Paeds · genetics-dysmorphology-and-metabolism
Hypoglycaemia due to inherited metabolic disease
Also known as Metabolic hypoglycaemia · Hypoketotic hypoglycaemia · Congenital hyperinsulinism · CHI · Fatty acid oxidation defect with hypoglycaemia · MCAD deficiency · Glycogen storage disease type I · GSD Ia
A fellowship approach to hypoglycaemia caused by inherited metabolic disease: recognise the fasted or febrile child with hypoketotic hypoglycaemia, seizures or hepatomegaly as an emergency, capture the critical sample before treating, then work through the three physiological failures — insulin-driven (congenital hyperinsulinism), glucose-production failure (glycogen storage disease, fructose-1,6-bisphosphatase deficiency), and fuel-oxidation block (fatty-acid oxidation defects) — each with its own acute and lifelong management.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who works in three layers at once. The first is the child in front of you — an encephalopathic neonate or a seizing infant — where the immediate questions are the glucose, the ketones, and the critical sample, not the gene. The second is the physiology: glucose homeostasis is a balance between supply and utilisation, and ketones are the brain's alternate fuel when glucose falls, so a child who cannot make ketones is doubly exposed. The third is the family: a monogenic cause such as congenital hyperinsulinism or a fatty-acid oxidation defect carries a recurrence risk, a risk to siblings, and a lifelong management obligation that begins the moment the diagnosis is named. [1] [2]
Overview & Definition
Hypoglycaemia due to inherited metabolic disease is a low blood glucose caused by a genetically determined failure of one of the metabolic pathways that maintain the blood sugar during fasting. The threshold that should trigger investigation is a plasma glucose below roughly 2.6 to 3.0 millimoles per litre in a symptomatic child, with the Pediatric Endocrine Society recommending a structured evaluation of any child with persistent or recurrent hypoglycaemia rather than accepting a single normal value. The inherited causes matter because they are recurrent, potentially lethal, and — uniquely among the causes of hypoglycaemia — they offer a disease-specific treatment once the diagnosis is made. [1]
Clinically, these disorders sit within the family of intoxication-type and energy-failure-type inborn errors of metabolism: conditions in which a blocked pathway produces either a toxic accumulation or an energy deficit that is itself the disease. The framing matters because it dictates management — the priority is to restore substrate and capture the diagnostic window during the decompensation, because the abnormal metabolites and hormone profile are only visible while the child is hypoglycaemic. Once glucose is corrected the biochemical fingerprint disappears, which is why a missed critical sample can delay the diagnosis for years. [9] [10]
Classification
The classification that earns marks is physiological, because the position of the failure determines the clinical pattern, the critical-sample signature, and the treatment. A child whose pancreas secretes insulin inappropriately, a child whose liver cannot release glucose, and a child whose fat cannot be oxidised all present with hypoglycaemia — but their ketones, fatty acids, lactate and insulin tell them apart. This three-way split — insulin-driven, glucose-production failure, and fuel-oxidation block — is the framework the Pediatric Endocrine Society and the Saudubray pathophysiological classification both converge on, and it is the structure that makes the critical sample interpretable. [1] [10]

The insulin-driven group is dominated by congenital hyperinsulinism, in which a genetic defect — most often in the KATP-channel genes ABCC8 or KCNJ11 — keeps the pancreatic beta cell depolarised and secreting insulin regardless of the glucose level. The hallmark biochemical signature is a measurable insulin, a suppressed beta-hydroxybutyrate, and a suppressed free fatty acid at the moment of hypoglycaemia, because insulin is an anabolic hormone that simultaneously drives glucose uptake and switches off both glycogenolysis and lipolysis. The glucose-production group centres on glycogen storage disease type I, where glucose-6-phosphatase deficiency blocks the final step of both glycogenolysis and gluconeogenesis, producing fasting hypoglycaemia with a lactic acidosis, hyperuricaemia and hepatomegaly. The fuel-oxidation group is the fatty-acid oxidation defects, of which medium-chain acyl-CoA dehydrogenase deficiency is the prototype: the block in beta-oxidation means the liver cannot make ketones, so the brain loses its alternate fuel and the child presents with hypoketotic hypoglycaemia after a fast. [2] [4] [6]
Epidemiology & Risk Factors
Congenital hyperinsulinism is the commonest cause of severe persistent hypoglycaemia in infancy, with an overall incidence of roughly one in 25,000 to 50,000 live births in outbred populations and much higher — up to one in 2,500 — in communities with consanguinity, because the recessive KATP-channel defects concentrate in founder populations. The Kapoor series of 300 molecularly characterised patients and the Snider series of 417 children together established that KATP-channel mutations underlie the majority of severe cases. The diffuse versus focal histological distinction predicts both the response to medical therapy and the need for surgery, and the genotype correlates with severity and with the chance of a focal lesion amenable to curative resection. [2] [3]
Among the fatty-acid oxidation defects, medium-chain acyl-CoA dehydrogenase deficiency is by far the most common, with an incidence around one in 10,000 to 20,000 live births and a recognition that has shifted dramatically since newborn screening by tandem mass spectrometry became routine. Before screening, the typical presentation was a previously well toddler found comatose or dead after a viral illness with vomiting, with a mortality that could approach 25 percent in the first recognised episode — a figure that underpins why a hypoketotic hypoglycaemic child is never sent home without a work-up. Glycogen storage disease type I is rarer, at roughly one in 100,000, but it is the archetype of the glucose-production group and the gateway to understanding the gluconeogenesis disorders. [4] [6]
The risk factors that raise the pre-test probability are consanguinity and a family history of unexplained neonatal or infant death, recurrent hypoglycaemia, or seizures — and these are the questions that must be asked explicitly because the inherited causes are autosomal recessive or, less commonly, dominant. The metabolic trigger is almost always a fast, an intercurrent illness that reduces intake, or a period of increased metabolic demand, which is why the history of fasting tolerance is the single most useful clinical anchor. A neonate who is large for gestational age, or who has features of Beckwith-Wiedemann syndrome, carries a higher risk of congenital hyperinsulinism. [2] [1]
Pathophysiology
The molecular story is about substrate balance and the brain's dependence on two fuels. During fasting, a normal child maintains glucose through glycogenolysis for the first few hours and then through gluconeogenesis, while simultaneously mobilising fat from adipose tissue and oxidising it in the liver to make ketones. The ketones — beta-hydroxybutyrate and acetoacetate — cross the blood-brain barrier and become the brain's principal energy source once glucose falls, which is why a fasting child tolerates a low glucose far better than an adult: the brain has an alternative. [9] [1]
Inherited hypoglycaemic disease breaks this system at one of three points. In congenital hyperinsulinism, a mutant beta cell secretes insulin regardless of the glucose, so the brain is hit from both sides: insulin drives peripheral glucose uptake, suppresses glycogenolysis and gluconeogenesis, and suppresses lipolysis and ketogenesis, so there is no glucose and no ketone alternative. In the glycogen and gluconeogenesis defects, the liver simply cannot release glucose, so fasting produces progressive hypoglycaemia — but fat oxidation is intact, so ketones rise and the picture is ketotic, with the exception of glycogen storage disease type I where the accumulated glucose-6-phosphate is shunted to lactate. In the fatty-acid oxidation defects, the block in beta-oxidation prevents ketone formation, so the brain is deprived of its alternate fuel exactly when it needs it most. [6] [10]
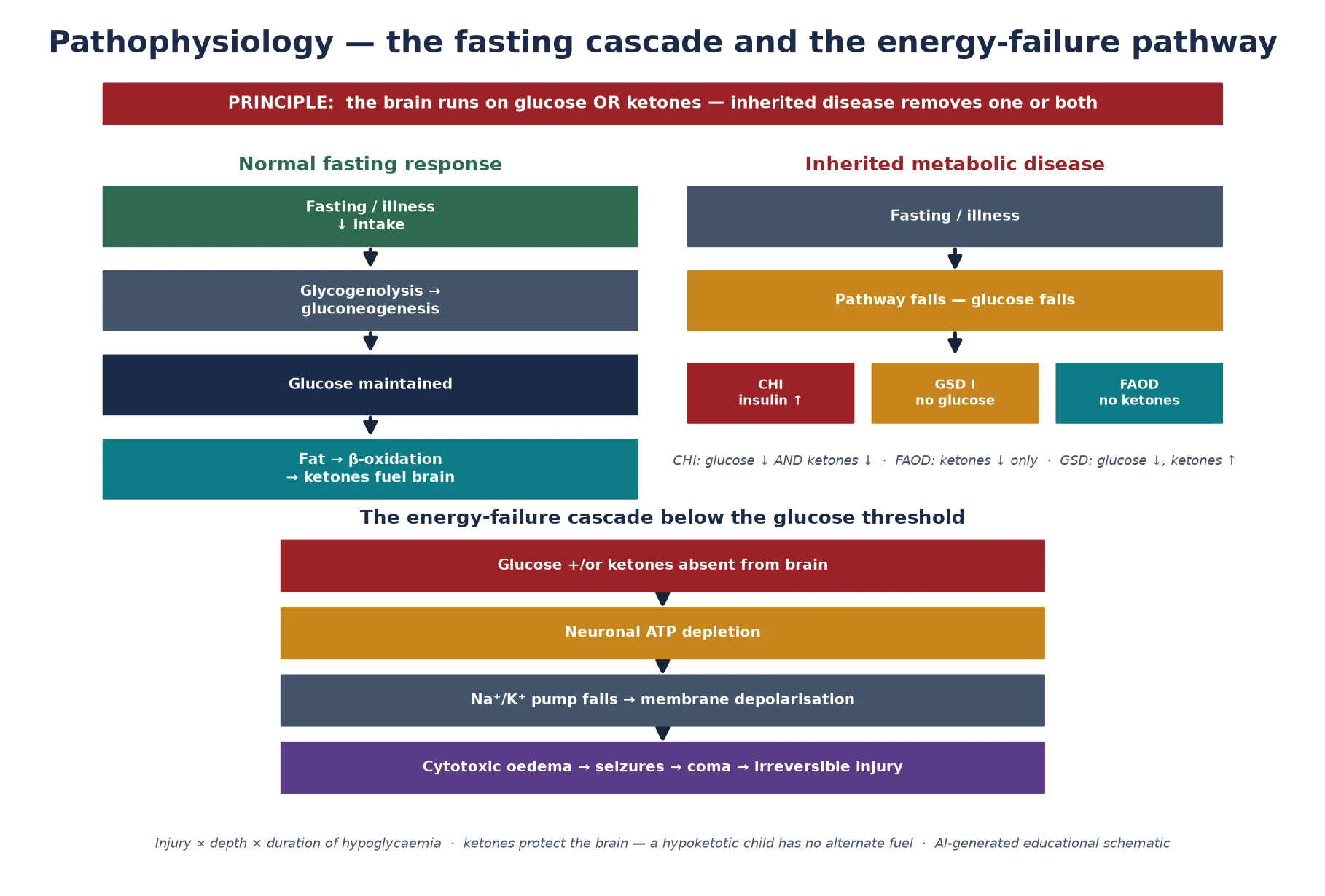
The brain injury that follows is an energy failure. Neurons depend on a continuous supply of ATP to maintain the sodium-potassium pump and the membrane potential; when fuel is absent, ATP depletes, the pump fails, water moves into the cell, and the result is cytotoxic oedema, neuronal depolarisation, seizures, and ultimately irreversible injury. The injury is proportional to both the depth and the duration of the hypoglycaemia. The Garg and Devaskar analysis of the long-term impacts of neonatal hypoglycaemia argues for an operational threshold that prompts treatment rather than waiting for symptoms, and the speed of correction of the first recognised episode is the dominant modifier of neurodevelopmental outcome. [12] [1]
Clinical Presentation
The presentation has a recognisable core that cuts across the specific diagnosis: a child who becomes unwell during a fast or an intercurrent illness, with symptoms of neuroglycopenia — jitteriness, irritability, poor feeding, hypotonia, seizures, apnoea, and coma. The tempo is usually hours, and the trigger is almost always reduced intake from a viral illness, a missed overnight feed, or a surgical procedure. The age at first presentation is a useful discriminator: congenital hyperinsulinism and glycogen storage disease declare themselves in the neonatal period or early infancy, the fatty-acid oxidation defects classically emerge in toddlers once overnight feeds are dropped, and idiopathic ketotic hypoglycaemia presents in preschool children with thin build and a history of poor fasting tolerance. [1] [9]
The physical examination adds discriminating signs. Hepatomegaly points to glycogen storage disease type I (often with a doll-like face, short stature and a protuberant abdomen), to the fatty-acid oxidation defects where the liver may be enlarged with steatosis, or to a gluconeogenesis disorder. Cardiomyopathy, muscle pain, or a history of dark urine after exercise raises a long-chain fatty-acid oxidation defect or a carnitine palmitoyltransferase deficiency. Macrosomia, hemihypertrophy, macroglossia or umbilical hernia suggests Beckwith-Wiedemann-associated hyperinsulinism. Pigmentation or hyperpigmentation with hypoglycaemia raises an adrenal cause, and the dysmorphic features of a syndromic hyperinsulinism should be sought. [4] [8]
Differential Diagnosis
The differential of childhood hypoglycaemia is broad, and the inherited metabolic diseases are one — important — slice of it. The first task is to separate the inherited metabolic causes from the non-metabolic mimics, because the management and the recurrence risk differ. Idiopathic ketotic hypoglycaemia is the commonest cause of hypoglycaemia beyond infancy, presenting in a thin preschool child after a fast, with appropriately raised ketones and a benign course that resolves with age — it is a diagnosis of exclusion made only after the inherited and endocrine causes are ruled out. Endocrine causes — growth hormone deficiency, cortisol deficiency from adrenal insufficiency or congenital adrenal hyperplasia, and hypopituitarism — produce hypoglycaemia that is usually ketotic, because the counter-regulatory response is intact but the substrate is deficient. [1] [11]
The toxic and pharmacological causes — oral hypoglycaemics, insulin administration, alcohol, beta-blockers, and salicylates — must be considered in any child with unexplained hypoglycaemia, particularly when the pattern is atypical or there is a safeguarding concern. Sepsis and systemic illness can produce hypoglycaemia through a combination of increased utilisation and impaired gluconeogenesis, and in the neonate the differential from transitional hyperinsulinaemia of the infant of a diabetic mother must be resolved by the time course and the insulin requirement. The discriminator is the critical sample and the fasting tolerance history: an inherited metabolic disease declares itself with a reproducible biochemical signature during a documented hypoglycaemic episode, whereas the mimics either resolve with the underlying illness or carry a different hormonal profile. [9] [1]
Clinical & Bedside Assessment
The bedside assessment runs in parallel with resuscitation, because the diagnostic information is only available while the child is hypoglycaemic. The immediate moves are to confirm the glucose with a laboratory measurement (capillary strips are inaccurate at low ranges), draw the critical sample, and begin treatment. The history, taken simultaneously, must capture the fasting tolerance, the precipitating illness, the age and mode of onset, and the family history of consanguinity, unexplained death, or known metabolic disease. Do not delay glucose correction for a complete history in a symptomatic child, but do not miss the critical sample — if the child is seizing, treat first and sample during the recovery. [1]
The examination looks for the stigmata that localise the diagnosis: hepatomegaly and the doll-like facies of glycogen storage disease, the cardiomyopathy of a long-chain fatty-acid oxidation defect, the macrosomia and hemihypertrophy of Beckwith-Wiedemann, the midline defects and optic hypoplasia of septo-optic dysplasia with hypopituitarism, and the hyperpigmentation of adrenal insufficiency. A glucagon stimulation test performed during the hypoglycaemic episode is both diagnostic and therapeutic. A glycaemic rise of more than 1.5 to 2.0 millimoles per litre after intramuscular or intravenous glucagon indicates that glycogen is present and mobilisable and that insulin is high, pointing to congenital hyperinsulinism, whereas no response suggests a glycogen storage or gluconeogenesis disorder. [1] [2]
The bedside assessment converts directly into the investigation plan. The critical sample is the panel that interprets the hypoglycaemia at the moment it occurs, and it is the single highest-yield diagnostic act. If the child arrives normoglycaemic after pre-hospital treatment, a supervised diagnostic fast may be required, but this is a specialist metabolic or endocrine procedure with a rescue protocol, not a ward exercise. The general paediatrician's role is to ensure that every hypoglycaemic episode is sampled, that the family is given a provisional plan, and that the child is referred to the metabolic or endocrine service for definitive characterisation. [1] [9]
Investigations
The critical sample is the cornerstone, and it must be drawn while the child is genuinely hypoglycaemic — a plasma glucose below 2.6 to 3.0 millimoles per litre — because the hormone and metabolite profile normalises the moment glucose is corrected. The panel that the Pediatric Endocrine Society recommends includes glucose, insulin, beta-hydroxybutyrate, free fatty acids, lactate, ammonia, and the counter-regulatory hormones growth hormone and cortisol, supplemented by plasma acylcarnitines and a urine sample for ketones and organic acids. The interpretation is relational, not absolute: insulin is "high" only in relation to a low glucose, and beta-hydroxybutyrate is "low" only in relation to the degree of hypoglycaemia — a measurably high insulin or a suppressed beta-hydroxybutyrate at the glucose nadir is itself the diagnosis. [1]
The pattern interpretation converts the panel into a working diagnosis. A measurable insulin with suppressed ketones and free fatty acids at a low glucose indicates congenital hyperinsulinism, and a positive glucagon response confirms it. A low insulin with high free fatty acids but inappropriately low ketones indicates a fatty-acid oxidation defect, and the acylcarnitine profile and urine organic acids identify the specific enzyme. Hypoglycaemia with a high lactate, hyperuricaemia and hepatomegaly indicates glycogen storage disease type I, confirmed by a deficient glucose-6-phosphatase response on a glucagon or lactate challenge and by molecular testing. A normal anion gap with ketosis and an appropriate counter-regulatory response points toward idiopathic ketotic hypoglycaemia once the inherited causes are excluded. [9] [6]
Why one normal glucose does not close the case
Most inherited hypoglycaemic diseases produce intermittent hypoglycaemia, so a single normal glucose drawn when the child is well and recently fed does not exclude the diagnosis. In a child with a suggestive history — episodic symptoms after fasting or illness, a positive family history, or prior unexplained hypoglycaemia — the critical sample must be captured during a documented episode, which may require a supervised diagnostic fast. Discharging a child without a critical sample and a provisional diagnosis is the error that allows a fatty-acid oxidation defect to present later as sudden death. [1] [11]
Molecular confirmation is then undertaken by sequencing the candidate gene suggested by the biochemical pattern — ABCC8 and KCNJ11 for congenital hyperinsulinism, ACADM for medium-chain acyl-CoA dehydrogenase deficiency, G6PC and SLC37A4 for glycogen storage disease type I — or by a metabolic gene panel or exome when the pattern is ambiguous. For congenital hyperinsulinism, distinguishing a diffuse from a focal lesion is the pivotal downstream step, because a focal lesion is curable by limited pancreatectomy while diffuse disease is managed medically or by near-total pancreatectomy; 18-fluorodopa positron-emission tomography and the genetic pattern together guide this distinction. Confirming the molecular defect enables cascade testing, prenatal or preimplantation diagnosis, and — for the fatty-acid oxidation defects — confirms whether a sibling is at risk. [2] [3]
Management — Resuscitation
Resuscitation is the section that determines outcome, because the brain injury is proportional to the depth and duration of the hypoglycaemia. The principle is to correct glucose immediately while preserving the diagnostic window: draw the critical sample first if it is safe, then treat. A child who is seizing or comatose is treated first, but every effort must be made to capture a sample during the episode or in the recovery window. The immediate move is an intravenous bolus of dextrose at 0.3 grams per kilogram — 2 millilitres per kilogram of 10 percent dextrose — followed by a continuous infusion at 6 to 9 milligrams per kilogram per minute, titrated upward until the glucose is stable above 3.5 millimoles per litre. [1]
In the hyperinsulinaemic child the glucose requirement is often far above the physiological range, and a need for more than 10 to 12 milligrams per kilogram per minute is itself a diagnostic clue to congenital hyperinsulinism. Glucagon, given intramuscularly or intravenously at 0.5 to 1 milligram (or 20 to 30 micrograms per kilogram), is both a rescue therapy that mobilises glycogen under the high insulin and a diagnostic test that confirms the insulin-driven mechanism. If intravenous access is delayed, intramuscular glucagon can stabilise the child while a line is placed. Octreotide, a somatostatin analogue that suppresses insulin secretion, is used as a bridge in the refractory case, and hydrocortisone covers the possibility of adrenal insufficiency until the critical sample returns. [1] [2]
Management — Definitive & Stepwise
Once the acute episode is controlled, definitive management is disease-specific and lifelong, and it is built around the physiological failure the critical sample has identified. The three pillars — suppress the insulin, sustain the glucose, or bypass the oxidation block — each carry their own therapy, their own monitoring, and their own emergency plan. The unifying principle is that recurrence is preventable, and that the family is the front line of that prevention through a written sick-day plan, home glucose and ketone monitoring where appropriate, and a clear threshold for presentation. [1] [9]
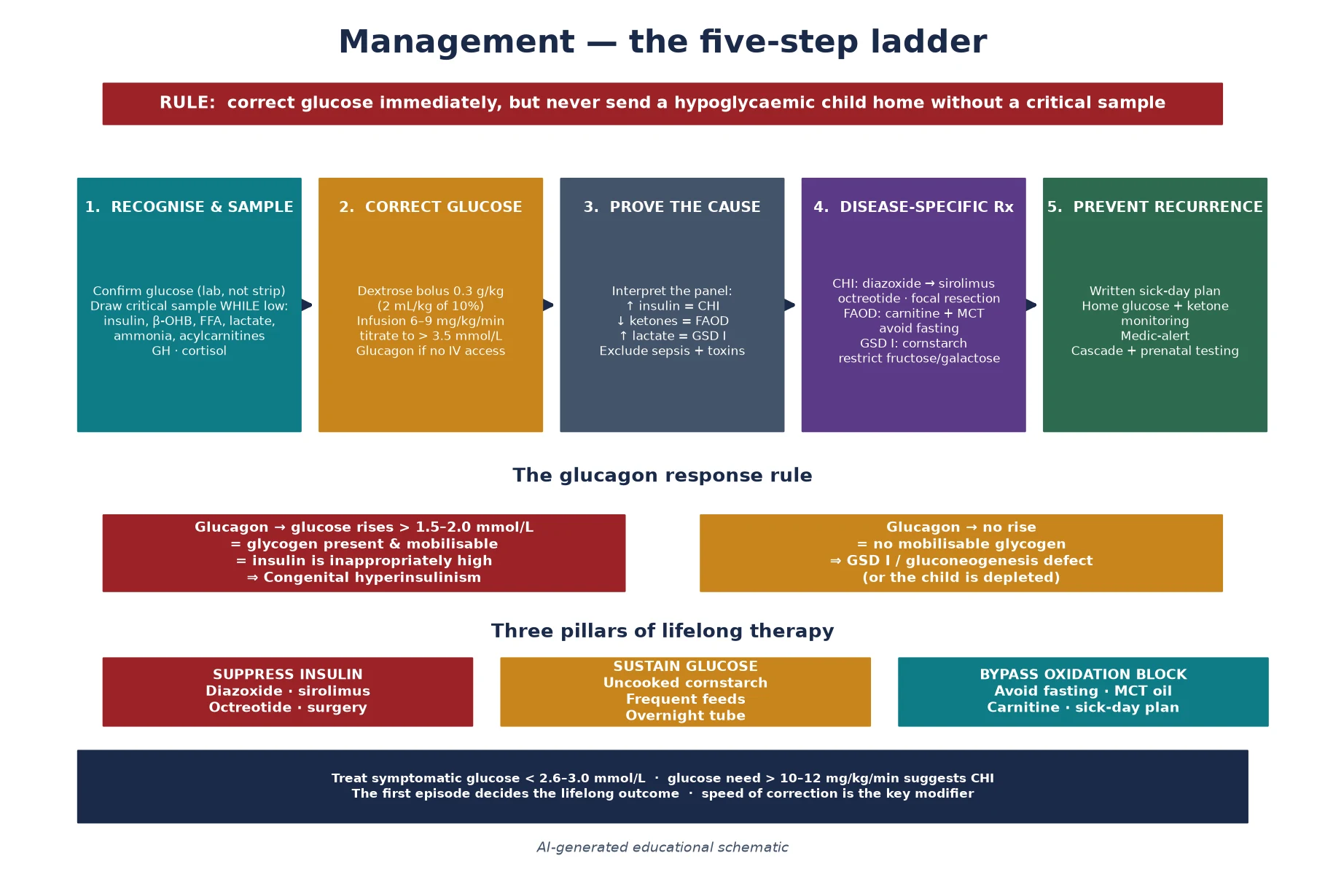
For congenital hyperinsulinism, the medical mainstay is diazoxide, a KATP-channel opener that hyperpolarises the beta cell and switches off insulin secretion; the KATP-channel recessive forms are typically diazoxide-unresponsive, while the milder dominant and GDH-driven forms respond well. In diazoxide-unresponsive disease, sirolimus and other mammalian-target-of-rapamycin inhibitors are increasingly used as a medical alternative to surgery, with octreotide or lanreotide as additional suppressants. When medical therapy fails to maintain safe glucose, near-total pancreatectomy is undertaken — but the focal lesion, identifiable by 18-fluorodopa positron-emission tomography and curable by limited resection, is the diagnosis that must not be missed, because limited surgery cures while near-total pancreatectomy trades hypoglycaemia for diabetes and exocrine insufficiency. [2] [3]
For the fatty-acid oxidation defects, the cornerstone is the prevention of catabolism: avoid fasting, ensure frequent feeds, and provide an emergency sick-day plan that switches to high-carbohydrate intake at the first sign of illness. Carnitine supplementation is used where there is a secondary carnitine deficiency, and in the long-chain disorders medium-chain triglyceride oil provides a substrate that bypasses the block and anaplerotic therapy may be considered. For glycogen storage disease type I, the therapy is the maintenance of a continuous glucose supply through frequent daytime feeds and overnight uncooked cornstarch — a slow-release glucose polymer — supplemented by a restriction of fructose and galactose. The disease is also the target of active gene-therapy development, reviewed by Koeberl and colleagues, which promises to address the enzyme defect rather than its downstream consequences. [6] [4] [5]
C.R.I.T.I.C.A.L. — the sample that makes the diagnosis
Specific Subtypes & Scenarios
Congenital hyperinsulinism is the prototype of the insulin-driven group and the commonest cause of severe persistent hypoglycaemia in infancy. The recessive KATP-channel defects (ABCC8, KCNJ11) produce severe diazoxide-unresponsive disease, either diffuse — requiring medical suppression or near-total pancreatectomy — or focal, where a paternally inherited mutation with somatic loss of the maternal allele in a single lesion is curable by limited resection. The dominant forms, including the glutamate dehydrogenase hyperinsulinism-hyperammonaemia syndrome (GLUD1), are milder and diazoxide-responsive, and the GDH form carries a characteristic mild persistent hyperammonaemia that is a high-yield exam clue. The Snider and Kapoor cohorts established that genotype predicts the histology, the diazoxide response, and the surgical strategy. [2] [3]
Medium-chain acyl-CoA dehydrogenase deficiency is the prototype of the fuel-oxidation group and the fatty-acid oxidation defect most likely to present as hypoketotic hypoglycaemia or sudden death. The typical history is a previously well toddler, between one and two years old, who develops vomiting during a viral illness, is found comatose or seizing after a fast of several hours, and has a hypoketotic hypoglycaemia with an abnormal acylcarnitine profile (elevated octanoylcarnitine). Newborn screening has transformed the prognosis by identifying at-risk infants before the first decompensation, but a screen-negative or missed case can still present catastrophically, which is why every hypoketotic hypoglycaemic child receives the full work-up. [6] [7]
Glycogen storage disease type I (von Gierke disease) is the prototype of the glucose-production group, caused by glucose-6-phosphatase deficiency (Ia) or its translocase (Ib). The clinical picture is fasting hypoglycaemia with lactic acidosis, hyperuricaemia, hyperlipidaemia, and hepatomegaly from infancy, and the long-term complications include growth failure, hepatic adenomas with malignant potential, and renal disease. Management with uncooked cornstarch and frequent feeds has transformed survival and development, and the disorder is a leading target for gene therapy. The rarer gluconeogenesis defects — fructose-1,6-bisphosphatase deficiency and the milder glycogenoses 0, III, VI and IX — share the fasting hypoglycaemia with ketosis and lactic acidosis but have a milder course and a better prognosis. [4] [5]
The carnitine transporter and carnitine palmitoyltransferase defects complete the fuel-oxidation picture and deserve specific recognition because primary carnitine deficiency is curable with carnitine supplementation. Carnitine palmitoyltransferase I deficiency presents with hypoketotic hypoglycaemia and hepatomegaly, the transporter defect with cardiomyopathy and hypotonia that responds dramatically to carnitine, and carnitine palmitoyltransferase II deficiency with exercise-induced rhabdomyolysis or, in the severe neonatal form, with multisystem disease. Stanley's review of the carnitine deficiency disorders remains the framework for distinguishing these treatable conditions from the irreversible beta-oxidation enzyme defects. [8]
Complications & Pitfalls
The complications divide into the acute neurological injuries, the chronic multisystem burden, and the cognitive traps that cost marks. The acute injuries are seizures, coma, cerebral oedema and death, and the risk of permanent neurodevelopmental impairment tracks the depth and duration of the hypoglycaemia — which is why the Garg and Devaskar analysis argues for treating at an operational threshold and why the first episode is the most important one to manage well. The chronic burden includes the neurodevelopmental sequelae of recurrent hypoglycaemia in poorly controlled congenital hyperinsulinism, the hepatic adenomas and renal disease of glycogen storage disease type I, the cardiomyopathy and rhabdomyolysis of the long-chain fatty-acid oxidation defects, and the diabetes and exocrine insufficiency that follow near-total pancreatectomy. [12] [4]
The chief cognitive trap is the missed critical sample, which takes three forms: treating before sampling (necessary in the seizing child, but the recovery window must be used), sampling when the child is no longer hypoglycaemic (the panel is uninterpretable), and discharging without a plan to capture the next episode. The second trap is misclassification — confusing a ketotic from a hypoketotic hypoglycaemia, which changes the entire work-up and the recurrence counselling. The third is missing the family, because a recessive fatty-acid oxidation defect or hyperinsulinism gene carries a recurrence risk in future pregnancies and a risk to siblings that must be addressed by cascade testing and prenatal or preimplantation diagnosis. [1] [2]
Prognosis & Disposition
Prognosis is determined by three factors: the specific disease and how completely it can be controlled, the severity and duration of the hypoglycaemia at presentation, and the consistency of the long-term management. A child with a diazoxide-responsive congenital hyperinsulinism or a well-managed fatty-acid oxidation defect detected on newborn screening can expect near-normal development, whereas severe KATP-channel diffuse hyperinsulinism with recurrent hypoglycaemia despite therapy, or a fatty-acid oxidation defect presenting with a prolonged coma, carries a significant neurodevelopmental burden. The speed and completeness of the acute response is the single most powerful modifier of the lifelong outcome, which is why the first episode is the one that matters most. [2] [12]
Disposition is shared, lifelong, and multidisciplinary. The specialist metabolic or endocrine service owns the diagnostic characterisation, the disease-specific therapy, and the surveillance for the long-term complications. The general paediatrician or general practitioner owns coordination, immunisation, growth and developmental monitoring, and the front-line response to intercurrent illness. The family owns the day-to-day vigilance that prevents recurrence — the sick-day plan, the home glucose and ketone monitoring, the medic-alert, and the early presentation at the first sign of a catabolic trigger. Every transition — into solid foods, into overnight fasting, into school, and into adolescence — is a high-risk point, so the emergency plan and the contact pathway must be re-taught at each stage. [1] [4]
Special Populations
The same inherited hypoglycaemic disease behaves differently across populations because access, recognition, and service models are unevenly distributed. In remote and Indigenous communities, later presentation, distance from a metabolic or endocrine service, and the need for aeromedical retrieval during a crisis mean that the window between onset and treatment is longer and outcomes are worse. A written, location-specific emergency plan and a low threshold to measure glucose and ketones in any unwell child are therefore disproportionately important. In migrant, refugee, and asylum-seeking families, consanguinity raises the pre-test probability of the autosomal recessive KATP-channel and fatty-acid oxidation defects, language barriers complicate the teaching of a sick-day plan, and a trained interpreter is mandatory for any discussion of recurrence risk and reproductive options. [2] [6]
In neonates, the inherited causes must be distinguished from the transitional hyperinsulinaemia of the infant of a diabetic mother and from the common benign hypoglycaemia of the first days of life — persistence beyond 48 hours, a high glucose requirement, or a hypoketotic profile all warrant a formal evaluation. In adolescents transitioning to adult metabolic or endocrine care, the move is a high-risk point: dietary adherence and cornstarch compliance may slip, the emergency plan may not transfer, and pregnancy in a woman with a fatty-acid oxidation defect or hyperinsulinism requires a metabolic obstetric plan. In families managing complex chronic metabolic disease, fragmentation of care is the chief threat, and a written, shared, reconciled care plan is the intervention that matters most. [1] [11]
Evidence, Guidelines & Regional Differences
The evidence base rests on three pillars: consensus clinical guidelines, longitudinal cohort and registry data, and emerging molecular and therapeutic evidence. The Thornton-led Pediatric Endocrine Society guidelines are the current international standard for the evaluation and management of persistent hypoglycaemia, setting the operational threshold, the critical-sample panel, and the framework that most national programmes adopt. The Saudubray pathophysiological classification of inherited metabolic disease translates the biochemistry into a practical clinical framework, and the Saudubray IEM overview in Pediatric Clinics consolidates the evaluation pathway across the intoxication-type and energy-failure-type disorders. [1] [10] [9]
The cohort data rest on large characterised series. The Kapoor 300-patient and Snider 417-patient congenital hyperinsulinism cohorts established the genotype-phenotype-histology correlations and the diazoxide-response patterns. The Chou and Koeberl glycogen storage disease reviews defined the natural history and the gene-therapy frontier, and the Spiekerkoetter and Wanders fatty-acid oxidation defect reviews set the treatment principles. The Garg and Devaskar analysis of the long-term impacts of neonatal hypoglycaemia grounds the threshold debate in outcome data. [2] [3] [12]
In Australia and New Zealand, childhood hypoglycaemia is managed through the state-based metabolic and endocrine services based in the major children's hospitals and at Starship in New Zealand, with newborn screening by tandem mass spectrometry detecting most fatty-acid oxidation defects before the first decompensation. A normal newborn screen does not exclude congenital hyperinsulinism or the glycogen storage diseases, which are not reliably captured on the bloodspot. Aeromedical retrieval to a tertiary centre is the expected pathway for a severe or refractory presentation, and 18-fluorodopa positron-emission tomography for focal-lesion localisation in congenital hyperinsulinism is available at the quaternary centres that receive these patients. Genetic counselling, carrier testing, and prenatal or preimplantation diagnosis are coordinated through the clinical genetics services. [1] [6]
Exam Pearls
A fellowship candidate answering on hypoglycaemia due to inherited metabolic disease should land six anchor points and avoid three classic traps. The anchors are the three physiological failures (insulin-driven, glucose-production, fuel-oxidation), the hypoketotic-versus-ketotic distinction, the critical sample and how insulin, beta-hydroxybutyrate, free fatty acids and lactate localise the defect, the glucagon response as a diagnostic and therapeutic tool, the disease-specific therapy (diazoxide and sirolimus for hyperinsulinism, carnitine and fasting avoidance for FAOD, cornstarch for GSD), and the recurrence and sudden-death counselling. The traps are the missed critical sample, the misclassification of a hypoketotic as a ketotic hypoglycaemia, and the failure to warn the family of the recurrence risk and the emergency plan. The candidate who can name the hyperinsulinism-hyperammonaemia syndrome, the focal-versus-diffuse distinction, and the protective role of ketones will defend the topic at viva. [1] [9]
References
- [1]Thornton PS, Stanley CA, De Leon DD, Harris D, Haymond MW, Hussain K, et al. Recommendations from the Pediatric Endocrine Society for Evaluation and Management of Persistent Hypoglycemia in Neonates, Infants, and Children. J Pediatr, 2015.PMID 25957977
- [2]Kapoor RR, Flanagan SE, Arya VB, Shield JPH, Ellard S, Hussain K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol, 2013.PMID 23345197
- [3]Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, et al. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab, 2013.PMID 23275527
- [4]Chou JY, Jun HS, Mansfield BC. Glycogen storage disease type I and G6Pase-β deficiency: etiology and therapy. Nat Rev Endocrinol, 2010.PMID 20975743
- [5]Koeberl DD, Koch RL, Lim JA, et al. Gene therapy for glycogen storage diseases. J Inherit Metab Dis, 2024.PMID 37421310
- [6]Spiekerkoetter U, Bastin J, Gillingham M, Duran M, Wanders RJA, Marsden D, et al. Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis, 2010.PMID 20830526
- [7]Wanders RJA, Visser G, Ferdinandusse S, Waterham HR, Vaz FM, Houten SM. Mitochondrial Fatty Acid Oxidation Disorders: Laboratory Diagnosis, Pathogenesis, and the Complicated Route to Treatment. J Lipid Atheroscler, 2020.PMID 33024728
- [8]Stanley CA. Carnitine deficiency disorders in children. Ann N Y Acad Sci, 2004.PMID 15591002
- [9]Saudubray JM, Garcia-Cazorla À. Inborn Errors of Metabolism Overview: Pathophysiology, Manifestations, Evaluation, and Management. Pediatr Clin North Am, 2018.PMID 29502909
- [10]Saudubray JM, Mochel F, Lamari F, Sedel F, Legrand A, Giral P, et al. Proposal for a simplified classification of IMD based on a pathophysiological approach: A practical guide for clinicians. J Inherit Metab Dis, 2019.PMID 30883825
- [11]Douillard C, Mention K, Dobbelaere D, Wemeau JL, Saudubray JM, Vantyghem MC. Hypoglycaemia related to inherited metabolic diseases in adults. Orphanet J Rare Dis, 2012.PMID 22587661
- [12]Garg M, Devaskar SU. Exploring the long-term impacts of neonatal hypoglycemia to determine a safe threshold for glucose concentrations. Eur J Pediatr, 2025.PMID 40119223