a gap-junction protein, a motor protein, an ion channel, or a retinal visual-cycle enzyme, each pointing to a specific gene and inheritance pattern. The third is the hidden complication: a long QT interval, a renal anomaly, a goitre, or a progressive retinal degeneration that has not yet declared itself. Missing any layer is the classic examination error. [2] [4]
Overview & Definition
Genetic deafness and blindness syndromes are inherited disorders in which a single pathogenic gene variant produces sensorineural hearing loss, retinal or ocular blindness, or both. They are the dominant cause of permanent childhood sensory impairment once environmental factors such as congenital cytomegalovirus and prematurity are excluded, and they cluster into syndromic forms, where the sensory loss is accompanied by a second organ system, and nonsyndromic forms, where the ear or eye is the only site. [1] [2]
Hearing loss is the entry point for most of these children, because universal newborn hearing screening identifies congenital deafness within the first month of life. Roughly half of permanent childhood hearing loss is genetic, and of the genetic cases around 70 per cent are nonsyndromic with GJB2 as the single largest contributor, while 30 per cent are syndromic and carry the extra-organ findings that make the diagnosis. [1] [3]
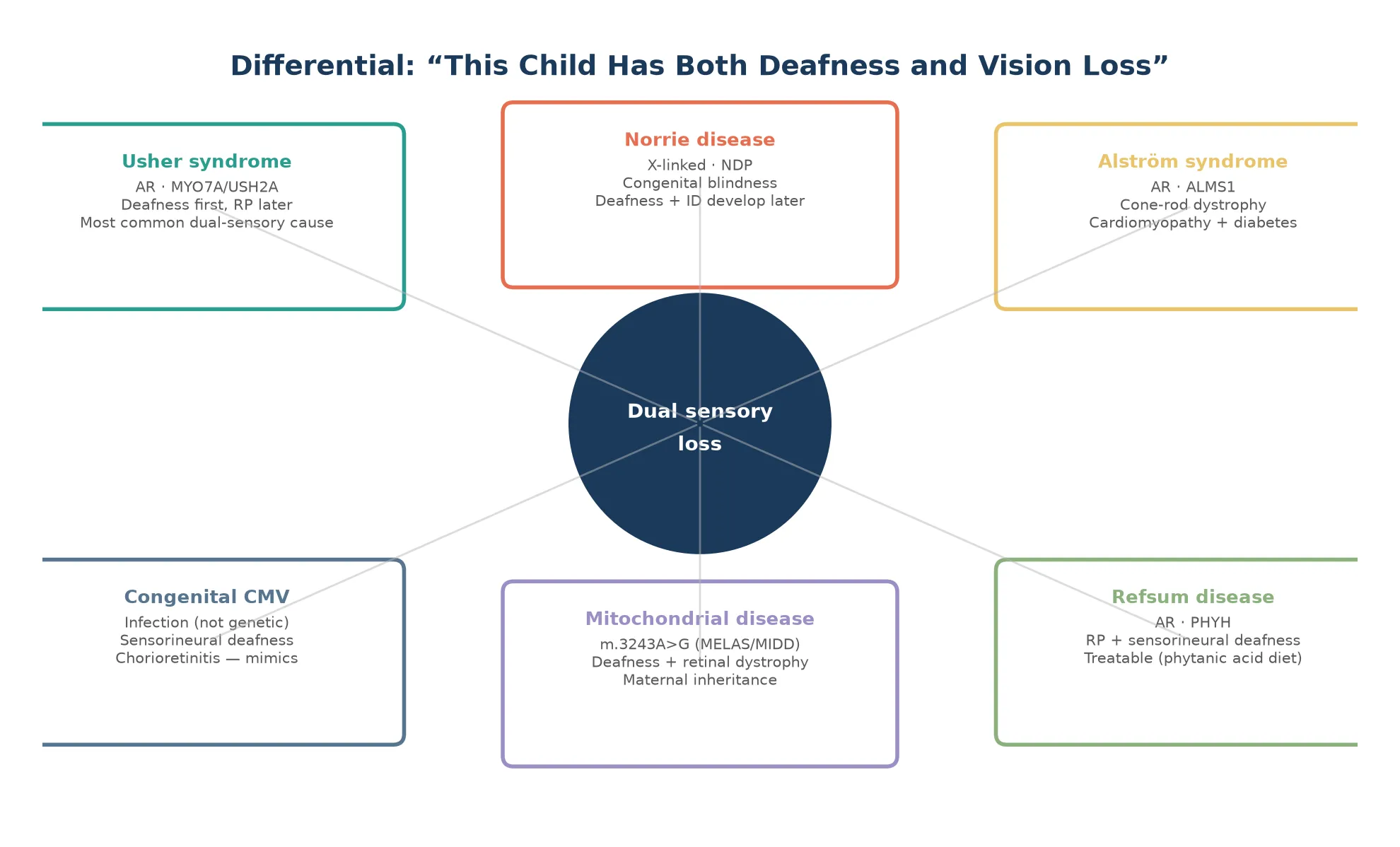
Vision loss enters through the paediatric eye clinic, where the combination of congenital nystagmus, poor visual fixation, and a roving eye movement pattern raises Leber congenital amaurosis or Norrie disease. The crossover — a child who is both deaf and blind — is Usher syndrome territory, and recognising the crossover early is the single highest-yield clinical decision in this whole family of conditions. [4] [9]
Classification
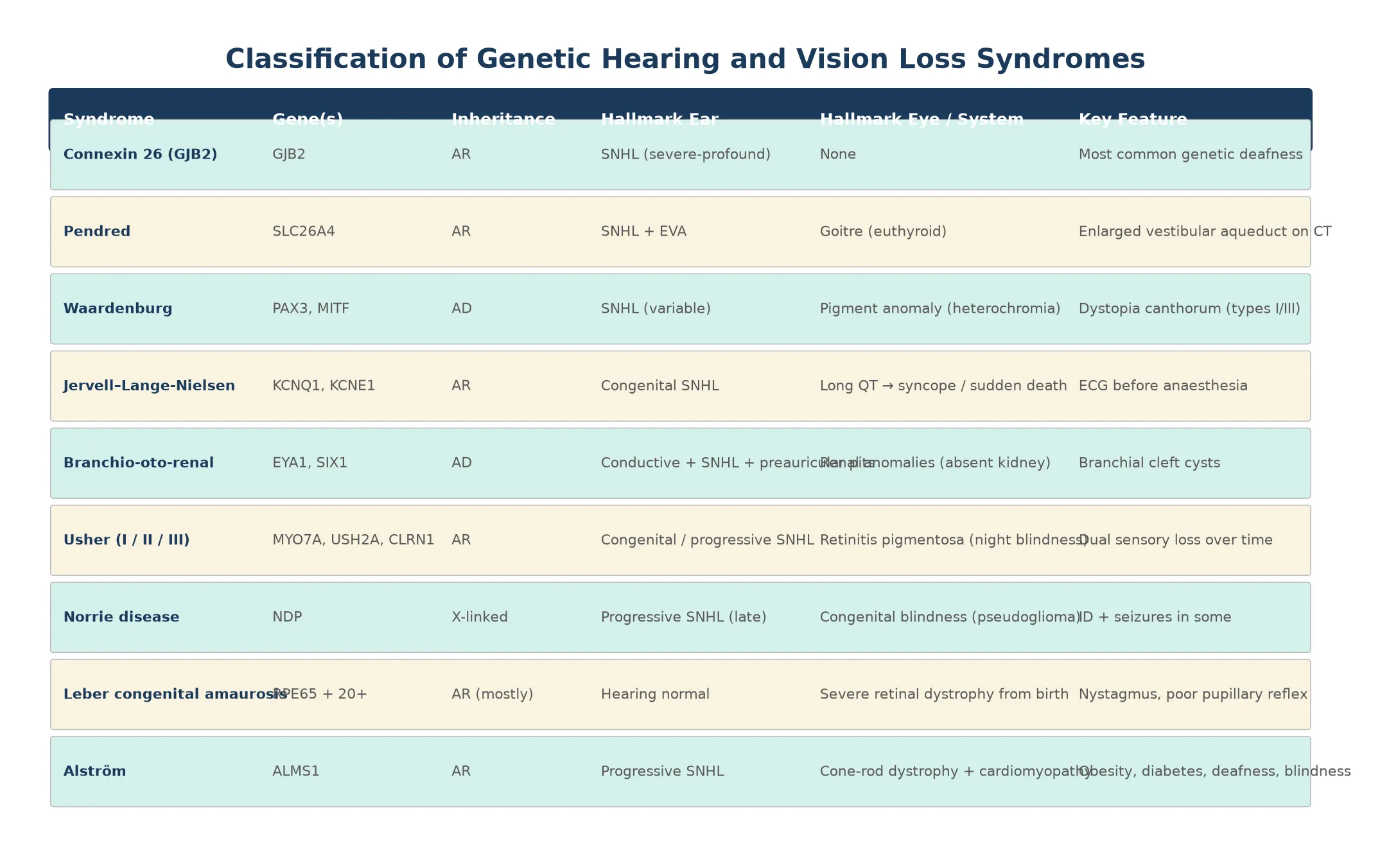
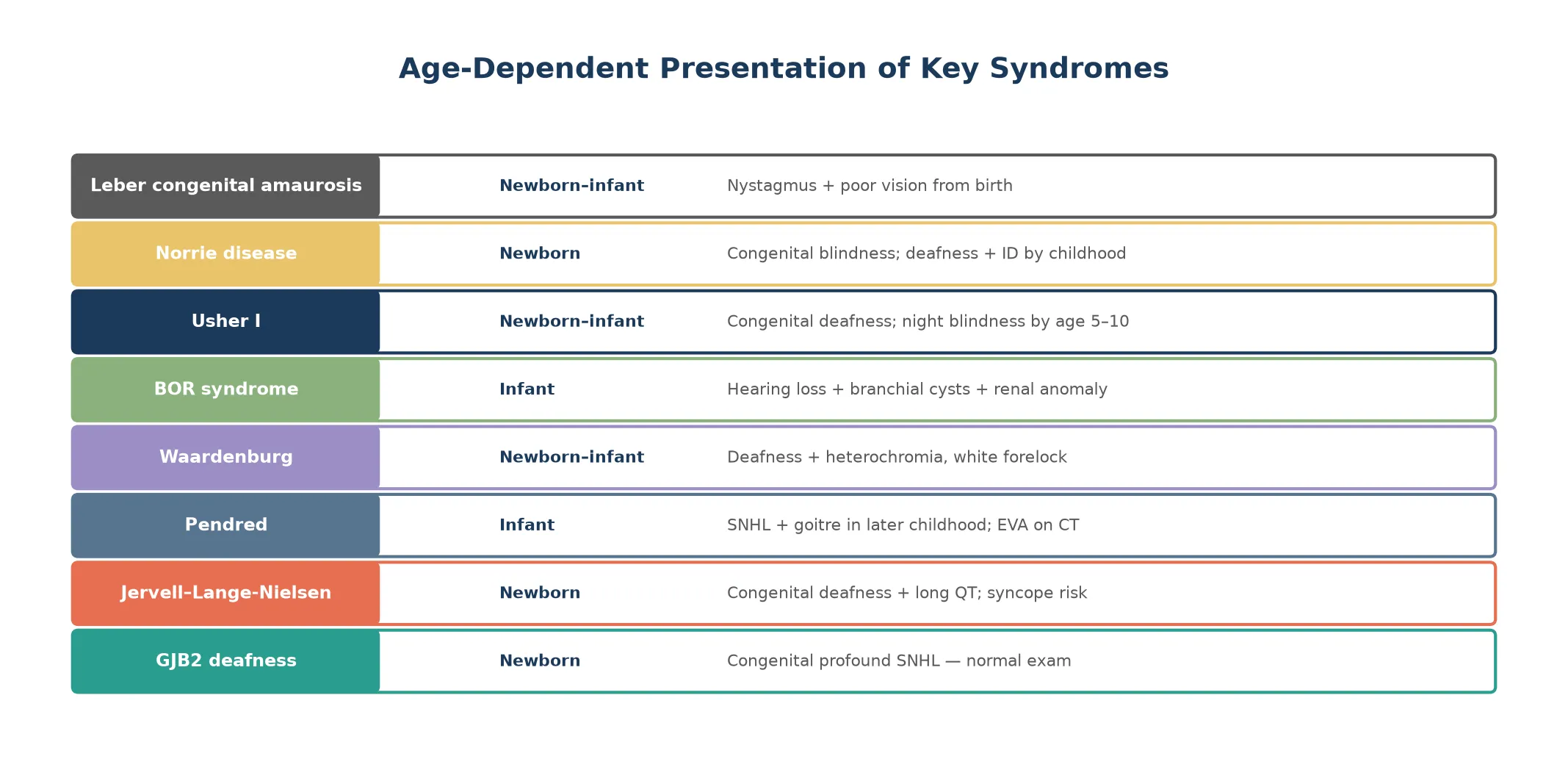
These syndromes separate cleanly into three clinical groups. The first is nonsyndromic genetic deafness, led by GJB2 connexin-26 variants, which cause isolated congenital sensorineural hearing loss with no other organ involvement and normal imaging. The second is syndromic deafness, where the hearing loss travels with a second system: Pendred with thyroid goitre and enlarged vestibular aqueduct, Waardenburg with pigmentary anomalies, Jervell and Lange-Nielsen with long QT, and branchio-oto-renal with branchial clefts and kidney anomalies. The third is genetic blindness, led by Leber congenital amaurosis and Norrie disease, where retinal or ocular maldevelopment causes congenital visual impairment. [2] [6]
Usher syndrome spans the boundary between deafness and blindness, which is why it deserves its own category in clinical reasoning. A child who presents with congenital hearing loss from an Usher type I variant will not show retinitis pigmentosa until age five to ten, yet the molecular diagnosis is available from birth. The classification that governs practice is therefore molecular rather than purely clinical, because the gene name sets both the surveillance plan and the reproductive counselling. [4]
Syndromic hearing loss — the Go PITWaJB set
Epidemiology & Risk Factors
Permanent childhood hearing loss affects roughly one to three per thousand newborns, and in high-risk groups — neonatal intensive care graduates, those with a family history, and consanguineous families — the rate is tenfold higher. Genetic causes account for at least half of all cases, and of the genetic fraction, autosomal-recessive inheritance dominates because most pathogenic variants are carried silently in the population at a combined carrier frequency that makes recessive deafness the most common Mendelian disorder in many populations. [1] [3]
The single largest contributor to nonsyndromic genetic deafness is GJB2, which encodes connexin 26 and underlies up to 20 per cent of all congenital sensorineural hearing loss in many populations. GJB2 deafness is typically prelingual, severe to profound, and non-progressive, and because it is autosomal recessive the recurrence risk for a couple with one affected child is 25 per cent. The ClinGen Hearing Loss Expert Panel has refined variant interpretation so that clinically actionable GJB2 variants can now be reported with confidence. [3] [2]
Usher syndrome has an estimated prevalence of about one in 6,000 to one in 10,000 and accounts for roughly half of the deaf-blind population. Pendred syndrome is the most common syndromic form of deafness with goitre. Waardenburg syndrome affects roughly one in 40,000 and is the commonest cause of syndromic deafness with pigmentary anomalies. The branchio-oto-renal and Jervell and Lange-Nielsen syndromes are rarer, but their extra-organ complications — renal failure and sudden cardiac death respectively — make early detection life-saving. [4] [8]
In remote, rural, and Indigenous communities, and in migrant and refugee families, several barriers compound: newborn hearing screening follow-up rates are lower, access to audiology and genetic services is geographically constrained, and language and cultural differences between the Deaf community and the hearing medical establishment can impede counselling. These children are diagnosed later, receive cochlear implants outside the critical neuroplastic window, and miss cascade testing — which widens the outcome gap that early identification is designed to close. Telehealth-enabled genetic counselling and mobile audiology outreach are narrowing this gap in Australia and New Zealand. [1]
Pathophysiology
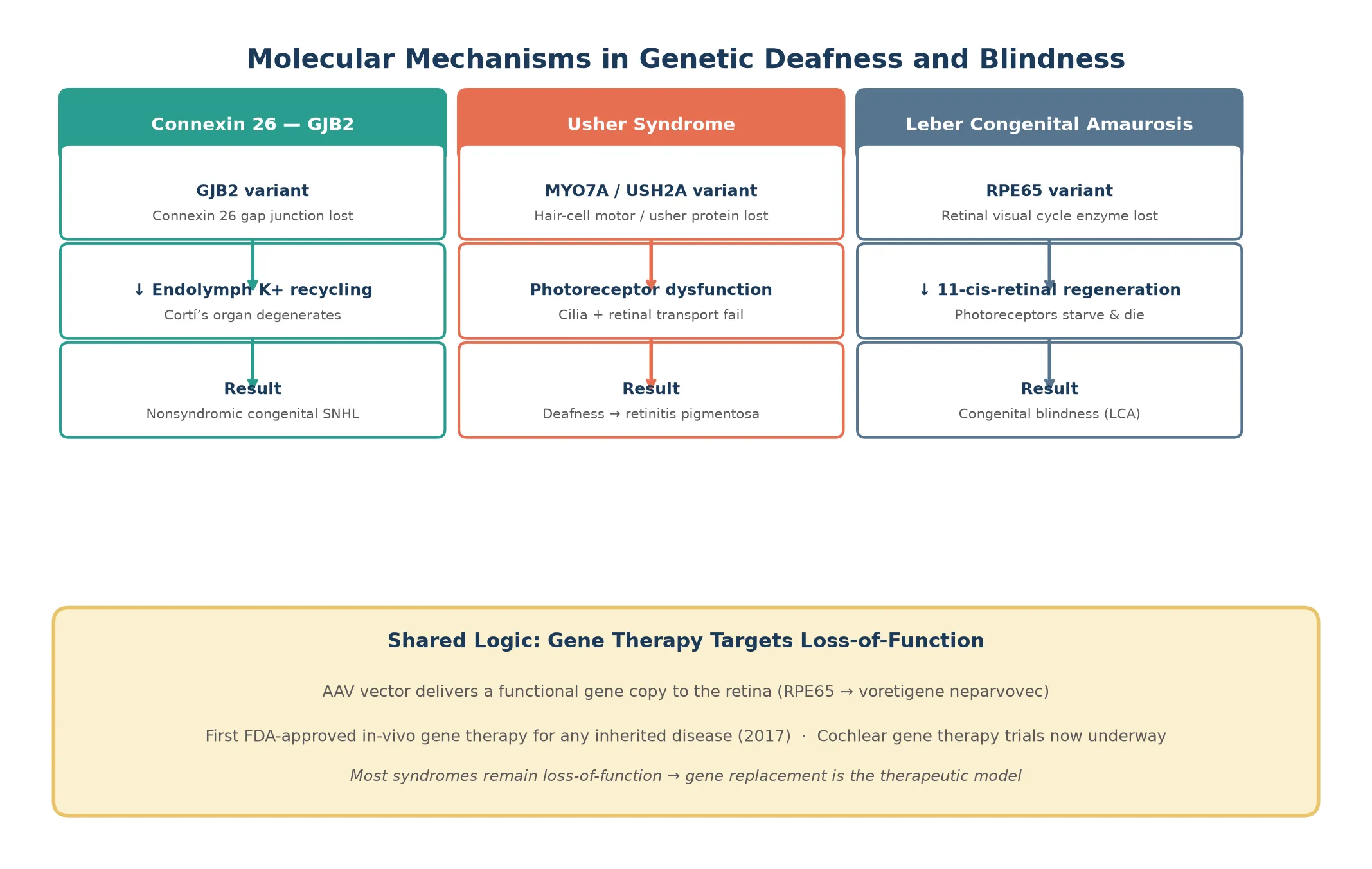
Each syndrome reflects what the affected protein does in the sensory organ. Connexin 26, the product of GJB2, forms gap junctions between cochlear supporting cells that recycle potassium ions away from the hair-cell synapse after each sound stimulus. When connexin 26 is absent, potassium accumulates and the organ of Corti degenerates, producing congenital profound hearing loss without any structural abnormality on imaging. [3]
Pendrin, the product of SLC26A4, is an iodide and bicarbonate transporter expressed in the thyroid, the inner ear, and the endolymphatic sac. Loss of pendrin disrupts endolymph homeostasis, enlarging the vestibular aqueduct and causing progressive or fluctuating sensorineural hearing loss, while in the thyroid it impairs organification of iodide and produces a goitre that is usually euthyroid in childhood. The enlarged vestibular aqueduct seen on temporal bone CT is the radiographic hallmark. [5]
Waardenburg syndrome arises from variants in transcription factors that govern melanocyte development, principally PAX3 and MITF. Melanocytes are required both for pigmentation of skin, hair and iris and for normal function of the stria vascularis in the inner ear, so their loss produces the characteristic pigmentary anomalies — white forelock, heterochromia iridis, dystopia canthorum — alongside variable sensorineural hearing loss. [6]
Jervell and Lange-Nielsen syndrome is a cardiac-channelopathy disguised as a deafness syndrome. Variants in KCNQ1 or KCNE1 disable the slow delayed-rectifier potassium current (IKs) in both the heart and the stria vascularis, producing congenital profound deafness and a prolonged QT interval that carries a high risk of torsades de pointes and sudden death. The cardiac risk is the reason every infant with congenital hearing loss of unknown cause should have an ECG. [7]
The genetic blindness syndromes follow the same loss-of-function logic. In Leber congenital amaurosis, variants in any of more than 20 genes disrupt the retinal visual cycle or photoreceptor development, most classically RPE65, whose enzyme regenerates 11-cis-retinal after phototransduction. Without RPE65, the photoreceptors cannot respond to light and progressively die, producing nystagmus and severe visual impairment from birth. Norrie disease is caused by NDP variants on the X chromosome and disrupts norrin, a signalling protein essential for retinal and cochlear vascular development, producing congenital blindness with later-onset hearing loss and intellectual disability in some boys. [9] [10]
Clinical Presentation
The presentation is age-dependent and the age of onset is itself diagnostic. A child with GJB2 deafness presents at the newborn hearing screen with a refer result and a completely normal physical examination, which is why a normal examination never excludes genetic deafness. The diagnostic auditory brainstem response confirms severe-to-profound bilateral sensorineural hearing loss, and temporal bone imaging is normal. [1] [3]
Pendred syndrome presents with hearing loss that is often prelingual but can be progressive or fluctuating, particularly after minor head trauma. The thyroid goitre does not appear until later childhood or adolescence and is usually biochemically euthyroid, so its absence in a young child never excludes the diagnosis. The discriminating investigation is temporal bone imaging showing an enlarged vestibular aqueduct or Mondini dysplasia. [5]
Waardenburg syndrome is recognisable at the bedside from the pigmentary phenotype. A child with a white forelock, heterochromia iridis, or widely spaced inner canthi (dystopia canthorum) who also has hearing loss carries the diagnosis until proven otherwise. The hearing loss is variable — present in some, absent in others within the same family — which reflects the incomplete penetrance and variable expressivity of the melanocyte developmental pathway. [6]
Jervell and Lange-Nielsen syndrome presents as congenital profound deafness with episodes of syncope or seizure triggered by exertion or emotion. The syncope reflects the prolonged QT interval and the risk of ventricular arrhythmia, and sudden death may be the first presentation in an undiagnosed child. This is why an ECG belongs in the initial workup of every congenitally deaf infant. [7]
Branchio-oto-renal syndrome presents with a triad of branchial cleft cysts or sinuses, ear malformations including preauricular pits and hearing loss of any type, and renal anomalies that range from mild hypoplasia to complete renal agenesis. The renal component may be clinically silent, so a renal ultrasound is mandatory when the branchial and ear findings are present. [8]
Usher syndrome presents in two waves. The first is congenital or early-onset sensorineural hearing loss, which may be profound (type I), moderate (type II), or progressive (type III). The second wave is retinitis pigmentosa, which appears years to decades later as night blindness, progressive visual field constriction, and eventually tunnel vision and blindness. The electroretinogram is abnormal before symptoms appear, which is why it belongs in the surveillance of every deaf child of unknown cause. [4]
Leber congenital amaurosis presents from birth with nystagmus, poor visual fixation, eye pressing or poking (the oculo-digital sign), and severely reduced or absent pupillary reflexes. The electroretinogram is extinguished or markedly reduced, and fundus examination may be normal early on. Norrie disease presents in boys with congenital blindness from retinal dysplasia or pseudoglioma, followed by progressive hearing loss and, in some, intellectual disability and seizures. [9] [10]
Differential Diagnosis
The differential of congenital sensorineural hearing loss is dominated by two entities that must be separated early: GJB2-related genetic deafness and congenital cytomegalovirus infection. Congenital CMV is the most common non-genetic cause of sensorineural hearing loss, and because it is treatable with antiviral therapy when identified in the first month of life, CMV PCR on saliva or urine belongs in the early workup of every infant who fails screening. [2]
When the hearing loss is syndromic, the differential turns on the extra-organ finding. A goitre and enlarged vestibular aqueduct point to Pendred; pigmentary anomalies point to Waardenburg; syncope or a family history of sudden death points to Jervell and Lange-Nielsen; branchial remnants and preauricular pits point to BOR; and progressive visual loss points to Usher. Each finding narrows the differential to one or two genes and directs the targeted panel. [5] [6]
Congenital CMV versus GJB2 deafness
The differential of the deaf-blind child converges on Usher syndrome but must exclude Alstrom syndrome (cone-rod dystrophy with cardiomyopathy and diabetes), Refsum disease (treatable retinitis pigmentosa with deafness and neuropathy), mitochondrial disorders such as the m.3243A greater than G variant, and congenital rubella syndrome in unvaccinated populations. Norrie disease enters when the blindness is congenital and X-linked. [4] [10]
Clinical & Bedside Assessment
A structured assessment begins with the audiology and ends with the whole child. Confirm the hearing loss with auditory brainstem response testing in infants and behavioural audiometry in older children, characterise it as sensorineural or conductive, and grade its severity. Examine the external ears for malformations and preauricular pits, the neck for branchial remnants, the eyes for pigmentary anomalies, and the skin for café-au-lait or depigmented patches. [2] [8]
The ECG is non-negotiable in any infant with congenital sensorineural hearing loss of unknown cause, because a prolonged QT interval identifies Jervell and Lange-Nielsen syndrome and prevents sudden cardiac death. Plot the corrected QT interval against age-appropriate norms, and remember that the cardiac risk persists even after the hearing loss is managed. [7]
Ophthalmology assessment includes fundoscopy, electroretinography when retinal dystrophy is suspected, and optical coherence tomography for structural detail. Every deaf child of unknown cause should have a baseline ophthalmology review by school age, because the retinitis pigmentosa of Usher syndrome is clinically silent for years but detectable on the electroretinogram. [4]
Take a three-generation pedigree focusing on hearing loss, vision loss, sudden death, thyroid disease, and renal problems, and examine the parents for subtle features such as dystopia canthorum or preauricular pits. Consanguinity raises the prior probability of autosomal-recessive disease and should be asked about directly and respectfully. [1]
Investigations
The investigation ladder moves from bedside to molecular. Begin with diagnostic ABR and OAE to confirm the type and degree of hearing loss, then temporal bone CT or MRI to look for enlarged vestibular aqueduct or cochlear malformation. Send CMV PCR on saliva or urine within the first three weeks of life, because after that congenital infection cannot be distinguished from postnatal acquisition. [2]

GJB2 sequencing is the highest-yield single genetic test for congenital nonsyndromic sensorineural hearing loss. When GJB2 is negative, a targeted multi-gene hearing-loss panel — covering SLC26A4, MYO7A, USH2A, PAX3, MITF, KCNQ1, EYA1 and others — is the next step, and whole-exome or whole-genome sequencing is reserved for panel-negative cases with a strong genetic suspicion. For vision loss, a targeted retinal dystrophy gene panel including RPE65, NDP, and the Usher genes is the molecular front line. [3] [9]
The thyroid workup for suspected Pendred syndrome includes thyroid function tests and thyroid ultrasound, but the goitre may be absent in childhood, so a normal thyroid examination does not exclude the diagnosis. For BOR syndrome, renal ultrasound identifies the renal component, and an ECG excludes the long QT of Jervell and Lange-Nielsen syndrome in any child with congenital deafness. [5] [8]
Management — Resuscitation
The resuscitation scenarios in this family of conditions are few but life-threatening. The first is the syncopal or cardiac arrest episode in an undiagnosed Jervell and Lange-Nielsen child, which requires standard paediatric advanced life support with attention to avoiding QT-prolonging drugs, followed by beta-blockade and consideration of an implantable cardioverter-defibrillator. Any anaesthetic in a child with congenital hearing loss of unknown cause must be preceded by an ECG. [7]
The second scenario is the acute visual deterioration in a child with known retinal dystrophy, which may reflect cystoid macular oedema or retinal detachment rather than disease progression, and warrants urgent ophthalmology review. The third is the recognition of a metabolic decompensation in syndromes such as Alstrom or Refsum where systemic disease accompanies the sensory loss, which requires the standard metabolic emergency protocol of fluids, glucose, and specialist consultation. [4]
Management — Definitive & Stepwise
The definitive management framework runs in five steps. Confirm the sensory loss and grade it, identify the molecular cause through tiered genetic testing, deliver the syndrome-specific intervention, screen for the extra-organ complication, and provide family-centred support and genetic counselling. The first three steps have a time-critical window because neural plasticity for language and vision is greatest in the first two to three years of life. [1] [2]
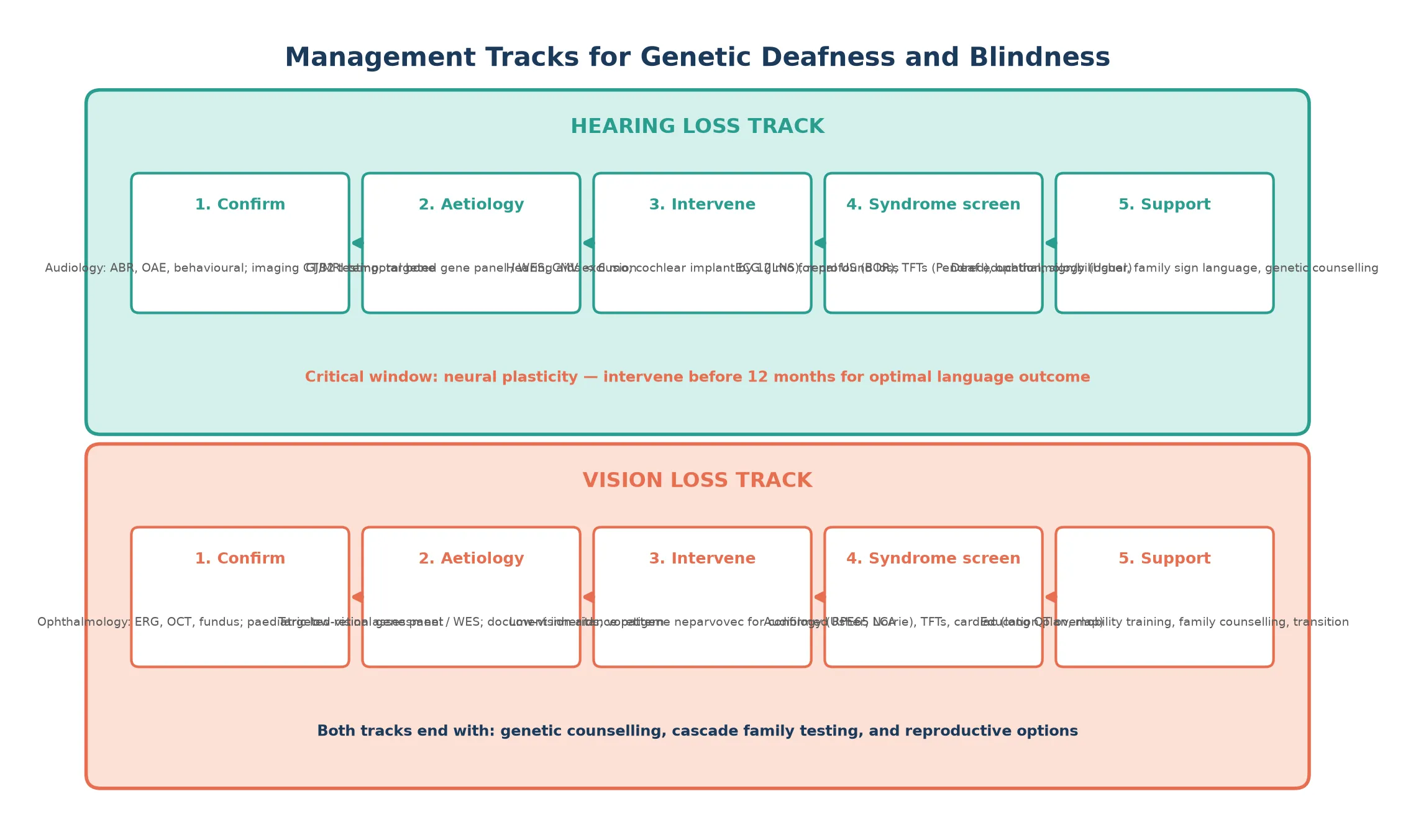
For hearing loss, the definitive interventions are hearing aids for moderate loss fitted before six months of age, and cochlear implantation for severe-to-profound loss undertaken by twelve months for optimal language outcome. The choice between spoken-language and sign-bilingual communication is a family-centred decision that should be supported, not steered, and both pathways can produce excellent outcomes with early intervention. Cochlear implant candidacy is broad and includes most syndromic forms of deafness provided the auditory nerve is intact. [2]
For vision loss from confirmed RPE65-related Leber congenital amaurosis, voretigene neparvovec is the definitive therapy, delivered as a single subretinal injection in both eyes, and is most effective in children who still have surviving photoreceptors. For other genetic retinal dystrophies, management is low-vision support, mobility training, and enrolment in clinical trial pathways. For Norrie disease, management is supportive and surveillance-focused, monitoring for progressive hearing loss and developmental needs. [11] [10]
Jervell and Lange-Nielsen syndrome requires lifelong beta-blocker therapy and, in many cases, an implantable cardioverter-defibrillator. Pendred syndrome may require thyroid replacement if hypothyroidism develops. BOR syndrome requires renal surveillance and, in some cases, surgical management of branchial remnants. In every case, an annual multidisciplinary review coordinates audiology, ophthalmology, genetics, and developmental support. [7] [8]
Specific Subtypes & Scenarios
Usher syndrome type I is the most severe subtype, with congenital profound deafness, vestibular areflexia causing delayed motor milestones, and prepubertal retinitis pigmentosa. These children are candidates for early cochlear implantation and intensive vestibular rehabilitation, and the family must be counselled that progressive blindness is expected. Type II has moderate stable hearing loss with normal vestibular function and later-onset retinitis pigmentosa, and type III has progressive hearing loss with variable vestibular function and adult-onset vision loss. [4]
GJB2 connexin-26 deafness is the prototype of nonsyndromic genetic deafness and the condition most likely to appear in a written examination. It is autosomal recessive, congenital, severe to profound, non-progressive, and has a structurally normal inner ear on imaging. The carrier frequency for common pathogenic GJB2 variants is high in many populations, which explains why the majority of affected children have unaffected parents. The ClinGen expert panel has clarified the pathogenicity of borderline variants such as p.Val37Ile and p.Met34Thr, allowing confident counselling. [3]
Leber congenital amaurosis is genetically heterogeneous, with more than 20 causative genes, but RPE65-related LCA is the therapeutically important subtype because it is the target of voretigene neparvovec. The genotype determines whether gene therapy is an option, which is why molecular diagnosis now carries therapeutic urgency rather than being a confirmatory afterthought. [9] [11]
Complications & Pitfalls
The classic pitfall is attributing congenital deafness to GJB2 or nonsyndromic causes without checking for the syndromic possibilities. A child discharged as nonsyndromic who later collapses from a long QT arrhythmia is the failure that an ECG would have prevented. Equally dangerous is the child with Usher syndrome whose retinitis pigmentosa goes undetected until vision is irreversibly lost, when an electroretinogram at school age would have made the diagnosis years earlier. [4] [7]
A second pitfall is assuming that a normal thyroid examination excludes Pendred syndrome. The goitre develops late and is often euthyroid, so the diagnosis rests on the enlarged vestibular aqueduct and SLC26A4 testing, not on the thyroid. A third is missing the renal component of branchio-oto-renal syndrome, which can lead to undiagnosed renal hypoplasia progressing to renal failure. [5] [8]
The fourth and most subtle pitfall is failing to offer cascade testing. In an autosomal-recessive condition with a 25 per cent recurrence risk, and in autosomal-dominant conditions with a 50 per cent risk, every first-degree relative of a confirmed case deserves genetic assessment. Missing this step means the next affected sibling is born without the benefit of early diagnosis. [1]
Prognosis & Disposition
The prognosis is overwhelmingly determined by the age at diagnosis and intervention. Children with GJB2 deafness who receive cochlear implants within the first year achieve spoken-language outcomes within one standard deviation of hearing peers, and those enrolled in bilingual sign-and-spoken-language programmes have equally strong developmental trajectories. Children diagnosed after age three face a steeper climb regardless of the intervention chosen. [2]
For the genetic blindness syndromes, the prognosis depends on the gene and the availability of therapy. RPE65-related Leber congenital amaurosis treated with voretigene neparvovec shows measurable and durable improvement in visual function and mobility. Other retinal dystrophies follow a progressive course, but early diagnosis enables education planning, mobility training, and enrolment in emerging gene-therapy trials. Norrie disease carries a guarded prognosis for vision but supportive management of hearing and development can substantially improve quality of life. [11] [10]
Jervell and Lange-Nielsen syndrome carries the gravest prognosis among the syndromic forms because of the cardiac risk, but beta-blockade and implantable defibrillators substantially reduce sudden death. Pendred syndrome has an excellent prognosis with appropriate hearing intervention and thyroid surveillance, and BOR syndrome's prognosis is determined by the renal component. In every case, the disposition is lifelong multidisciplinary care with a planned transition to adult genetics and sensory-loss services. [7] [8]
Special Populations
In remote, rural, and Indigenous communities, the barriers to early diagnosis compound. Newborn hearing screening may not be available or follow-up may be incomplete, audiology services are geographically distant, and genetic counselling may not be offered in the family's first language. These children are diagnosed later, receive intervention outside the critical window, and miss cascade testing. Telehealth-enabled genetic counselling, mobile audiology outreach, and partnerships with Indigenous health workers are narrowing this gap in Australia and New Zealand. [1]
Children in migrant and refugee families face similar barriers compounded by displacement, interrupted medical records, and cultural and linguistic diversity in how deafness is understood and discussed. The Deaf community itself is a cultural and linguistic minority, and culturally competent care respects sign language as a complete language and Deaf identity as a valid cultural identity rather than a deficit to be repaired. Genetic counselling must be conducted in the family's preferred language with a qualified interpreter, including sign-language interpreters for Deaf parents. [2]
Socioeconomic disadvantage intersects with all of these factors. Families who cannot afford the time, transport, or co-payments for repeated specialist visits fall out of care, and the children who drop out are the ones most likely to have progressive or syndromic disease. Wrap-around care coordination and funded access to cochlear implantation and genetic services are the structural interventions that close the outcome gap. [1]
Evidence, Guidelines & Regional Differences
The evidence base for newborn hearing screening and early cochlear implantation is robust and derives from large cohort studies and the landmark review by Morton and Nance, which established that universal screening could identify congenital hearing loss in the first months of life and that early intervention transformed language outcomes. The JAMA review by Lieu and colleagues provides the current framework for childhood hearing loss management, including the role of genetic testing and the timing of intervention. [1] [2]
The gene-therapy evidence for voretigene neparvovec is built on phase III trial data showing sustained improvement in multi-luminance mobility testing in RPE65-related LCA patients, and it established the regulatory pathway for in-vivo gene therapy that is now being extended to other retinal dystrophies and, in early-phase trials, to the cochlea. The ClinGen Hearing Loss Expert Panel's variant-curation work on GJB2 has resolved years of ambiguity about borderline pathogenic variants and standardised clinical reporting. [11] [3]
Regional differences are most visible in screening infrastructure and service access. Australia and New Zealand have well-established universal newborn hearing screening programmes, publicly funded cochlear implantation, and increasingly integrated genomic testing pathways, but the gap between metropolitan and remote services remains. The United States, the United Kingdom, and Canada have comparable screening programmes with variations in funding models for genetic testing and gene therapy. In all regions, the principle is the same: early identification, early molecular diagnosis, early intervention, and cascade family testing. [1]
Exam Pearls
The five examination anchors for genetic deafness and blindness syndromes are: the newborn hearing screen and GJB2, the ECG for Jervell and Lange-Nielsen, Usher syndrome as the dual-sensory diagnosis, Leber congenital amaurosis and voretigene neparvovec, and cascade family testing. If you can speak fluently to these five, you have covered the territory the examiner will probe. [2]
The four classic traps are: attributing deafness to GJB2 without an ECG and missing long QT, discharging a deaf child without an ophthalmology review and missing Usher, assuming a normal thyroid excludes Pendred, and forgetting cascade testing so the next sibling is born without the benefit of early diagnosis. Each trap is avoided by a single protocolised step. [4] [7]
The highest-yield discriminating facts are that GJB2 is autosomal recessive and the most common genetic deafness, that Jervell and Lange-Nielsen is the only one that kills suddenly, that Usher is the only one that causes both deafness and progressive blindness, and that RPE65 LCA is the only one with approved gene therapy. Commit these four to memory and the short-case or viva question is yours. [3] [11]
References
- [1]Morton CC, Nance WE. Newborn hearing screening--a silent revolution. N Engl J Med, 2006.PMID 16707752
- [2]Lieu JEC, Kenna M, Anne S, Davidson L. Hearing Loss in Children: A Review. JAMA, 2020.PMID 33258894
- [3]Shen J, Oza AM, Del Castillo I, Duzkale H, Matsunaga T, Pandya A, et al. Consensus interpretation of the p.Met34Thr and p.Val37Ile variants in GJB2 by the ClinGen Hearing Loss Expert Panel. Genet Med, 2019.PMID 31160754
- [4]Castiglione A, Moller C. Usher Syndrome. Audiol Res, 2022.PMID 35076463
- [5]Wemeau JL, Kopp P. Pendred syndrome. Best Pract Res Clin Endocrinol Metab, 2017.PMID 28648509
- [6]Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat, 2010.PMID 20127975
- [7]Bitner-Glindzicz M, Tranebjaerg L. The Jervell and Lange-Nielsen syndrome. Adv Otorhinolaryngol, 2000.PMID 10868213
- [8]Kochhar A, Fischer SM, Kimberling WJ, Smith RJ. Branchio-oto-renal syndrome. Am J Med Genet A, 2007.PMID 17238186
- [9]Huang CH, Yang CM, Yang CH, Hou YC, Chen TC. Leber's Congenital Amaurosis: Current Concepts of Genotype-Phenotype Correlations. Genes, 2021.PMID 34440435
- [10]De Silva SR, Arno G, Robson AG, Fakin A, Pontikos N, Mohamed MD, et al. The X-linked retinopathies: Physiological insights, pathogenic mechanisms, phenotypic features and novel therapies. Prog Retin Eye Res, 2021.PMID 32860923
- [11]Padhy SK, Takkar B, Narayanan R, Venkatesh P, Jalali S. Voretigene Neparvovec and Gene Therapy for Leber's Congenital Amaurosis: Review of Evidence to Date. Clin Ophthalmol, 2020.PMID 33268999