Paeds · genetics-dysmorphology-and-metabolism
Glycogen-storage and carbohydrate metabolism disorders
Also known as Glycogen storage diseases · GSD · Glycogenoses · Von Gierke disease (GSD I) · Pompe disease (GSD II) · Cori / Forbes disease (GSD III) · McArdle disease (GSD V) · Galactosaemia · Hereditary fructose intolerance
A fellowship approach to the glycogen-storage and carbohydrate metabolism disorders: recognise the hepatic glycogenoses through the signature of hepatomegaly with fasting hypoglycaemia and lactic acidosis, separate Pompe disease by its hypertrophic cardiomyopathy and hypotonia, place the muscle glycogenoses by exercise intolerance with a second wind, and hold galactosaemia and hereditary fructose intolerance as the toxic-sugar disorders — then deliver the unifying treatment principle of preventing fasting and catabolism, with enzyme replacement for Pompe and emerging gene and transplant therapy.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who frames these disorders around one physiological idea and three clinical patterns. The idea is that glycogen is the liver's battery: it stores glucose between feeds and releases it as free glucose during fasting, and a defect anywhere along that release pathway starves the brain while the liver swells with the stored glycogen it cannot use. The first pattern is the hepatic energy-failure pattern — a child with a big liver, a low glucose, a high lactate and, in GSD I, the tell-tale urate and lipid rises. The second is the lysosomal muscle pattern of Pompe — a floppy baby with a thick heart. The third is the exercise-intolerance pattern of the muscle glycogenoses — a child or young adult who cramps and "warms up" into a second wind. Holding these three patterns together is what turns a confusing list of numbered diseases into a recognisable clinical framework. [1] [7]
Overview & Definition
The glycogen-storage and carbohydrate metabolism disorders are a family of inherited metabolic diseases in which the body cannot make, break down or correctly use stored glycogen or dietary sugars. The largest group, the glycogen storage diseases (GSDs, the glycogenoses), are numbered 0 to XIII by the enzyme or transporter affected, and they share a common biochemistry: a defect in the synthesis, breakdown or intracellular handling of glycogen that either starves the body of glucose during fasting or swells tissues with glycogen that cannot be processed. Alongside them sit the carbohydrate (sugar) metabolism disorders, in which a specific dietary sugar cannot be metabolised and its toxic intermediates accumulate. Classic galactosaemia from galactose-1-phosphate uridylyltransferase deficiency, hereditary fructose intolerance from aldolase B deficiency, and fructose-1,6-bisphosphatase deficiency. [1] [12]
The single most useful organising principle is where the block sits and which tissue it cripples. The hepatic, fuel-regulating enzyme defects — glycogen synthase (GSD 0), glucose-6-phosphatase and its translocase (GSD I), the debrancher enzyme (GSD III), liver phosphorylase (GSD VI) and phosphorylase kinase (GSD IX). All produce fasting hypoglycaemia because the liver cannot release free glucose. Pompe disease (GSD II) is biochemically different: acid α-glucosidase works inside the lysosome, so its deficiency floods the lysosome with glycogen and damages cardiac and skeletal muscle rather than blood glucose. The muscle glycogenoses (GSD V McArdle, GSD VII Tarui) block glycolysis within exercising muscle and produce exercise intolerance. Recognising these three clinical destinations — liver energy failure, lysosomal muscle disease, and exertional muscle pain — is the foundation of the whole topic. [1] [5] [11]
A fellow should leave this overview with a working definition, not a numbered list. A glycogen storage disease is an inherited defect of glycogen synthesis, breakdown or handling that produces a recognisable tissue-specific syndrome. Fasting energy failure in the liver, cardiomyopathy and hypotonia in the lysosome, or exercise intolerance in muscle. The carbohydrate metabolism disorders are the closely related group in which a dietary sugar cannot be metabolised, causing toxic-metabolite injury. The two groups are taught together because their presentation overlaps (hepatomegaly, hypoglycaemia, lactic acidosis), their investigation overlaps (the fasting metabolic panel), and. Critically — their management overlaps in the principle of preventing the metabolic stress that unmasks the enzyme block. [1] [7]
Classification
The classification that earns marks is the pathway and tissue classification, because the position of the block determines the clinical syndrome and the treatment. The hepatic glycogenoses are grouped by their position along glycogenolysis. GSD 0 (glycogen synthase deficiency) reduces glycogen synthesis. GSD I blocks the final step — glucose-6-phosphatase (GSD Ia, von Gierke) or its microsomal translocase (GSD Ib). GSD III blocks the debrancher enzyme that unties glycogen's branch points. GSD VI (liver phosphorylase) and GSD IX (phosphorylase kinase, the commonest mild hepatic GSD) block the early phosphorylytic release of glucose-1-phosphate. Each hepatic defect shares the fasting-hypoglycaemia phenotype but differs in severity, the metabolic fingerprint, and the complications. [1] [9]
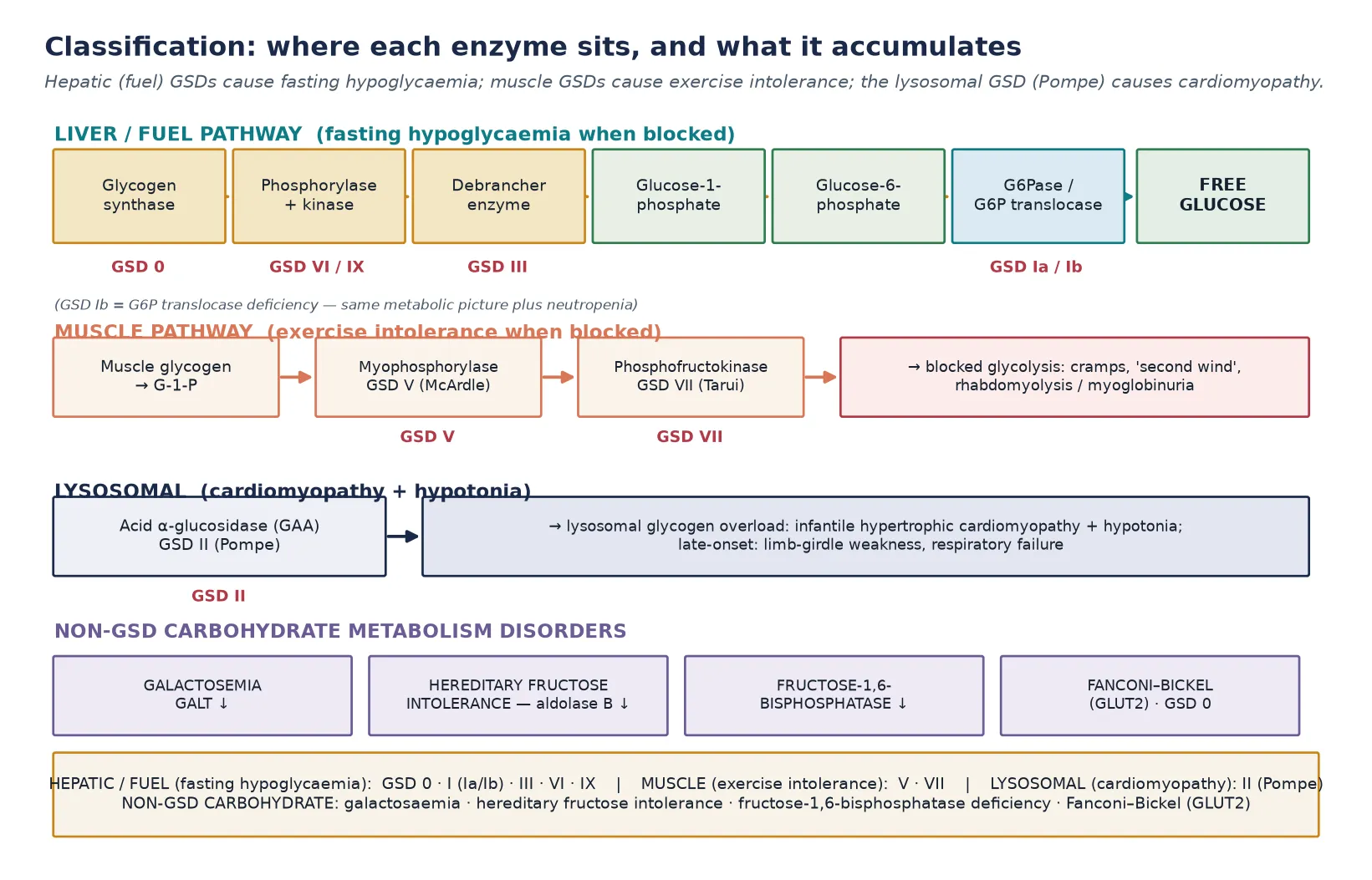
Pompe disease (GSD II) sits in a class of its own because acid α-glucosidase (acid maltase, GAA) is a lysosomal enzyme: it degrades the small fraction of glycogen that enters the lysosome by autophagy. Its deficiency does not cause hypoglycaemia — blood glucose regulation is intact — but instead causes progressive lysosomal glycogen accumulation in cardiac, skeletal and respiratory muscle. The infantile form is dominated by hypertrophic cardiomyopathy; the late-onset form presents with proximal and respiratory muscle weakness without cardiac involvement. This lysosomal-versus-cytosolic distinction is why Pompe is treated with enzyme replacement rather than dietary glucose management, and it is the single most important classification point in the topic. [5] [6]
The non-GSD carbohydrate disorders complete the classification. Classic galactosaemia (galactose-1-phosphate uridylyltransferase, GALT, deficiency) and hereditary fructose intolerance (aldolase B deficiency) share a toxic-intermediate mechanism. The offending sugar is partly metabolised to a phosphorylated intermediate that accumulates and injures the liver, kidney and other organs. So the clinical syndrome appears only once the sugar is introduced — galactose with lactose-containing milk in the neonate, fructose with fruit, sucrose or formula in the weaning infant. Fructose-1,6-bisphosphatase deficiency blocks gluconeogenesis and presents with fasting lactic acidosis and hypoglycaemia. Fanconi–Bickel syndrome (GLUT2 deficiency) straddles both groups, causing galactose and glucose handling defects with glycogen accumulation and renal tubular dysfunction. [12] [13] [14]
Epidemiology & Risk Factors
The combined incidence of the glycogen storage diseases is roughly one in 20,000 to 25,000 live births, with the hepatic forms. Particularly GSD I at around one in 50,000 to 100,000 and GSD III at a similar frequency — making up the bulk. Pompe disease occurs in approximately one in 40,000 live births, though late-onset Pompe is under-recognised and may be considerably more common than the classic infantile form. GSD IX, caused by phosphorylase kinase deficiency and most often X-linked, is probably the most common mild hepatic GSD but is frequently missed because its phenotype is mild and its metabolic fingerprint subtle. With the exception of the X-linked forms (GSD IX and some GSD VI variants), all the glycogenoses are autosomal recessive, which makes consanguinity and a family history of unexplained infant death or hepatomegaly the highest-yield risk factors. [1] [9]
Classic galactosaemia is the most common of the carbohydrate disorders, with an incidence around one in 30,000 to 60,000 live births in populations with newborn screening, and it is included on the bloodspot panel in most developed programmes. Including Australia and New Zealand — so many cases are now detected before symptoms. Hereditary fructose intolerance is rarer, around one in 20,000, and because it is not screened for it presents only once fructose, sucrose or sorbitol is introduced into the diet, typically at weaning. The epidemiological engine for the late-onset and recurrent presentations of all these disorders is the catabolic trigger: intercurrent infection, fasting, surgery, or a dietary indiscretion unmasks a partial enzyme defect that has been silent, and a child previously labelled "well" presents with hypoglycaemia, metabolic acidosis or myoglobinuria. [12] [13]
Recognition and access are unevenly distributed. In remote and Indigenous communities, and in migrant and refugee families, the interval between symptom onset and a measured fasting metabolic panel is longer, the dietary and cornstarch regimens are harder to sustain, and the aeromedical retrieval of a hypoglycaemic infant adds time that the developing brain cannot afford. A family history of consanguinity, unexplained neonatal or infant death, recurrent "sepsis" with negative cultures, or siblings with hepatomegaly should raise the index of suspicion for an autosomal recessive glycogen or sugar disorder and prompt early testing. Because early diagnosis and metabolic control are the single most powerful modifiers of long-term outcome. [1] [15]
Pathophysiology
The pathophysiology of the hepatic glycogenoses is the pathophysiology of fasting energy failure. In health, the liver stores glucose as glycogen between feeds and, during fasting, breaks it back down to glucose-6-phosphate, which glucose-6-phosphatase then converts to free glucose for release into the blood. When this final step is blocked — as in GSD Ia (glucose-6-phosphatase deficiency) or GSD Ib (glucose-6-phosphate translocase deficiency) — glucose-6-phosphate cannot leave the liver as glucose. So the blood glucose falls (fasting hypoglycaemia). The trapped glucose-6-phosphate is shunted down glycolysis to lactate (lactic acidosis), and the substrates that would have fed gluconeogenesis back up into glycogen synthesis and cause hepatomegaly. The same trapped intermediates feed glycerol and purine metabolism, producing the hypertriglyceridaemia and hyperuricaemia that complete the biochemical tetrad of GSD I. [1] [3]
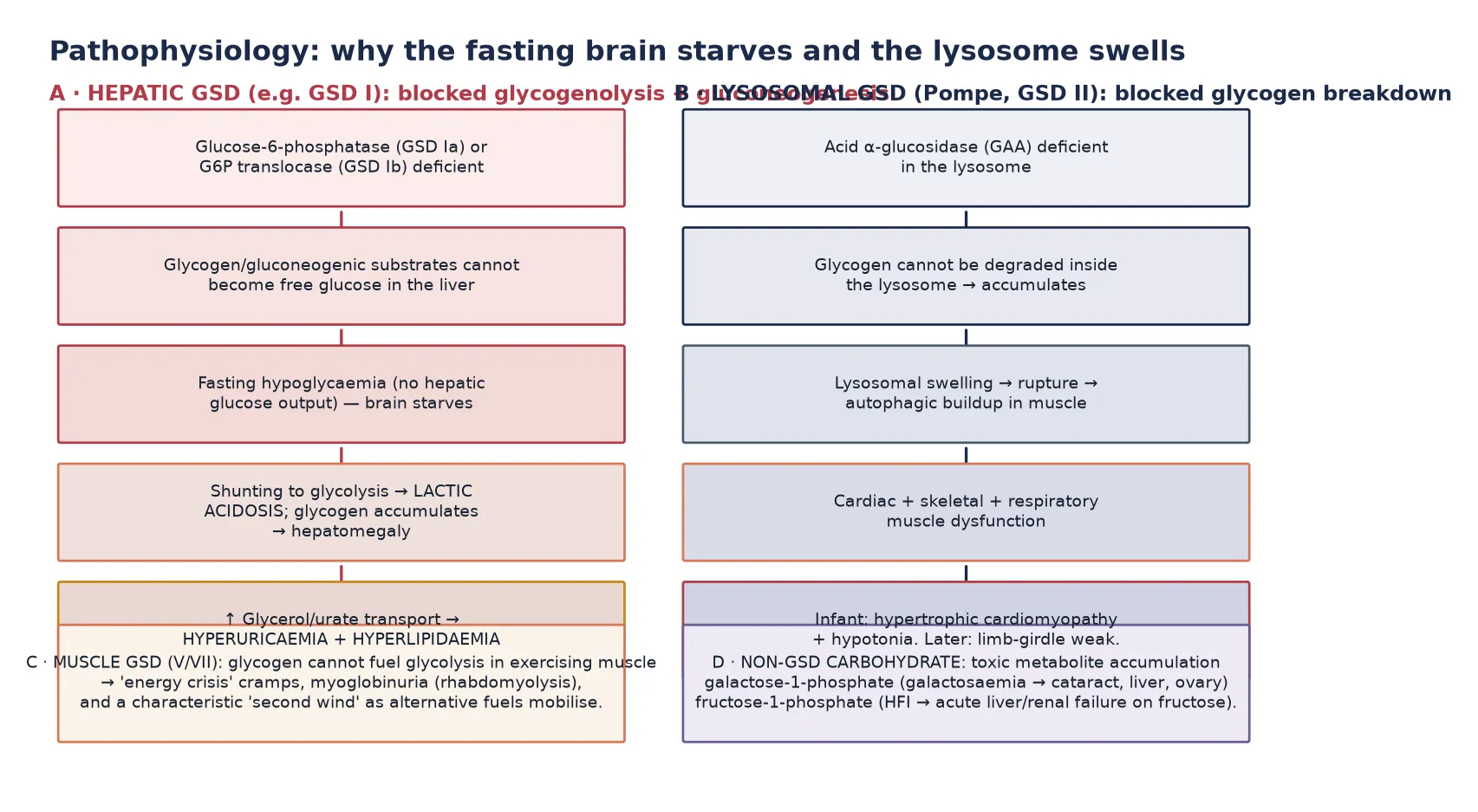
In GSD III (debrancher deficiency) the pathophysiology is slightly different: glycogen can be phosphorylysed down to its outer branch points but the branch points themselves cannot be cleaved, so a limit-dextrin glycogen accumulates and the liver, and sometimes skeletal and cardiac muscle, are affected. The hypoglycaemia is usually milder than in GSD I and is ketotic rather than lactic, because gluconeogenesis remains intact. A distinction that is both a diagnostic clue and a management point, since GSD III tolerates fasting slightly better and responds well to protein and continuous glucose. In the milder hepatic forms (GSD VI, IX) the same phosphorylytic block is partial, so hypoglycaemia and hepatomegaly are present but the severe biochemical tetrad of GSD I is absent, and growth and outcome are usually near-normal with modest dietary adjustment. [7] [8]
Pompe disease follows a completely different mechanism because acid α-glucosidase is a lysosomal hydrolase. Glycogen that enters the lysosome by autophagy cannot be degraded, so it accumulates, distends the lysosome, and ultimately ruptures it, releasing glycogen and lysosomal enzymes into the cytoplasm and triggering a cascade of autophagic buildup, mitochondrial dysfunction and muscle fibre destruction. The result is progressive dysfunction of the tissues most dependent on glycogen turnover — the heart (massive hypertrophic cardiomyopathy in the infant), the skeletal muscles (severe hypotonia), and the respiratory muscles (diaphragmatic weakness). This lysosomal mechanism is why Pompe responds to enzyme replacement therapy, which delivers functional enzyme to the lysosome via mannose-6-phosphate receptors — a treatment that has no equivalent in the cytosolic hepatic GSDs. [5] [6]
The muscle glycogenoses (GSD V, VII) starve exercising muscle of the glycolytic substrate it needs for bursts of activity. When myophosphorylase (GSD V) or phosphofructokinase (GSD VII) is deficient, muscle cannot mobilise or metabolise its glycogen during exertion, so an energy crisis produces cramps, contractures and, with exertion, breakdown of muscle with myoglobinuria and a risk of acute kidney injury. The characteristic second wind of McArdle disease arises because, after several minutes, the muscle switches to circulating free fatty acids and glucose mobilised from the liver, which partially restores energy supply. A phenomenon that is both pathognomonic and a teaching point about fuel flexibility. The sugar disorders, finally, cause toxic-metabolite injury: galactose-1-phosphate and fructose-1-phosphate accumulate and poison the liver, kidney and (in galactosaemia) the lens and ovary, which is why removal of the offending sugar is both diagnostic and therapeutic. [11] [12] [13]
Clinical Presentation
The clinical presentation divides cleanly into the three patterns, and a fellow should be able to generate each one from the pathophysiology. The hepatic pattern presents in infancy, often once the interval between feeds lengthens, with hepatomegaly (the parents usually notice the distended abdomen), fasting hypoglycaemia manifesting as lethargy, seizures or early-morning twitching, and the biochemical fingerprint of lactic acidosis. In GSD I the child also has a doll-like facies from fat deposition, growth failure, xanthomata, and recurrent symptoms that are strikingly relieved by feeding. In GSD Ib the same metabolic picture is accompanied by neutropenia with recurrent infections and an inflammatory bowel disease-like enteropathy that is itself a major source of morbidity. [1] [4]
The Pompe pattern presents in one of two ways. The classic infantile form declares itself in the first months of life with severe hypotonia (the "floppy infant"), macroglossia, feeding difficulty, and a massively hypertrophied heart that causes heart failure. The chest X-ray shows an enormous cardiac silhouette, the electrocardiogram shows huge QRS voltages, and the creatine kinase is markedly raised. Without treatment, infantile Pompe is fatal within the first one to two years from cardiorespiratory failure. The late-onset form presents at any age from childhood to adulthood with progressive proximal (limb-girdle) weakness, often beginning in the hips and shoulders, and — critically — with respiratory muscle weakness that may precede limb weakness. The heart is usually spared. A child or young adult with unexplained limb-girdle weakness, a raised creatine kinase, or ventilatory failure deserves a Pompe work-up. [5] [6]
The muscle pattern presents in older children, adolescents or young adults with exercise intolerance: cramps, muscle pain and stiffness during exertion, often with a recognisable second wind after a few minutes, and episodes of red-brown urine (myoglobinuria) after strenuous activity that may precipitate acute kidney injury. The carbohydrate disorders present with their own signatures. Classic galactosaemia appears in the neonate who has started milk feeds, with jaundice, liver dysfunction, cataracts, failure to thrive and a striking predisposition to Escherichia coli sepsis. Hereditary fructose intolerance appears in the weaning infant who develops vomiting, hypoglycaemia, jaundice and liver or renal failure within minutes to hours of ingesting fructose, sucrose or sorbitol. Often after the first taste of fruit or a sucrose-containing formula. A careful dietary history, anchored to the introduction of the offending sugar, is diagnostic. [11] [12] [13]
Differential Diagnosis
The differential depends on which pattern the child presents with. For the hepatic pattern, the differential of hepatomegaly with fasting hypoglycaemia includes the other inborn errors that disturb fasting adaptation: the fatty-acid oxidation defects (which cause hypoketotic hypoglycaemia without lactic acidosis and often with myopathy), the gluconeogenic defects such as fructose-1,6-bisphosphatase deficiency, and the ketotic hypoglycaemias of childhood. The critical discriminator is the metabolic fingerprint: GSD I alone produces the tetrad of hypoglycaemia, lactic acidosis, hyperuricaemia and hypertriglyceridaemia, and GSD III produces ketotic hypoglycaemia with a raised creatine kinase when muscle is involved. A precise fasting panel — glucose, lactate, ketones (beta-hydroxybutyrate), urate, lipids, liver function, creatine kinase, and a blood gas — usually localises the disorder before the enzyme result returns. [1] [8]
For the Pompe pattern, the differential of the floppy infant with cardiomyopathy includes the other causes of infantile hypotonia with cardiac involvement: mitochondrial disease, spinal muscular atrophy (which does not cause cardiomyrophy but may coexist in the differential of profound weakness), congenital myopathies, and the glycogen and lysosomal storage disorders generally. The discriminating features are the marked creatine kinase elevation and the hypertrophic (not dilated) cardiomyopathy with huge voltages; definitive separation comes from acid α-glucosidase enzyme assay and GAA sequencing. For the muscle pattern, the differential of exercise intolerance with myoglobinuria includes the fatty-acid oxidation defects (which cause a different, longer-duration exercise intolerance), mitochondrial myopathies, and other metabolic myopathies. The second wind and the contractures on maximal exertion point toward McArdle disease. [5] [11]
For the sugar disorders, the differential of neonatal liver dysfunction with cataracts is essentially galactosaemia versus congenital infection and biliary atresia (which cause cholestasis without the hypoglycaemia and cataracts), while the differential of acute liver failure after a dietary change is hereditary fructose intolerance versus tyrosinaemia, viral hepatitis and toxin exposure. The galactosaemia clue is the combination of E. coli sepsis, cataracts and a positive reducing-substance urine test after a galactose load; the hereditary fructose intolerance clue is the precise temporal link between fructose ingestion and symptoms, and the resolution on withdrawal. In all three patterns the principle is the same: use the metabolic panel and the dietary and family history to localise, then confirm with enzyme and molecular testing. [12] [13]
Clinical & Bedside Assessment
The bedside assessment has two speeds. In the acute hypoglycaemic presentation, the immediate question is the glucose and the safety of the brain: take a rapid history anchored to fasting, the relation of symptoms to feeds, and any catabolic trigger, while drawing the fasting panel (glucose, lactate, beta-hydroxybutyrate, free fatty acids, urate, triglycerides, cholesterol, cortisol, growth hormone, insulin, liver function, creatine kinase, ammonia, and a blood gas) before correcting the glucose with intravenous dextrose. The critical point — and a common exam trap — is that the diagnostic information lives in the sample drawn at the moment of hypoglycaemia, so a hypoglycaemic child must have a full "hypo screen" taken before the glucose is pushed up, even while resuscitation proceeds. [1] [7]
The examination of the child with a suspected hepatic GSD should document the liver size and consistency (large and smooth in GSD I, with a renomegaly often palpable alongside), growth parameters (growth failure is almost universal in untreated GSD I), the doll-like facies, xanthomata, and any signs of neutropenic infection or perioral inflammation in GSD Ib. For the child with suspected Pompe, the examination centres on the cardiorespiratory and neuromuscular systems: heart rate, perfusion and signs of cardiac failure, the degree and distribution of hypotonia, the deep tendon reflexes (reduced), macroglossia, and the respiratory effort. Because diaphragmatic weakness may be the dominant and life-threatening feature. A careful developmental and growth history, and a three-generation pedigree, complete the bedside assessment. [5] [4]
The problem representation that follows a complete assessment should be one sentence that captures the pattern. For GSD I: "A previously well infant with hepatomegaly, fasting hypoglycaemia, lactic acidosis, hyperuricaemia and hypertriglyceridaemia consistent with glucose-6-phosphatase deficiency." For Pompe: "A hypotonic infant with macroglossia, a markedly raised creatine kinase and hypertrophic cardiomyopathy consistent with infantile-onset Pompe disease." For galactosaemia: "A neonate on milk feeds with jaundice, liver dysfunction, cataracts and E. coli sepsis consistent with classic galactosaemia." These one-sentence framings are what the examiner listens for in a long case, and they are the bridge between the bedside and the confirmatory testing. [1] [5]
Investigations
The investigation strategy is tiered. The first tier is the fasting metabolic panel, which in the hepatic GSDs distinguishes the lactic, non-ketotic hypoglycaemia of GSD I (with raised urate and triglycerides) from the ketotic hypoglycaemia of GSD III and the milder pattern of GSD VI and IX. A controlled fast, conducted in a supervised setting with frequent glucose monitoring, may be needed to provoke the diagnostic biochemistry, but many centres now proceed directly to enzyme and molecular testing once the panel suggests a GSD, because the fast carries a risk of symptomatic hypoglycaemia and the molecular answer is more precise. Lactate, urate, lipids, liver function, creatine kinase, and a blood gas together form the metabolic fingerprint. [1] [3]
The second tier is the confirmatory testing. For the hepatic GSDs, enzyme assay on a liver biopsy was historically the gold standard. Molecular genetic testing is now the primary confirmatory tool. It sequences the relevant gene (G6PC for GSD Ia, SLC37A4 for GSD Ib, AGL for GSD III, PHKA2 and related genes for GSD IX), and it is less invasive, enables cascade family testing, and supports prenatal and preimplantation diagnosis. For Pompe, the confirmatory test is acid α-glucosidase enzyme activity in dried blood spot, leucocytes or fibroblasts. A low activity on the dried blood spot is a sensitive screen. This is followed by GAA sequencing to confirm and define the variant. Where available, Pompe is now detected through newborn screening programmes that measure GAA activity on the bloodspot. [5] [9]
Why a single normal glucose does not exclude a hepatic glycogen storage disease
The hypoglycaemia and metabolic fingerprint of a hepatic GSD are fasting-dependent: a sample drawn after a feed, or during a period of regular intake, may look completely normal. In a child with a suggestive history — hepatomegaly, early-morning symptoms, growth failure — repeat the panel during a symptomatic episode or after a supervised fast, and supplement it with urate, triglycerides and lactate at the same moment, because the diagnostic pattern is only visible when the liver is under metabolic stress. The same principle applies to hereditary fructose intolerance, where the biochemistry is normal until the offending sugar is ingested. [1] [13]
For the sugar disorders, the investigations are specific. Classic galactosaemia is suggested by raised galactose-1-phosphate and galactitol, a positive reducing substance in the urine (though this is nonspecific and depends on recent intake), and confirmed by GALT enzyme activity in erythrocytes and GALT gene sequencing. Many cases are now detected on newborn screening. Hereditary fructose intolerance is suggested by the clinical syndrome and confirmed by aldolase B enzyme activity (historically on a liver biopsy) and ALDOB sequencing, with a fructose challenge now avoided because it is dangerous. Fructose-1,6-bisphosphatase deficiency shows fasting hypoglycaemia with lactic acidosis and is confirmed by FBP1 sequencing. Across all the disorders, the principle is to confirm the enzyme or gene defect, because the diagnosis commits the family to lifelong management, reproductive counselling, and cascade testing. [12] [13] [14]
Management — Resuscitation
The resuscitation of a child with a suspected hepatic glycogen storage disease is built on one physiological move: give glucose and stop catabolism. The moment fasting hypoglycaemia is identified, establish intravenous access and give a bolus of intravenous dextrose (2 mL/kg of 10 percent glucose), then continue with a glucose infusion rate sufficient to suppress counter-regulation and switch the child into anabolism. Typically 8 to 12 mg/kg/min, titrated upward until the glucose is stable and the lactate falls. Stopping the fast is the single most important intervention, because the brain injury of these disorders is driven by recurrent, prolonged hypoglycaemia, and the metabolic acidosis resolves as the glycolytic shunt shuts down. [1] [7]
Correction of the biochemistry matters as much as the glucose. In GSD I, the lactate, urate and lipid abnormalities are markers of ongoing metabolic stress, and they normalise only when the glucose supply is continuous and sufficient. So the resuscitation is not finished when the glucose is 5 mmol/L, but when the lactate has fallen and the metabolic fingerprint has improved. Concurrent acidosis should be monitored and, if severe, cautiously corrected; hyperuricaemia and dyslipidaemia are addressed once the acute episode is controlled. Throughout, the child should be kept nil by mouth for the offending sugars (fructose and galactose in GSD I) and any intercurrent infection treated aggressively, because infection is the commonest reason for decompensation. [1] [2]
For the child with suspected Pompe in cardiorespiratory failure, the resuscitation is cardiac and respiratory rather than metabolic: supportive management of the heart failure, attention to the airway and ventilation (the diaphragmatic weakness makes respiratory failure the proximate threat), and prompt referral for enzyme replacement therapy once the diagnosis is confirmed or strongly suspected. For galactosaemia presenting with liver failure or sepsis, the resuscitation includes stopping galactose (switch to a galactose-free formula), treating the E. coli sepsis aggressively, and supportive management of the coagulopathy and liver dysfunction. For hereditary fructose intolerance, the resuscitation is the removal of all fructose, sucrose and sorbitol from the diet and intravenous glucose support — which, by itself, often produces dramatic improvement. [5] [12] [13]
Management — Definitive & Stepwise
Once the acute crisis is controlled, the definitive management of the hepatic GSDs is built around preventing fasting — the continuous provision of glucose so that the liver is never asked to release a glucose it cannot make. The mainstay is frequent daytime feeds supplemented with uncooked cornstarch, a slowly digested complex carbohydrate that releases glucose over several hours and extends the fasting interval. In infants and young children, continuous overnight nasogastric or gastrostomy glucose is often required to prevent early-morning hypoglycaemia. The cornstarch regimen, refined over decades, has transformed the prognosis of GSD I from early death to survival into adulthood, and it is the cornerstone of long-term care. [1] [2]
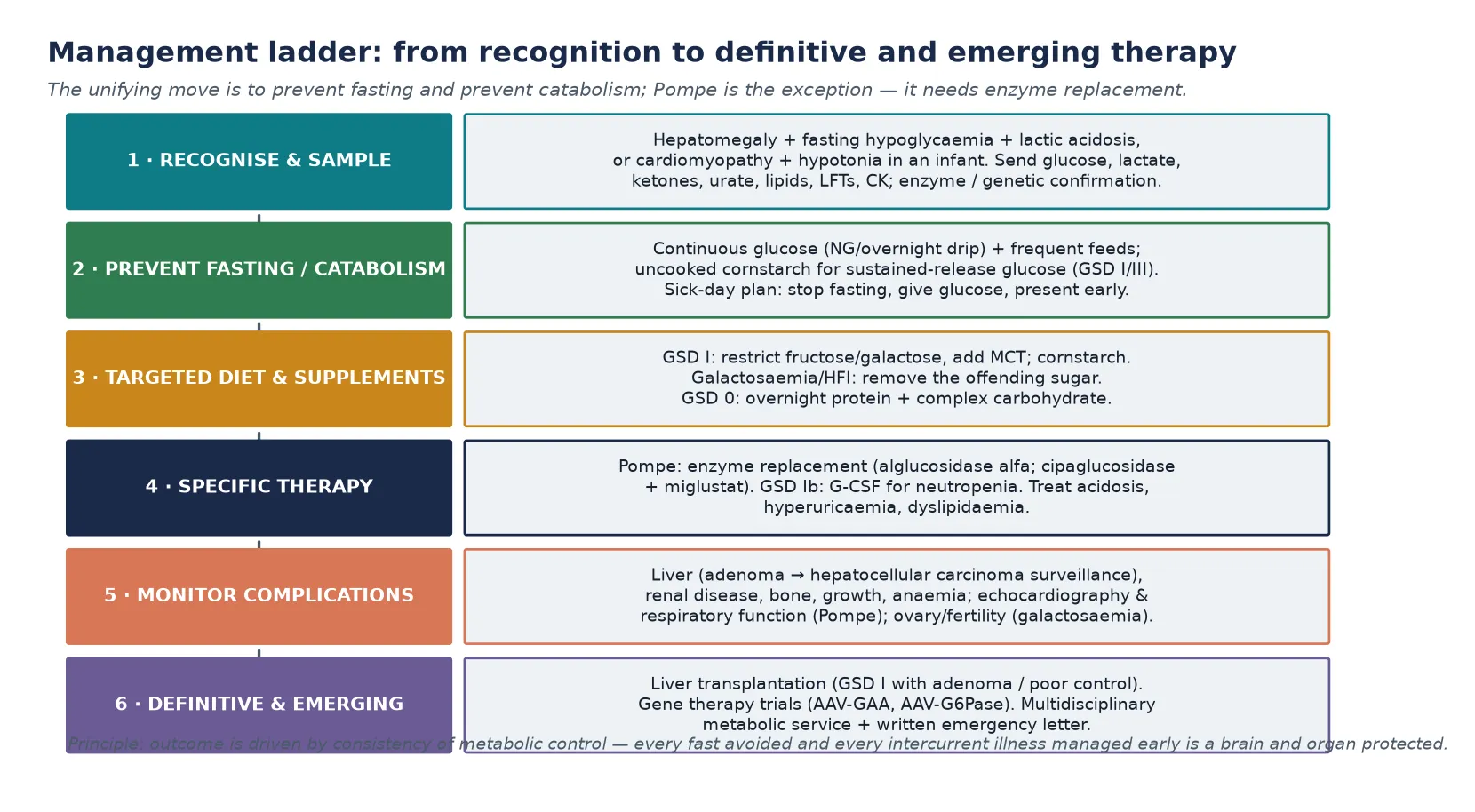
The diet is tailored to the specific defect. In GSD I, fructose and galactose are restricted because they feed the trapped glucose-6-phosphate pool, and medium-chain triglycerides may be added as an alternative fuel. In GSD III, a higher-protein diet with cornstarch is favoured because gluconeogenesis is intact and protein supports muscle; in the milder GSD VI and IX, modest dietary adjustment is often sufficient. Every child needs a written sick-day emergency plan: at the first sign of illness, fasting or reduced intake, the family stops fasting, gives glucose or cornstarch, and presents early. Because catabolism is the enemy, and a minor illness can become a metabolic emergency within hours. A medic alert and a metabolic letter travel with the child everywhere. [1] [8]
P.R.E.V.E.N.T. the fast — the hepatic GSD management principle
The definitive management of Pompe disease is enzyme replacement therapy with recombinant acid α-glucosidase (alglucosidase alfa), which delivers functional enzyme to the lysosome and has transformed the prognosis of infantile Pompe from uniformly fatal to survivable with long-term motor and cardiac benefit. A newer regimen — cipaglucosidase alfa plus the enzyme stabiliser miglustat, demonstrated in the PROPEL trial — offers improved efficacy in late-onset disease. Treatment must be started early, before irreversible muscle damage, and continued lifelong, alongside supportive respiratory and rehabilitation care. For galactosaemia and hereditary fructose intolerance, the definitive management is lifelong strict avoidance of the offending sugar — galactose (including lactose) and fructose/sucrose/sorbitol respectively. This is supplemented by calcium and nutrient replacement. Even with excellent adherence, the long-term outcomes of galactosaemia (cognitive, ovarian and bone) are imperfect and remain an active area of research. [6] [12] [13]
Specific Subtypes & Scenarios
Glycogen storage disease type I is the prototype and the most demanding hepatic form. GSD Ia (glucose-6-phosphatase deficiency, von Gierke disease) and GSD Ib (glucose-6-phosphate translocase deficiency) share the severe fasting-hypoglycaemia phenotype and the biochemical tetrad, but GSD Ib carries the additional burden of neutropenia with recurrent infections and a Crohn-like inflammatory bowel disease that significantly affects quality of life and requires granulocyte colony-stimulating factor (G-CSF) in many patients. Long-term complications include hepatic adenomas (with a risk of malignant transformation to hepatocellular carcinoma that mandates regular imaging), renal disease (glomerular hyperfiltration progressing to proteinuria and renal failure), osteoporosis, anaemia, and growth failure that improves with metabolic control. The European Study on Glycogen Storage Disease Type I guidelines set the structure of management that most programmes adopt. [1] [4] [15]
Pompe disease (GSD II) is the lysosomal exception. The infantile form presents with hypertrophic cardiomyopathy and severe hypotonia and, untreated, is fatal in infancy. Enzyme replacement therapy, particularly when started before irreversible damage (including in presymptomatic infants detected by newborn screening), markedly improves survival, cardiac function and motor outcomes. The late-onset form presents with proximal weakness and respiratory involvement at any age, and the diagnostic delay is often years. A missed diagnosis that a fellow should preempt by checking creatine kinase and GAA activity in any unexplained limb-girdle weakness or ventilatory failure. The PROPEL trial established cipaglucosidase alfa plus miglustat as a next-generation option with superior efficacy in late-onset disease. [5] [6]
Glycogen storage disease type III (Cori/Forbes disease), from debrancher enzyme deficiency, is the second commonest hepatic GSD and is distinguished from GSD I by its ketotic hypoglycaemia, intact gluconeogenesis, and frequent muscle and cardiac involvement that may dominate in adulthood. Management centres on a high-protein diet with cornstarch, and surveillance for the cardiomyopathy and hepatic fibrosis that can develop over time. GSD VI and IX (liver phosphorylase and phosphorylase kinase deficiencies) are the commonest mild hepatic GSDs: they present with hepatomegaly and mild ketotic hypoglycaemia, grow well with minimal intervention, and generally resolve toward normal through childhood. Though ongoing follow-up for growth, liver and (in some) muscle involvement is advised. The milder course is a reassurance for families but does not excuse missing the diagnosis. [7] [8] [9]
The muscle glycogenoses (GSD V McArdle, GSD VII Tarui) present with exercise intolerance, cramps, the second wind, and episodes of myoglobinuria with a risk of acute kidney injury. Management is built around moderated exercise (avoiding maximal isometric exertion), a pre-exercise sucrose boost in McArdle disease (which transiently improves exercise tolerance by providing circulating glucose), and prompt management of rhabdomyolysis episodes to protect the kidney. GSD IV (branching enzyme deficiency, Andersen disease) is rare and severe in its infantile hepatic form (progressive liver failure, often needing transplant) but also includes adult polyglucosan body disease, a late-onset neurogenic disorder. A spectrum that reminds the fellow that the same enzyme defect can present very differently across the life course. [10] [11]
Complications & Pitfalls
The complications divide into the metabolic, the organ-specific, and the cognitive traps. The metabolic complications are the recurrent hypoglycaemic crises driven by fasting, illness or dietary indiscretion, which carry a cumulative risk of neurological injury and, in the acute presentation, seizures and coma. The organ-specific complications of GSD I are the most important to know: hepatic adenomas (appearing in childhood or adolescence, with a real risk of hepatocellular carcinoma that mandates annual or biennial imaging), renal disease (glomerular hyperfiltration, albuminuria, Fanconi-like tubular dysfunction, and progressive renal failure), osteoporosis, anaemia, hyperuricaemic gout and growth failure. Pompe adds cardiorespiratory failure, and the muscle GSDs add rhabdomyolysis and renal injury. [1] [5]
The long-term outcomes of the sugar disorders are themselves a complication. In classic galactosaemia, despite early diagnosis and strict galactose restriction, a substantial proportion of patients develop cognitive and speech difficulties, motor coordination problems, and — most notably — premature ovarian insufficiency, so that reproductive counselling and ovarian function surveillance are part of long-term care. The bone mineral density is also reduced. In hereditary fructose intolerance, strict fructose avoidance prevents the acute crises but requires meticulous lifelong dietary vigilance, because fructose, sucrose and sorbitol are ubiquitous in processed foods, medicines and even intravenous preparations. The recognition that "diet alone" does not fully normalise outcome is a key teaching point. [12] [13]
Prognosis & Disposition
Prognosis is determined by three factors: the specific disorder, the speed of diagnosis and the quality and consistency of long-term metabolic control. The hepatic GSDs, once uniformly fatal in early childhood, now carry a prognosis of survival into adulthood with near-normal cognitive outcome when metabolic control is excellent. The quality of that control is the dominant modifier. Children who experience recurrent hypoglycaemia, or who cannot sustain the cornstarch and overnight glucose regimen, carry a higher burden of growth failure, renal disease and hepatic adenoma. GSD III and the milder GSD VI and IX have a generally favourable prognosis, with the caveat of progressive muscle and cardiac involvement in some GSD III patients. [1] [8]
Pompe disease has undergone the most dramatic prognostic transformation: untreated infantile Pompe is fatal in infancy, but early enzyme replacement therapy — particularly in presymptomatic newborn-screened infants — achieves long-term survival with meaningful motor and cardiac function. The late-onset form is a chronic, slowly progressive disorder in which treatment stabilises or slows decline but does not reverse established muscle loss, so early diagnosis and treatment are again the dominant modifiers. The muscle glycogenoses carry a normal life expectancy complicated by episodes of rhabdomyolysis and renal injury that must be anticipated and managed. The disposition for all of these is lifelong multidisciplinary care through a metabolic service, with a clear and written emergency pathway. [5] [6]
The sugar disorders carry a prognosis shaped by the imperfect outcomes of galactosaemia and the dietary burden of hereditary fructose intolerance. Classic galactosaemia, even with newborn-screened early diagnosis and strict galactose restriction, is associated with long-term cognitive, speech, motor and ovarian sequelae in a substantial proportion of patients, so the prognosis is "survival with disability-modified quality of life" rather than cure. Hereditary fructose intolerance, with meticulous lifelong fructose avoidance, carries an excellent prognosis, but the constant dietary vigilance and the risk of inadvertent fructose exposure (including from medicines) are lifelong burdens. For all the disorders, the disposition is a coordinated network of metabolic, genetics, dietetics, primary care and — for GSD I — hepatology and nephrology services. [12] [13]
Special Populations
The same disorder behaves differently across populations because access, recognition and service models are unevenly distributed. In remote and Indigenous communities, and in migrant and refugee families, the interval between symptom onset and a measured fasting metabolic panel is longer, the dietary and cornstarch regimens are harder to sustain, and the aeromedical retrieval of a hypoglycaemic infant or a cardiorespiratory-compromised Pompe baby adds time that the developing brain cannot afford. A written, location-specific emergency plan, a low threshold to measure the metabolic panel in any child with hepatomegaly or unexplained hypoglycaemia, and a close partnership with the primary care service and retrieval system are the practical responses. And they are the difference between a good and a poor outcome. [1] [15]
In the adolescent and young-adult transition population, the challenge is the handover from the paediatric metabolic service to adult care, with the risks of dietary non-adherence, loss of emergency planning, and the emergence of the long-term complications (adenoma surveillance, renal protection, reproductive counselling in galactosaemia) that dominate adult care. A structured transition, with the young person taking ownership of the sick-day plan and the medic alert, and a warm handover to an adult metabolic physician, is essential. For the technology-dependent child — the GSD I patient on continuous overnight glucose or the Pompe patient on lifelong enzyme replacement and ventilatory support. The family is the unit of care, and the planning must include the home, the school and the emergency services. [1] [6]
For families facing a new diagnosis, the burden is both practical and genetic. Because most of these disorders are autosomal recessive, the diagnosis carries a one-in-four recurrence risk for siblings and obliges cascade carrier testing. The X-linked GSD IX obliges maternal carrier assessment; and the severity of the infantile presentations (Pompe, GSD I) makes reproductive counselling, prenatal and preimplantation genetic diagnosis a central part of the service. The genetic counsellor, the metabolic nurse specialist and the dietitian are as important to the outcome as the physician, and the written plan — diet, sick-day rules, medic alert, school letter — is the family's lifeline. [5] [16]
Evidence, Guidelines & Regional Differences
The evidence base rests on international consensus guidelines, longitudinal cohort data, randomised trials of therapy, and emerging molecular treatment. For the hepatic GSDs, the European Study on Glycogen Storage Disease Type I guidelines (Rake 2002) remain the structural foundation of GSD I management. These are complemented by the cornstarch long-term outcome data (Weinstein 2002) and the genotype-phenotype correlation work (Matern 2002). The liver transplantation evidence is summarised by Boers and colleagues (2014), and gene therapy is reviewed by Koeberl and colleagues (2024). For GSD III, the Kishnani (2010) guidelines and the Sentner (2016) genotype and outcome review define current practice. For GSD VI and IX, the American College of Medical Genetics resource (Kishnani 2019) is the standard; and for GSD IV, the Koch (2023) clinical practice resource covers the full spectrum including adult polyglucosan body disease. [1] [9] [10]
For Pompe disease, the Kishnani diagnosis and management guideline (2006) established the framework for enzyme replacement, and the PROPEL trial (Schoser 2022) established cipaglucosidase alfa plus miglustat as a next-generation regimen with superior efficacy in late-onset disease. A rare randomised controlled trial in a rare disease. The muscle glycogenoses are covered by the Lucia (2021) international practice guidelines. For the sugar disorders, the galactosaemia evidence includes the Van Calcar (2014) re-evaluation of lifelong galactose restriction, which has shifted practice toward a less draconian diet while acknowledging the persistent long-term outcomes. Hereditary fructose intolerance is reviewed by Ali (1998); and fructose-1,6-bisphosphatase deficiency by Yi (2022). [5] [6] [11]
In Australia and New Zealand, the glycogen storage and carbohydrate metabolism disorders are managed through the state-based metabolic services. These are coordinated through the major children's hospitals in each state and Starship in New Zealand. They use a shared-care model that places the general paediatrician, the metabolic physician, the dietitian and the genetic counsellor in a single network. Newborn screening detects classic galactosaemia and, increasingly, Pompe disease (GAA on the bloodspot) in most jurisdictions, so several of these conditions now present as presymptomatic screen-positive results rather than acutely unwell children. A shift that has improved outcome but that obliges the clinician to confirm promptly and to start enzyme replacement (Pompe) before irreversible damage. Aeromedical retrieval to a tertiary metabolic centre, a written sick-day plan, and access to uncooked cornstarch and continuous overnight glucose are the practical pillars of rural and remote care. [1] [5]
Exam Pearls
A fellowship candidate answering on the glycogen-storage and carbohydrate metabolism disorders should land six anchor points and avoid three classic traps. The anchors are the three clinical patterns (hepatic energy failure, lysosomal muscle disease of Pompe, exertional muscle pain of the muscle GSDs). Next is the biochemical tetrad of GSD I (hypoglycaemia, lactic acidosis, hyperuricaemia, hypertriglyceridaemia). The third anchor is the fasting panel taken before glucose correction, and how it localises the defect. The fourth is the unifying management principle of preventing fasting and catabolism with cornstarch and continuous glucose. The fifth is enzyme replacement therapy that transforms Pompe. The last is the long-term complications (adenoma, renal disease, the imperfect galactosaemia outcomes) that drive surveillance. The sugar disorders are held as toxic-metabolite diseases that resolve on removing the offending sugar. [1] [5]
The three traps to avoid are the misclassification of GSD I as "ketotic hypoglycaemia" (it is lactic and non-ketotic), and the failure to think of Pompe in a floppy infant with a big heart (measure the creatine kinase and echo). The third is the reassurance that galactosaemia is "cured" by diet, when the long-term cognitive, ovarian and bone outcomes remain imperfect. A candidate who pairs the three patterns with the metabolic fingerprint, who can deliver the cornstarch and enzyme-replacement management with a written sick-day plan, and who knows the surveillance agenda for adenoma, kidney, bone and ovary will answer this topic at fellowship standard across a written, viva or long-case format. [7] [12]
References
- [1]Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GPA. Guidelines for management of glycogen storage disease type I - European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr, 2002.PMID 12373584
- [2]Weinstein DA, Sommer M, Stevens S, Wolfsdorf JI. Effect of continuous glucose therapy with uncooked cornstarch on the long-term clinical course of type 1a glycogen storage disease. Eur J Pediatr, 2002.PMID 12373568
- [3]Matern D, Seydewitz HH, Bali D, Lang C, Chen YT. Glycogen storage disease type I: diagnosis and phenotype/genotype correlation. Eur J Pediatr, 2002.PMID 12373566
- [4]Sim SW, Lee YC, Liu YF, Lin CH, Yang CC, Hwu WL, Lee NC. Glycogen storage disease type Ib: role of glucose-6-phosphate transporter in cell metabolism and function. FEBS Lett, 2020.PMID 31705665
- [5]Kishnani PS, Howell RR, Mandel H, Corzo D, Leslie N, Watson MS, et al. Pompe disease diagnosis and management guideline. Genet Med, 2006.PMID 16702877
- [6]Schoser B, Stewart F, Behin A, Bastaki L, Bhatia P, Bhattacharya K, et al. Safety and efficacy of cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo in late-onset Pompe disease (PROPEL): an international, randomised, double-blind, parallel-group, phase 3 trial. Lancet Neurol, 2021.PMID 34800400
- [7]Kishnani PS, Austin SL, Arn P, Bali DS, Boney A, Case LE, et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med, 2010.PMID 20631546
- [8]Sentner CP, Hoogeveen IJ, Weinstein DA, Santra S, Bhavsar C, Smit GPA, et al. Glycogen storage disease type III: diagnosis, genotype, management, clinical course and outcome. J Inherit Metab Dis, 2016.PMID 27106217
- [9]Kishnani PS, Goldstein J, Austin SL, Arn P, Bachrach B, Bali DS, et al. Diagnosis and management of glycogen storage diseases type VI and IX: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med, 2019.PMID 30659246
- [10]Koch RL, Bhattacharya K, Bhattacharjee A, Chen MA, Dessoffy K, Ekstein J, et al. Diagnosis and management of glycogen storage disease type IV, including adult polyglucosan body disease: a clinical practice resource. Mol Genet Metab, 2023.PMID 36796138
- [11]Lucia A, Quinlivan R, Nogales-Gadea G, Martinuzzi A, Arenas J, Marín-Quiles F, et al. Clinical practice guidelines for glycogen storage disease V & VII (McArdle disease and Tarui disease) from an international study group. Neuromuscul Disord, 2021.PMID 34848128
- [12]Van Calcar SC, Bernstein DL, Rohr F, Waisbren SE, Berry GT, Yannicelli S, et al. A re-evaluation of life-long severe galactose restriction for the nutrition management of classic galactosemia. Mol Genet Metab, 2014.PMID 24857409
- [13]Ali M, Rellos P, Cox TM. Hereditary fructose intolerance. J Med Genet, 1998.PMID 9610797
- [14]Yi C. Fructose-1,6-bisphosphatase deficiency. Endokrynol Pol, 2022.PMID 35971930
- [15]Boers SJ, Visser G, Smit PG, Fuchs SA. Liver transplantation in glycogen storage disease type I. Orphanet J Rare Dis, 2014.PMID 24716823
- [16]Koeberl DD, Kishnani PS, Chen YT. Gene therapy for glycogen storage diseases. J Inherit Metab Dis, 2024.PMID 37421310