Paeds · genetics-dysmorphology-and-metabolism
Genomic testing, variant interpretation and counselling
Also known as Genomic medicine · Next-generation sequencing in paediatrics · ACMG/AMP variant classification · Secondary findings in genomic sequencing · Pre- and post-test genetic counselling
A fellowship approach to genomic testing in paediatrics: choose the right test from the hierarchy of karyotype, chromosomal microarray, gene panel, exome, and whole-genome sequencing; apply the ACMG/AMP five-tier variant classification framework to interpret results; manage variants of uncertain significance and secondary findings; and deliver pre- and post-test genetic counselling that equips families for diagnosis, uncertainty, and cascade testing — because the diagnostic yield of genome sequencing in rare paediatric disease now reaches 40 to 50 percent.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who thinks in three layers at once. The first layer is the test and its yield: what question is being asked, which test answers it, and what the diagnostic probability is before the sample is sent. The second is the variant and its classification: the ACMG/AMP framework that converts raw sequence data into a probabilistic call, and the distinction between a pathogenic finding that drives management and a VUS that does not. The third is the family: the consent conversation that precedes testing, the disclosure that follows, the cascade testing of relatives, and the re-analysis that keeps the result alive as knowledge grows. [1] [3]
Overview & Definition
Genomic testing in paediatrics encompasses the range of laboratory techniques used to identify genetic variation underlying congenital anomalies, developmental disorders, and inherited diseases. The hierarchy runs from the traditional karyotype, which detects large-scale chromosomal rearrangements visible under a microscope, through chromosomal microarray (CMA), which detects sub-microscopic copy-number variants (CNVs) at far higher resolution, to next-generation sequencing technologies — gene panels, whole-exome sequencing (WES), and whole-genome sequencing (WGS) — that read the DNA letter by letter and can identify single-nucleotide variants, small insertions and deletions, and some structural rearrangements. [2] [9]
The clinical application of these technologies depends on a structured variant interpretation process that converts millions of sequence differences into a small number of clinically actionable calls. The American College of Medical Genetics and Genomics and the Association for Molecular Pathology published the consensus standards for this process in 2015, establishing a five-tier classification framework — pathogenic, likely pathogenic, variant of uncertain significance, likely benign, and benign — that every clinical genomics laboratory now applies. This framework, combined with structured pre- and post-test genetic counselling, defines the modern practice of genomic medicine in paediatrics. [1] [10]
Classification
The clinical classification that matters most is the ACMG/AMP five-tier variant classification, because it determines whether a variant drives clinical management, surveillance, reproductive counselling, or none of these. The framework assigns each variant to one of five tiers based on the accumulated weight of evidence from population data, computational prediction, functional studies, and segregation analysis. [1]
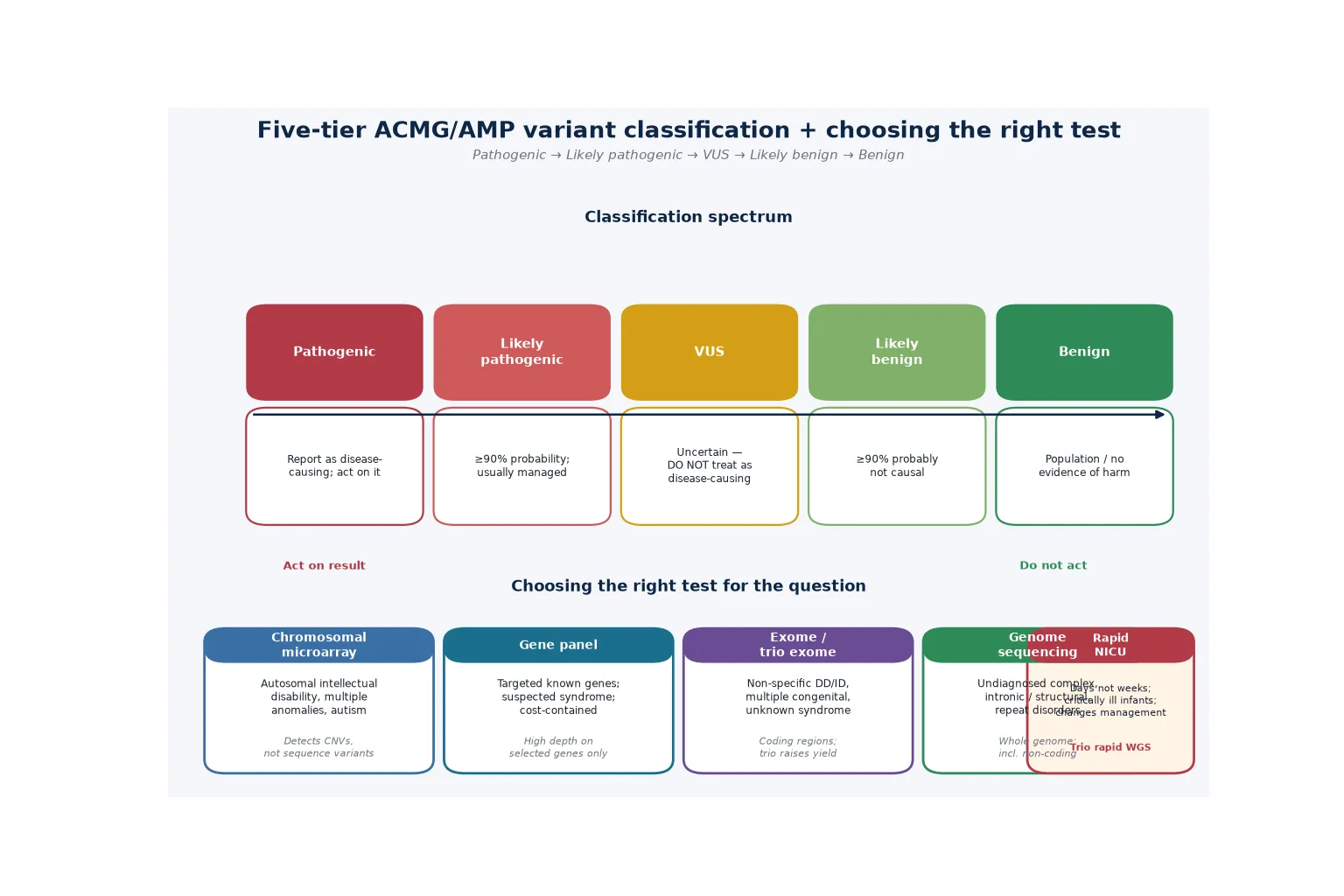
A pathogenic variant carries greater than 99 percent probability of causing disease and typically combines very strong evidence (such as a loss-of-function variant in a gene where haploinsufficiency is the known mechanism) with supporting evidence from multiple independent domains. A likely pathogenic variant carries 90 to 99 percent probability and usually combines strong evidence with moderate or supporting evidence. A variant of uncertain significance (VUS) sits in the middle zone — 5 to 90 percent probability — where the available evidence is insufficient to classify the variant as either pathogenic or benign. A likely benign variant carries 1 to 5 percent probability, and a benign variant carries less than 1 percent probability. [1]
When a laboratory identifies a candidate variant, the question is whether the accumulated evidence tips the balance decisively toward pathogenicity or benignity — and the ACMG/AMP framework answers it with a structured set of evidence codes. Pathogenic evidence codes include PVS1 (very strong, typically a predicted null variant in a gene with a loss-of-function mechanism), PS1 to PS4 (strong), PM1 to PM6 (moderate), and PP1 to PP5 (supporting). Benign evidence codes include BA1 (stand-alone, an allele frequency above 5 percent in a general population database), BS1 to BS4 (strong), and BP1 to BP7 (supporting). The combining rules specify which combinations of codes produce each tier — for example, one very strong plus one strong, or one very strong plus two moderate, or one very strong plus one moderate plus one supporting, all yield a pathogenic classification. [1] [10]
Epidemiology & Risk Factors
Approximately 3 to 5 percent of the paediatric population has a congenital anomaly, developmental delay, or intellectual disability with a significant genetic contribution, making genomic testing one of the highest-yield investigations in paediatrics. The diagnostic yield rises with the sophistication of the test: chromosomal microarray identifies a pathogenic CNV in 10 to 20 percent of children with unexplained developmental disability, exome sequencing raises the yield to 25 to 40 percent, and whole-genome sequencing reaches 40 to 50 percent by capturing coding variants, non-coding variants, and some structural rearrangements that exome misses. [2] [6] [8]
The 100,000 Genomes Project Pilot, reported by Smedley and colleagues in the New England Journal of Medicine in 2021, provided the first large-scale, population-level evidence for the diagnostic yield of genome sequencing in a national health system, demonstrating diagnoses in a meaningful proportion of previously undiagnosed rare-disease families and illustrating the clinical utility of ending the diagnostic odyssey. The yield is highest in children with multiple congenital anomalies, severe intellectual disability, and a clear family history consistent with Mendelian inheritance. [6]
The major risk factor for a positive genomic result is the phenotype: multiple congenital anomalies in a newborn, global developmental delay or intellectual disability with dysmorphic features, and unexplained neurodegenerative or metabolic decline carry the highest pre-test probability. Trio sequencing — sequencing both parents alongside the proband — increases the yield by identifying de novo variants and reduces the burden of VUS reporting by filtering the vast number of rare inherited variants that are unlikely to be causative. [9]
Pathophysiology
The variant interpretation process is the analytical engine that converts raw sequencing data into a clinically actionable call, and understanding it is essential to using genomic testing safely. The process begins with the identification of all variants in the sequenced genome or exome, typically millions of differences from the reference sequence. The laboratory then filters these against population databases (gnomAD), retains rare variants below a frequency threshold consistent with the disease prevalence, and prioritises those in genes with an established disease association and a plausible mechanism. [1]
Each surviving candidate variant is then assessed against five evidence domains. Population data asks whether the variant is absent or very rare in a population database (supporting pathogenicity) or common enough to be排除d as a benign polymorphism. Computational and predictive data uses in silico tools (such as CADD, REVEL, and splicing predictors) to assess the likely functional impact. Functional data draws on established assays or published studies demonstrating the effect of the variant or class of variant on protein function. Segregation data asks whether the variant co-segregates with the disease in affected family members. De novo status assesses whether the variant arose newly in the proband and is not present in either parent, a powerful indicator when the phenotype fits a known disease. [1] [10]
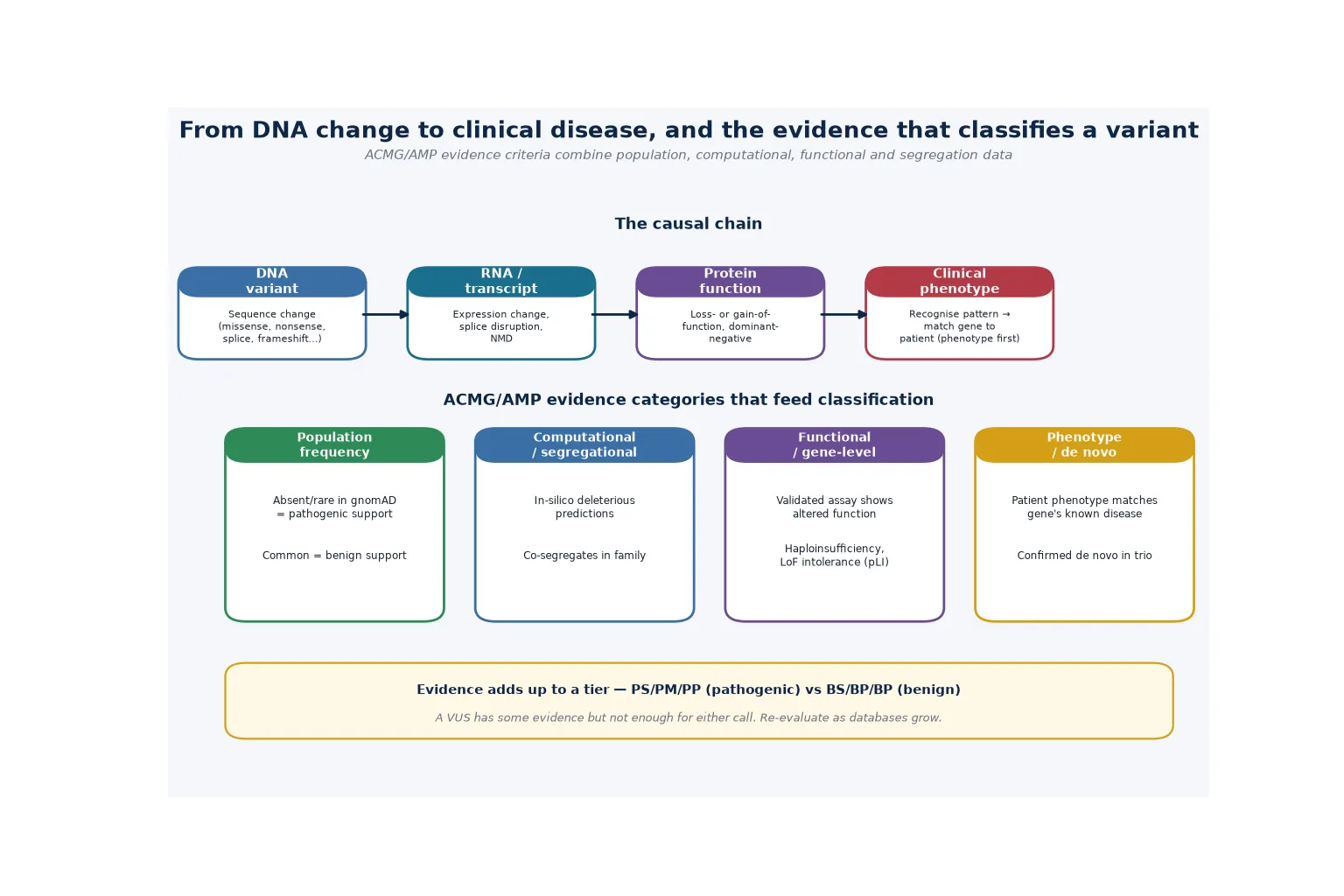
The Tavtigian Bayesian point system, published in 2020, refined the ACMG/AMP combining rules by assigning each evidence code a quantitative point value on a naturally scaled odds-of-pathogenicity axis. This made the combination of evidence codes more transparent and reproducible, reducing the inter-laboratory discordance that plagued the original qualitative framework. The system converts the qualitative categories into a continuous log-odds scale, so that, for example, a very strong code contributes approximately 8 points, a strong code contributes 4, a moderate code contributes 2, and a supporting code contributes 1, with the total mapping to the five-tier classification. [10]
The technical limitations of each test determine what the test can and cannot detect, and they are essential counselling points. Chromosomal microarray detects copy-number variants but misses balanced rearrangements (such as reciprocal translocations and inversions) and single-nucleotide variants. Exome sequencing captures the coding regions (approximately 1 to 2 percent of the genome) but misses deep intronic variants, most structural variants, repeat expansions, and epigenetic modifications. Whole-genome sequencing addresses some of these gaps by sequencing the entire genome, but it still misses repeat expansions (such as the fragile X CGG expansion), methylation defects, and some complex structural rearrangements. [2] [8]
Clinical Presentation
The clinical presentations that prompt genomic testing in paediatrics fall into several broad categories. The commonest indication in outpatient practice is unexplained global developmental delay or intellectual disability, particularly when accompanied by dysmorphic features, autism, seizures, or a family history of developmental disorder. Chromosomal microarray is the first-tier test in this group, followed by exome or genome sequencing if the microarray is non-diagnostic. [2] [9]
Multiple congenital anomalies in a newborn or child — particularly when two or more major anomalies coexist, or when a anomaly pattern suggests a syndromic diagnosis — are a high-yield indication for both microarray and exome or genome sequencing. The ACMG evidence-based clinical guideline on exome and genome sequencing for paediatric patients, published by Manickam and colleagues in 2021, established that exome and genome sequencing are clinically indicated as a first- or second-tier test for children with congenital anomalies or intellectual disability, with the strongest evidence supporting their use in this population. [9]
In the acute setting, a critically ill infant in the NICU or PICU with a suspected but undiagnosed genetic disorder is a candidate for rapid trio genome sequencing, which delivers a result within 7 to 14 days and can directly change acute management — guiding decisions about surgery, transplantation, escalation or withdrawal of intensive care, and end-of-life planning. The study by Willig, Petrikin, and colleagues demonstrated the feasibility and clinical impact of rapid whole-genome sequencing in this population, establishing it as a transformative tool in neonatal and paediatric intensive care. [7]
Differential Diagnosis
The differential of a non-diagnostic genomic test result is broader than many clinicians assume, and it must be actively worked through before concluding that a genetic diagnosis is excluded. The possibilities include a test limitation (the relevant variant type was not detected — for example, a repeat expansion missed by exome sequencing), an incomplete coverage (the relevant gene was not captured or was poorly covered), a VUS that cannot be resolved in either direction, a non-genetic cause (perinatal injury, infection, environmental exposure), a phenocopy (a clinical picture that mimics but is not caused by the suspected gene), or an oligogenic or polygenic inheritance pattern that does not fit a single-gene model. [8]
A true negative must be distinguished from a false negative. A true negative means the relevant gene and variant type were analysed and no pathogenic finding was present. A false negative means the test did not analyse the relevant variant — for example, an exome that does not reliably size the fragile X CGG repeat, a microarray that does not detect balanced translocations, or a sequencing pipeline with a technical blind spot in a specific gene (such as the SMN1 gene in spinal muscular atrophy, where gene conversion makes standard sequencing unreliable). [2] [8]
When a standard exome or genome returns non-diagnostic, additional testing may be warranted: mitochondrial DNA sequencing (for suspected mitochondrial disease), methylation analysis or methylation-specific testing (for imprinting disorders such as Prader-Willi and Angelman syndromes), RNA sequencing (to detect splicing defects not predicted from DNA alone), repeat-primed PCR (for repeat expansions such as fragile X), or skin biopsy with karyotyping (for mosaic conditions not detectable in blood). The clinical geneticist is the key decision-maker in escalating beyond standard testing. [9]
Clinical & Bedside Assessment
The pre-test clinical assessment is the foundation of accurate genomic testing, and it has three components: a detailed three-generation pedigree that explicitly documents consanguinity, ethnicity, family history of developmental disorders, miscarriages, and unexplained deaths; deep phenotyping using structured terminology (ideally Human Phenotype Ontology, HPO, terms) to communicate the clinical picture to the laboratory; and a review of prior genetic tests to avoid duplication and to identify what has already been excluded. [9]
The history discriminators that raise pre-test probability include consanguinity (increasing the likelihood of autosomal recessive disease), a family history consistent with X-linked or autosomal dominant inheritance, a perinatal history that may suggest an environmental contribution, and the temporal course of the child's presentation (static versus regressive versus progressive). The examination looks for multiple congenital anomalies, dysmorphic features, neurocutaneous stigmata (such as ash-leaf macules or cafe-au-lait spots suggesting tuberous sclerosis or neurofibromatosis), organomegaly (suggesting a metabolic or storage disorder), and tone abnormalities. [2] [9]
The pre-test assessment also includes a counselling conversation that assesses the family's understanding, health literacy, psychosocial context, and readiness for testing and results. The clinician documents the consent for secondary findings, the family's preference on VUS disclosure, data-sharing consent, and the storage of samples for future re-analysis. The testing order then specifies the correct test (microarray, panel, exome, genome, or trio), the phenotype in HPO terms, and the specific clinical question the test is intended to answer — because a well-framed clinical question is the single most important determinant of a useful laboratory report. [3] [9]
Investigations
The investigation pathway follows a stepwise logic built on diagnostic yield and phenotype specificity. Chromosomal microarray is the first-tier test for unexplained intellectual disability, developmental delay, autism with dysmorphic features, and multiple congenital anomalies, established by the Miller consensus statement in 2010 as the replacement for karyotype in this population. CMA detects copy-number variants across the genome at far higher resolution than karyotype and has a diagnostic yield of 10 to 20 percent. [2]
When the microarray is non-diagnostic and the phenotype is consistent with a single-gene disorder, exome or genome sequencing is the next step. The ACMG evidence-based clinical guideline on exome and genome sequencing for paediatric patients (Manickam 2021) established that these tests are clinically indicated for children with congenital anomalies or intellectual disability, either as a first-tier test (when the presentation is non-specific or atypical) or as a second-tier test after a non-diagnostic microarray. The guideline recommends trio sequencing (proband plus both parents) wherever possible, because parental testing distinguishes de novo from inherited variants, increases the diagnostic yield, and reduces the burden of VUS reporting by filtering the vast number of rare inherited variants. [9]
The laboratory then applies the ACMG/AMP variant classification framework (Richards 2015) to each candidate variant, integrating population data, computational prediction, functional evidence, segregation, and de novo status into a five-tier classification. The candidate should understand the combining rules: for example, a pathogenic call typically requires one very strong code (such as PVS1 for a loss-of-function variant in a haploinsufficient gene) combined with additional evidence, while a likely pathogenic call requires strong evidence with moderate or supporting evidence. A benign call can be made by a single stand-alone code (BA1, allele frequency above 5 percent), while a likely benign call requires strong benign evidence. [1] [10]
Why a normal microarray does not exclude a genetic diagnosis
Chromosomal microarray detects copy-number variants — deletions and duplications of chromosomal material — at sub-microscopic resolution. It does not detect single-nucleotide variants (the point mutations that cause most single-gene disorders), repeat expansions (such as the fragile X CGG expansion), balanced rearrangements (such as reciprocal translocations and inversions), or epigenetic changes (such as the methylation defects in Prader-Willi and Angelman syndromes). A normal microarray has excluded pathogenic CNVs, not the broader universe of genetic disease. If the phenotype is consistent with a single-gene disorder, the next step is exome or genome sequencing — and if the phenotype fits fragile X, a dedicated FMR1 PCR is required regardless of what the microarray or exome shows. [2] [9]
Management — Resuscitation
The resuscitation context for genomic testing is the critically ill infant in the NICU or PICU with a suspected but undiagnosed genetic disorder. In this setting, rapid trio genome sequencing (rapid exome or rapid genome, with a turnaround time of typically 7 to 14 days) has become a transformative tool. The study by Willig, Petrikin, and colleagues demonstrated that rapid whole-genome sequencing in critically ill newborns could identify Mendelian disorders, change acute management, and reduce the iatrogenic harm of undirected investigation. [7]
A rapid diagnostic result can directly inform acute management decisions: it may guide the decision to pursue or withdraw intensive care, to proceed with or defer surgery, to initiate a disease-specific therapy (such as a metabolic formula or cofactor supplementation), or to plan for transplantation. It may also identify a condition with a known poor prognosis, supporting a palliative approach. A rapid result that excludes a genetic diagnosis is also valuable, because it may redirect the investigation pathway and reduce unnecessary testing and treatment. [7]
The ethical framework for rapid testing in acutely ill children is built on informed consent under time pressure, the management of secondary findings in the parents (who are sequenced as part of a trio), and the communication of complex results to an intensive care team and a family in crisis. The genetic counsellor plays a central role in supporting the family before, during, and after the result disclosure, and the result must be communicated with clarity, empathy, and a plan for ongoing support. [7] [3]
Management — Definitive & Stepwise
Definitive management of genomic testing and counselling follows a five-step framework that a fellowship candidate can recite and a general paediatrician can coordinate: confirm the clinical indication and choose the right test, apply the ACMG/AMP variant classification, counsel the family on the result, coordinate cascade testing, and plan periodic re-analysis. [1] [9]

Pre-test counselling is the backbone of the framework and must cover several domains: the estimated diagnostic yield and the possibility of a non-diagnostic result; the possibility of a VUS and what that means; the option of secondary findings (the ACMG SF list) and the family's right to opt in or out; the implications of a carrier status result; the potential impact on insurance and employment (with reference to relevant legislative protections); and consent for data sharing and sample storage. This conversation is ideally conducted by a genetic counsellor, and it must be documented in the medical record. [3] [5]
Post-test counselling is structured around the disclosure of the result in its full clinical context. A pathogenic or likely pathogenic result prompts a discussion of the diagnosis, the natural history, the available management and surveillance, the reproductive implications (including prenatal and preimplantation genetic diagnosis for future pregnancies), and the cascade-testing obligation to the wider family. A VUS prompts a discussion of the uncertainty, the plan for segregation testing and re-analysis, and the principle that management should be guided by the clinical picture, not by the variant call alone. A non-diagnostic result prompts a discussion of what has been excluded, what remains possible, and the plan for re-analysis and further investigation. [1] [9]
C.A.S.C.A. — the genomic counselling framework
Specific Subtypes & Scenarios
The trio exome — sequencing the proband and both parents simultaneously — is the gold-standard approach for paediatric genomic sequencing when both parents are available. Trio sequencing increases the diagnostic yield by identifying de novo variants (a powerful indicator when the phenotype fits a known disease), filters the vast number of rare inherited variants that are unlikely to be causative, and reduces the burden of VUS reporting by classifying inherited variants against the parental background. When only one parent is available, duo sequencing (proband plus one parent) provides partial benefit. [9]
The rapid genome in the critically ill infant delivers a result within 7 to 14 days and directly changes acute management in the NICU or PICU. The Willig and Petrikin study demonstrated the clinical impact of this approach, and rapid sequencing is now established as standard of care in many tertiary paediatric centres for infants with suspected genetic disorders in intensive care. The consent conversation is conducted under time pressure, and the genetic counsellor plays a central role in supporting the family. [7]
The secondary finding is a clinically actionable variant in a gene unrelated to the indication for testing, reported under the ACMG SF list. The original ACMG recommendations on incidental findings (Green 2013) established a list of genes for which actionable findings should be offered, and the list has been updated through the Kalia 2017 v2.0 update and the Miller 2021 v3.0 policy statement, which expanded the list to 73 genes. The patient or family must opt in to receiving secondary findings during the pre-test counselling conversation, and the return of a secondary finding triggers disease-specific surveillance and management. [3] [4] [5]
Complications & Pitfalls
The complications of genomic testing divide into the cognitive traps that cost marks and the practical errors that harm patients. The chief cognitive trap is over-interpreting a VUS as pathogenic, driving unnecessary cardiac screening, prophylactic surgery, or anxiety, when the evidence is insufficient to support a pathogenic call. The management of a VUS should be guided by the clinical picture, not by the variant call alone, and a VUS should never be the sole basis for a clinical decision. [1] [10]
The second trap is returning a secondary finding without prior consent. The ACMG SF list represents an opt-in framework, and a family must consent to receiving secondary findings during the pre-test counselling conversation. If an actionable variant is found incidentally and the family has not consented, the situation must be managed carefully — the clinical geneticist and genetic counsellor should discuss the finding with the family, explain its significance, and offer appropriate follow-up. [3] [5]
The third trap is failing to cascade-test the family once a pathogenic variant is found. A confirmed molecular diagnosis obliges testing of both parents (to confirm de novo versus inherited status and to establish the inheritance pattern), siblings, and at-risk maternal or paternal relatives depending on the inheritance pattern. Failing to cascade-test leaves carriers and affected relatives undiagnosed, missing the opportunity for reproductive counselling and surveillance. The fourth trap is failing to re-analyse a previously non-diagnostic result, given that new genes are discovered and variant classification evolves continuously. [9]
Prognosis & Disposition
The prognosis following a genomic diagnosis is determined by the specific condition, the severity of the phenotype, the availability of disease-specific therapy, and — most powerfully — the value of the answer itself. A molecular diagnosis delivers two forms of utility: clinical utility (a management change, such as disease-specific surveillance, treatment, or reproductive counselling) and personal utility (ending the diagnostic odyssey, removing parental guilt by establishing that the condition is genetic and not caused by anything the parents did or did not do, and connecting the family to patient communities and advocacy groups). [6] [9]
Even when no disease-specific therapy exists, a molecular diagnosis changes the trajectory by enabling entry into precision medicine trials, natural history studies, and gene-specific registries that may lead to future therapies. The disposition is shared, lifelong care: the clinical geneticist owns the variant interpretation and re-analysis, the genetic counsellor owns the family testing and reproductive counselling, the general paediatrician owns the coordination and preventive care, and disease-specific subspecialty services own the surveillance and treatment. [9]
Periodic re-analysis of non-diagnostic results is essential as new genes are discovered and variant classification evolves. The family should be offered a re-analysis every 2 to 3 years, and the result should be reviewed whenever the child's phenotype evolves or when a new clinically relevant question arises. The transition from paediatric to adult care is a high-risk point for loss of continuity in genomic surveillance, so the transition plan must explicitly hand over the genetic record and the re-analysis schedule. [6] [8]
Special Populations
The same genomic testing framework behaves differently across populations because access, equity, and representation in variant databases are unevenly distributed. In remote and Indigenous communities, the barriers include geographic distance from genetic services, cost, the need for culturally safe counselling, and the ethics of sample storage and data sharing (which may conflict with cultural beliefs about biological material). Culturally safe practice requires an interpreter or cultural broker where needed, respectful discussion of data-sharing consent, and coordination with Indigenous health services. [9]
In migrant, refugee, and asylum-seeking families, the testing pathway may be complicated by incomplete family histories, consanguinity (which raises the likelihood of autosomal recessive disease), different prior test records from the country of origin, and language barriers. An interpreter must be used at every key consultation, and the consent conversation must be adapted to the family's health literacy and cultural context. The under-representation of non-European populations in variant databases (such as gnomAD) is a particular concern, because a variant that is rare in European populations but common in the patient's ancestral population may be falsely classified as pathogenic, leading to a false-positive diagnosis. [1] [8]
In families managing complex disability, a confirmed molecular diagnosis can unlock access to coordinated care plans, disability funding (such as the NDIS in Australia and the equivalent in New Zealand), and patient communities that provide practical and emotional support. In adolescents transitioning to adult care, the move is a high-risk point for loss of continuity in genomic surveillance and re-analysis, so the transition plan must explicitly hand over the genetic record, the re-analysis schedule, and the family-testing obligations. [9]
Evidence, Guidelines & Regional Differences
The evidence base for genomic testing, variant interpretation, and counselling rests on three pillars: consensus clinical guidelines, population-scale diagnostic yield studies, and the variant interpretation framework. The Richards 2015 ACMG/AMP consensus established the five-tier variant classification framework and the evidence codes that every clinical genomics laboratory now applies, and the Tavtigian 2020 Bayesian point system refined the combining rules to make them more transparent and reproducible, reducing inter-laboratory discordance. [1] [10]
The Miller 2010 CMA consensus established chromosomal microarray as the first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies, and the Manickam 2021 ACMG paediatric ES/GS guideline extended the pathway by establishing that exome and genome sequencing are clinically indicated for children with congenital anomalies or intellectual disability, with the strongest evidence supporting their use as first- or second-tier tests in this population. [2] [9]
The ACMG secondary findings recommendations (Green 2013, Kalia 2017, Miller 2021) shaped the return of medically actionable findings through the SF list, which has evolved from the original 56-gene list to the current 73-gene v3.0 list. The 100,000 Genomes Project Pilot (2021) provided the first large-scale, population-level evidence for the diagnostic yield of genome sequencing in a national health system, and the Jansen and Vissers 2023 review of intellectual disability genetics synthesised the diagnostic yield of genomic testing across the spectrum of paediatric intellectual disability. [3] [4] [5] [6] [8]
In Australia and New Zealand, genomic testing is publicly funded through the Medicare Benefits Schedule (MBS) for specific indications, including chromosomal microarray for unexplained developmental disability and whole-exome sequencing for certain phenotypes. State clinical genetics services deliver genetic counselling and coordinate cascade testing, with access in metropolitan, regional, and (by outreach or telehealth) remote settings. The Disability Discrimination Act 1992 (Cth) in Australia and the Human Genetic Information Act in New Zealand provide legislative protections against genetic discrimination in insurance and employment, though these protections have specific thresholds and exclusions that the counsellor should be familiar with. Always confirm the current local eligibility criteria for publicly funded genomic testing, as MBS items and state funding arrangements change.
[2][9]Exam Pearls
A fellowship candidate answering on genomic testing should land five anchor points and avoid four classic traps. The anchors are the test hierarchy and diagnostic yield (microarray 10 to 20 percent, exome 25 to 40 percent, genome 40 to 50 percent), the ACMG/AMP five-tier variant classification (pathogenic, likely pathogenic, VUS, likely benign, benign) and its evidence codes (PVS, PS, PM, PP, BA, BS, BP), the management of secondary findings through the ACMG SF v3.0 list with informed consent, the pre- and post-test counselling framework, and the cascade-testing and re-analysis obligations. The traps are treating a VUS as a diagnosis, concluding a normal microarray excludes genetic disease, returning a secondary finding without consent, and failing to re-analyse non-diagnostic results. [1] [2]
References
- [1]Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015.PMID 25741868
- [2]Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet, 2010.PMID 20466091
- [3]Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med, 2013.PMID 23788249
- [4]Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med, 2017.PMID 27854360
- [5]Miller DT, Lee K, Chung WK, Gordon AS, Herman GE, O'Daniel JM, et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med, 2021.PMID 34012068
- [6]100,000 Genomes Project Pilot Investigators, Smedley D, Smith KR, Martin A, Welsh SA, Ellingford M, et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care - Preliminary Report. N Engl J Med, 2021.PMID 34758253
- [7]Willig LK, Petrikin JE, Smith LD, Saunders CJ, Thiffault I, Miller NA, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill newborns: a retrospective analysis of diagnostic and clinical trajectories. Lancet Respir Med, 2015.PMID 25937001
- [8]Jansen S, Vissers LELM, Coe BP, Kleefstra T, Gilissen C. The Genetics of Intellectual Disability. Brain Sci, 2023.PMID 36831774
- [9]Manickam K, McClain MR, Demmer LA, Hickey SE, Kunaciecki MB, Nathanson KM, et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med, 2021.PMID 34211152
- [10]Tavtigian SV, Harrison SM, Boucher LD, Biesecker LG, Harrison T, Azzariti DR, et al. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum Mutat, 2020.PMID 32720330