Paeds · genetics-dysmorphology-and-metabolism
Inborn errors presenting with neurological regression
Also known as Inborn errors presenting with neurological regression · Metabolic causes of developmental regression · Neuroregression from inborn errors of metabolism · Progressive encephalopathy of metabolic origin · Treatable intellectual disability
A fellowship approach to the inborn errors of metabolism that present with neurological regression: recognise loss of previously acquired milestones as a red flag, distinguish true progressive neurodegeneration from plateau and static loss, group the disorders by affected pathway (intoxicating small molecule, energy/mitochondrial, storage/lysosomal, lipid-traffic and metal), deploy a tiered metabolic-and-genomic investigation strategy, and crucially identify the treatable subset before labelling a child degenerative or palliative.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A toddler who was speaking in two-word phrases and running stops talking and begins to stumble, and a school-age boy whose teacher notices falling grades and new seizures turns out to have inflammatory white-matter disease on his scan. In both rooms the unifying question is the same: is this loss of milestones a progressive metabolic disorder, and is there a therapy whose window is closing right now. The fellowship task is to convert that bedside worry into a structured, time-aware workup that does not forfeit a treatable cause to a comforting but wrong label such as cerebral palsy or autism. [5] [8]
R · E · G · R · E · S · S
Overview & Definition
The clinician's first act is to confirm what kind of developmental problem this is, because the word "regression" is used loosely and the distinction changes everything. Developmental delay means milestones are not being met on time. A plateau means the child has stopped gaining new skills but has not lost old ones. Neurological regression - the entity this page owns - means the child has lost previously acquired skills. Only regression carries the weight of a presumed progressive process, and only regression mandates the search for a neurodegenerative cause at speed. [8]
An inborn error of metabolism is a monogenic disorder in which a deficient enzyme, transporter, cofactor, or structural protein disrupts a biochemical pathway, and the accumulating substrate or the energy deficit injures the cell. When the affected pathway runs through the central nervous system, the injury declares itself as neurological regression - a slide from a higher to a lower level of function. The slide may be rapid, over days to weeks, as in acute decompensation of a late-onset small-molecule disorder; or insidious, over months to years, as in a leukodystrophy or a lysosomal storage disorder. Reading the tempo is part of reading the disease. [5] [13]
What makes the IEM group worth knowing as a cause of regression is that a meaningful subset is treatable, and that treatment works best - or only - when it is begun before the brain injury becomes irreversible. The systematic work of van Karnebeek, Stockler, Saudubray and others reframed the field around this idea: among the IEM that cause intellectual disability and regression, a defined fraction respond to a disease-modifying intervention, and missing them is the cardinal avoidable error in paediatric neurology. [1] [2] [3]
Classification
The IEM that regress the brain are classified by the affected biochemical pathway, because the pathway predicts the clinical pattern, the first-line test, and whether a disease-modifying therapy exists. The figure below lays out the four pathway groups alongside the high-yield non-metabolic mimics that must be excluded at the same time. [4] [13]
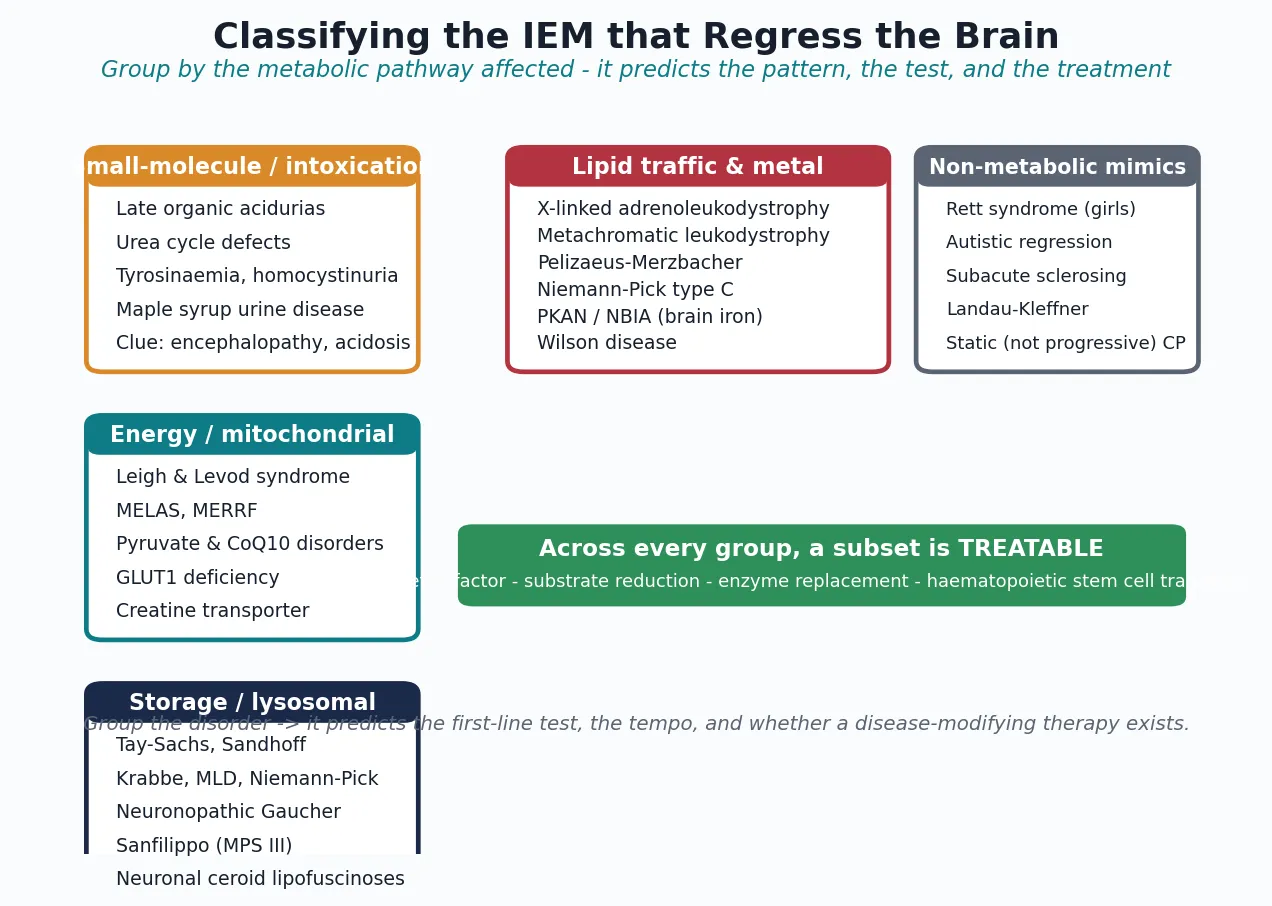
The four groups behave differently at the bedside. Intoxicating small-molecule disorders - the late-onset organic acidurias, urea-cycle defects, tyrosinaemia, homocystinuria, and maple syrup urine disease - produce episodic or subacute encephalopathy driven by a circulating toxin, often with acidosis, ketosis, or hyperammonaemia, and they declare themselves in metabolic blood and urine tests. Energy and mitochondrial disorders - Leigh syndrome, MELAS, MERRF, pyruvate and coenzyme Q10 disorders, GLUT1 deficiency, and creatine transporter deficiency - injure neurons through energy starvation, with characteristic basal-ganglia or stroke-like imaging and a raised lactate. Storage and lysosomal disorders store undegraded substrate in cells, producing regression with organomegaly, coarse facies, white-matter change, or a cherry-red spot. Lipid-traffic and metal disorders - the leukodystrophies, Niemann-Pick type C, the neurodegeneration-with-brain-iron-accumulation (NBIA) group, and Wilson disease - disrupt lipid handling or metal homeostasis and produce regression with white-matter lesions, a movement disorder, or iron deposition. [4] [5] [13]
A second, clinically decisive axis cuts across all four groups: treatable versus untreatable. The treatable subset is what the workup exists to find, and it spans every pathway group - the ketogenic diet for GLUT1 deficiency, cofactors for several metabolic epilepsies, diet for homocystinuria and the organic acidurias, substrate reduction and chaperones for selected storage disorders, enzyme replacement for non-neuronopathic viscera, and haematopoietic stem cell transplant for early cerebral adrenoleukodystrophy and selected neuronopathic storage disorders. Holding this binary in mind prevents the most consequential error in the field: labelling a treatable child untreatable. [1] [3]
Pathway groups at a glance - pattern, test, treatment
- Intoxicating small molecule (organic acidurias, urea-cycle, tyrosinaemia, homocystinuria, MSUD): episodic encephalopathy with acidosis, ketosis, or hyperammonaemia. First-line test: plasma amino acids, ammonia, urine organic acids, homocysteine. Treatment: diet, cofactors, ammonia scavengers; emergency protocol for decompensation.
- Energy / mitochondrial (Leigh, MELAS, MERRF, GLUT1, creatine transporter): epilepsy, stroke-like episodes, ataxia, basal-ganglia injury. First-line test: lactate, brain MRI with spectroscopy, molecular testing. Treatment: cofactors, ketogenic diet for GLUT1, supportive.
- Storage / lysosomal (Tay-Sachs, Krabbe, MLD, Niemann-Pick, neuronopathic Gaucher, Sanfilippo, NCL): regression with organomegaly, coarse facies, cherry-red spot, white-matter disease. First-line test: urine glycosaminoglycans and oligosaccharides, enzyme panel. Treatment: ERT for viscera, HSCT for neuronopathic forms given early.
- Lipid-traffic and metal (cerebral X-ALD, MLD, Pelizaeus-Merzbacher, Niemann-Pick C, PKAN/NBIA, Wilson): white-matter or basal-ganglia disease, movement disorder. First-line test: very-long-chain fatty acids, copper and caeruloplasmin, brain MRI, molecular testing. Treatment: HSCT for early cerebral ALD, chelation for Wilson, symptom management for many. [4] [13]
Epidemiology & Risk Factors
Neurological regression is uncommon as a presenting complaint, but when it occurs the probability of an underlying IEM is high enough to justify a systematic workup in every child. The yield depends on how strictly regression is defined and how hard the workup looks, but trio exome sequencing now identifies a diagnostic variant in a substantial fraction of children with unexplained developmental regression, and a defined minority of these are treatable IEM. [2] [8]
The strongest risk factors are genetic. Consanguinity, a previously affected sibling, and a family history of unexplained childhood death or developmental failure all raise the probability of an autosomal recessive IEM and should lower the threshold for screening in any child with a compatible phenotype. X-linked disorders change the counselling: adrenoleukodystrophy, creatine transporter deficiency, Hunter syndrome, and Pelizaeus-Merzbacher disease affect boys and reshape the screening of at-risk males and carrier mothers. Founder effects concentrate specific disorders in particular populations, and a careful three-generation pedigree is part of the workup, not an optional add-on. [5] [11]
The epidemiology is shifting because of genomic testing and newborn screening. Exome and genome sequencing have moved many children from the "degenerative, cause unknown" shelf to a named molecular diagnosis, and have brought treatable disorders to light that would once have been missed. Newborn-bloodspot panels have expanded to capture an increasing number of disorders before they regress the brain, which changes the presenting population from symptomatic children to asymptomatic neonates flagged by a screen. These advances raise the stakes on accurate, early diagnosis, because a named, treatable disorder that is recognised late is a missed opportunity. [2] [3]
Pathophysiology
A single metabolic defect becomes a whole-brain disease because the central nervous system is exquisitely dependent on every pathway the IEM disrupt: tight energy supply, clean substrate handling, intact myelin, and controlled metal and lipid traffic. The cascade runs from a gene variant to a deficient enzyme or transporter, to an accumulating substrate or an energy deficit, to neuronal injury expressed as lost skills. The accumulated substrate - not the missing gene - is what poisons the cell, which is why a Gaucher macrophage engorged with glucocerebroside behaves differently from a neuron starved by a defective respiratory chain. [5] [13]
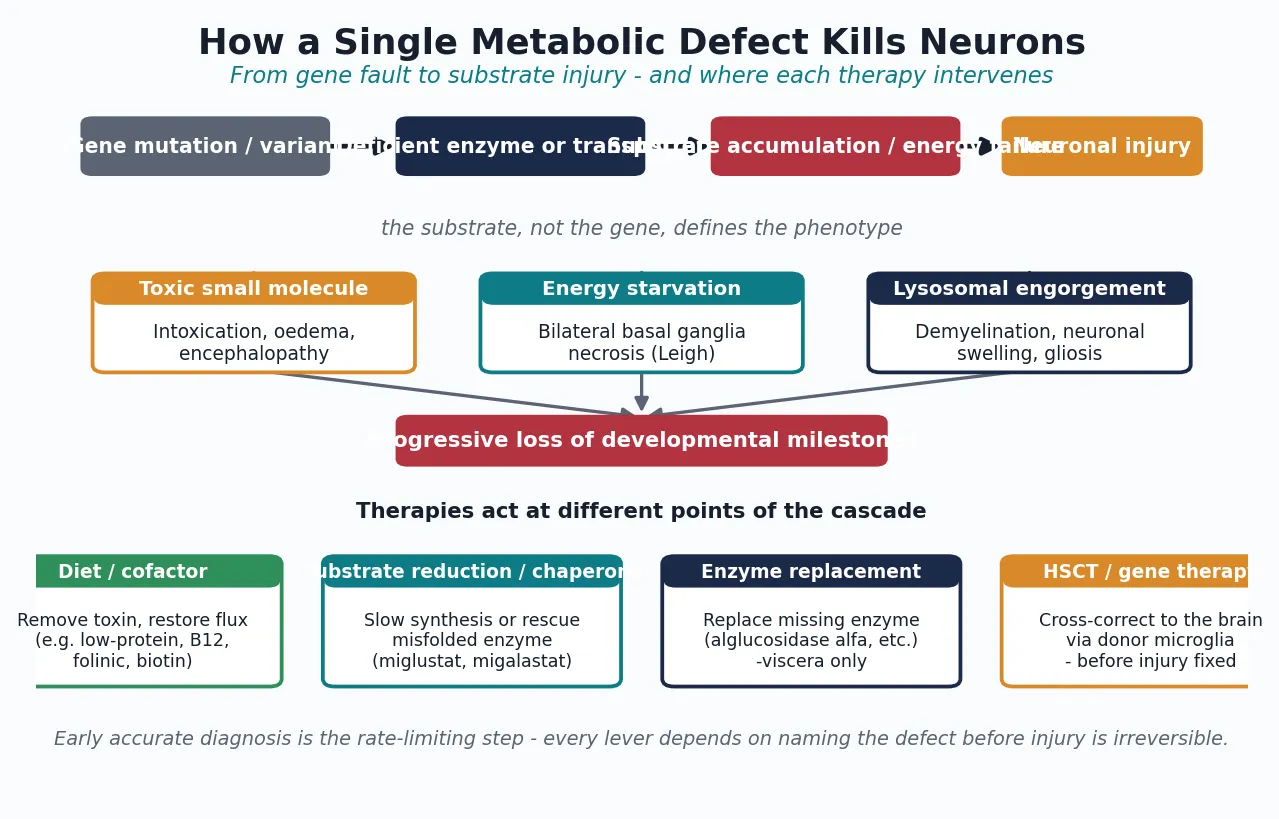
The mechanism of neuronal death differs by pathway group and explains the imaging and clinical patterns. Intoxicating small-molecule disorders poison neurons and glia directly, producing oedema and encephalopathy that may fluctuate with metabolic state. Energy and mitochondrial disorders starve high-demand structures - the basal ganglia, brainstem, and cortex - producing symmetric necrotic lesions (Leigh syndrome) or stroke-like episodes (MELAS). Storage and lysosomal disorders engorge neurons and macrophages with undegraded material, triggering demyelination, neuronal swelling, and gliosis. Lipid-traffic and metal disorders disrupt myelin synthesis or metal homeostasis, producing leukodystrophy, a movement disorder, or iron accumulation in the basal ganglia. [7] [10] [11]
The disease-modifying therapies exploit these mechanisms at different points, which is why one treatment does not fit all. Diet and cofactor therapy remove a toxic substrate or restore flux through a blocked pathway - the principle behind low-protein diets, biotin, thiamine, folinic acid, hydroxocobalamin, and the ketogenic diet for GLUT1 deficiency. Substrate reduction and pharmacological chaperones slow synthesis of the stored material or rescue a misfolded enzyme. Enzyme replacement therapy delivers a mannose-6-phosphate-tagged enzyme that reaches the viscera but does not cross the blood-brain barrier, which is why it works for non-neuronopathic disease and fails for the brain. Haematopoietic stem cell transplant supplies cross-correcting enzyme through donor-derived microglia and macrophages and can modify neuronopathic disease - but only before the injury is fixed. [5] [13]
Clinical Presentation
The presenting complaint is always the same in words - a child has lost skills - but the pattern of what is lost, what accompanies it, and how fast it happens is what points to the pathway group. Three patterns cover most of the bedside ground. The first is regression with organomegaly: a child who loses motor or language milestones and is found to have a large liver or spleen, coarse facies, or a cherry-red spot at the macula. This pattern points to a storage or lysosomal disorder, and the urgency is the transplant window for the neuronopathic forms. [5] [9]
The second pattern is regression with epilepsy. Seizures may be the first or the dominant feature, and their character matters: myoclonic epilepsy in a child who is losing skills suggests a storage disorder or a neuronal ceroid lipofuscinosis; absence-like or mixed seizures that resist standard anticonvulsants raise GLUT1 deficiency and creatine disorders; and epileptic encephalopathy with a raised lactate points to a mitochondrial disorder. The teaching point is that an epileptic encephalopathy that is not behaving as expected should prompt a metabolic workup rather than a fourth anticonvulsant. [7] [12]
The third pattern is regression with a movement disorder. Progressive dystonia, parkinsonism, chorea, or ataxia in a child who is losing skills points to a lipid-traffic or metal disorder: PKAN and other NBIAs produce dystonia with iron in the globus pallidus; Niemann-Pick type C produces a vertical supranuclear gaze palsy with cataplexy and cognitive decline; Wilson disease produces a movement disorder with psychiatric change and hepatic disease in the older child or adolescent; and X-linked adrenoleukodystrophy produces behavioural and cognitive decline with adrenal insufficiency and a characteristic parieto-occipital white-matter lesion. [10] [11]
Tempo - the fourth dimension
The tempo is itself diagnostic and triages urgency. Acute or subacute regression demands the emergency metabolic protocol and same-day biochemistry, because a late-onset small-molecule disorder in decompensation can be reversed if caught. Insidious regression demands a deliberate, tiered workup but with the same principle: name the disorder before a therapy window closes. [6] [13]
Differential Diagnosis
The differential of neurological regression is broader than the IEM alone, and several non-metabolic causes must be excluded because they change management entirely. Rett syndrome affects girls, classically after a period of normal development, with loss of hand skills and purposeful hand use, stereotypies, and deceleration of head growth. Autistic regression produces loss of language and social skills, usually without motor or systemic signs, and without the organomegaly, imaging change, or biochemistry of an IEM. These are diagnoses of pattern, supported by genetic testing (MECP2 for Rett), and they coexist with the search for an IEM rather than replacing it. [8]
A short list of acquired and treatable mimics must be actively excluded, because each changes the trajectory if missed. Subacute sclerosing panencephalitis, a late complication of measles, produces progressive regression with myoclonus and a characteristic periodic electroencephalogram. Landau-Kleffner syndrome produces acquired epileptic aphasia. Autoimmune encephalitis, including NMDA-receptor antibody encephalitis, can mimic a neurodegenerative disorder and is treatable with immunotherapy. Hypothyroidism, coeliac disease, nutritional deficiency, chronic illness, and medication effects can all mimic regression and must be considered and tested. Hearing and vision loss can masquerade as developmental loss, and a child who appears to have stopped engaging may simply have stopped seeing or hearing. [8]
Why does a normal initial metabolic screen not exclude an IEM? Because several disorders are intermittent or tissue-specific: lactate may be normal between mitochondrial flares, urine organic acids may be normal outside a decompensation, and a disorder that is not on the newborn-bloodspot panel will not be caught by the panel. When the clinical phenotype is highly suggestive, a normal screen lowers the probability but does not close the case, and the next step is targeted enzyme or metabolite assays and trio exome sequencing. [2] [8]
Clinical & Bedside Assessment
The single most informative bedside act is to chart the developmental trajectory against specific earlier milestones. Ask what the child could do at six months, at twelve months, at eighteen months, and now - could they sit, babble, use pincer grasp, walk, speak in phrases, follow a two-step instruction. A trajectory that rises then falls is regression; a line that flattens is plateau; a line that was always low is delay. Anchor each milestone to an age and a witness, because the trajectory is the diagnosis in a way that no single examination finding can be. [8]
Three questions frame every consultation. First, is the central nervous system involved and how fast - acute, subacute, or insidious? Second, which viscera are involved and how severely - is there organomegaly, cardiomyopathy, hepatic dysfunction, or skin change? Third, is there consanguinity, a previously affected sibling, or a family history of unexplained childhood death or developmental failure? These three questions convert a vague worry into a structured hypothesis about the pathway group and the urgency. [5] [8]
The systemic examination is then directed and recorded. Measure growth and head circumference and plot them against earlier values - deceleration of head growth suggests Rett or a storage disorder; macrocephaly suggests a leukodystrophy or a storage disorder. Palpate the liver and spleen and measure the edges in centimetres below the costal margin. Look for facial coarsening, corneal clouding, and cataracts. Examine the fundi for a cherry-red spot (storage disorders) and optic atrophy (leukodystrophies, mitochondrial). Assess tone and movement for dystonia, parkinsonism, and ataxia (lipid and metal disorders). Examine the skin for angiokeratomata (Fabry) and the heart for a gallop or murmur. [5] [13]
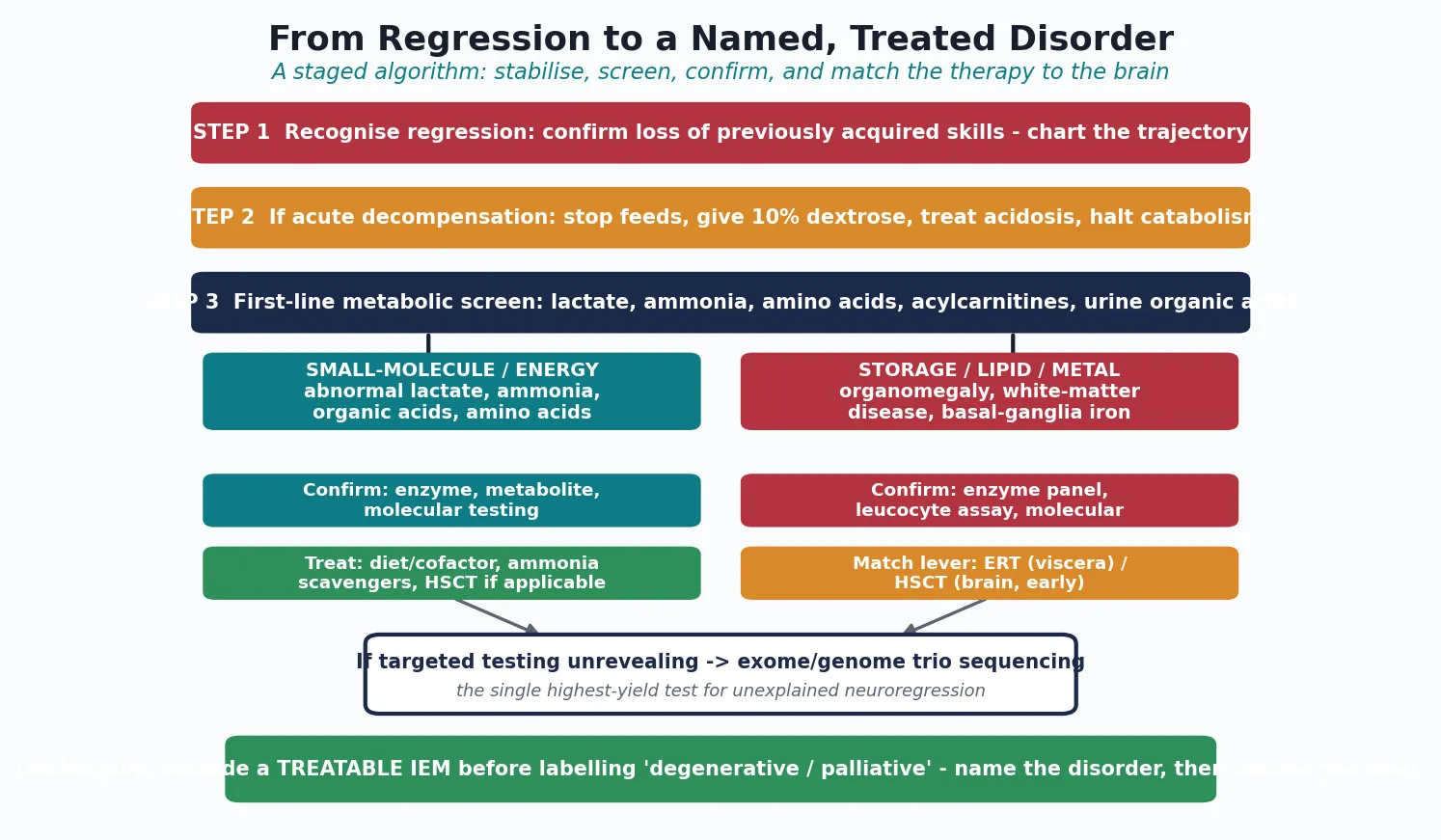
Investigations
The investigation strategy is tiered, and each tier either names the disorder or justifies the next. The first tier is the broad metabolic screen, sent on every child with unexplained regression: a venous lactate (drawn free-flowing, without a tourniquet), ammonia (on ice, processed immediately), plasma amino acids, acylcarnitine profile, urine organic acids, total homocysteine, and a creatine kinase. Add a full blood count, electrolytes, liver function, thyroid function, and a basic autoimmune and infective screen to exclude the mimics. Brain MRI, ideally with magnetic resonance spectroscopy, is central at this stage because the pattern of basal-ganglia, white-matter, cortical, or atrophic change points to a pathway group. [6] [13]
The second tier is targeted enzyme, metabolite, and functional assays, directed by the pattern. Suspected storage: urine glycosaminoglycans and oligosaccharides, then a lysosomal enzyme panel on leucocytes or dried blood spot. Suspected leukodystrophy: very-long-chain fatty acids (for peroxisomal disorders including X-ALD), arylsulfatase A and galactocerebrosidase (for MLD and Krabbe), and transferrin isoelectric focusing (for congenital disorders of glycosylation). Suspected NBIA: brain MRI for the eye-of-the-tiger sign and iron deposition. Suspected Wilson: copper, caeruloplasmin, and 24-hour urinary copper. Suspected creatine disorder: guanidinoacetate and creatine in plasma and urine. [4] [13]
The tiered workup
- Recognise regression - chart the trajectory; confirm loss of previously acquired skills.
- First-line metabolic screen - lactate, ammonia, plasma amino acids, acylcarnitines, urine organic acids, homocysteine, creatine kinase, plus brain MRI with spectroscopy.
- Targeted assays by pattern - storage (GAGs, oligosaccharides, enzyme panel), leukodystrophy (VLCFA, arylsulfatase A, transferrin IEF), NBIA and Wilson (copper, caeruloplasmin, iron-sensitive MRI), creatine (guanidinoacetate).
- Chromosomal microarray - first-tier genetic test for any child with developmental disorder, to detect copy-number variants.
- Trio exome or genome sequencing - the single highest-yield test for unexplained neuroregression when targeted testing is unrevealing. [2] [8]
The third tier is genomic, and it has transformed the field. Chromosomal microarray is the first-line genetic test for any child with a developmental disorder, detecting pathogenic copy-number variants. When the microarray and targeted assays are unrevealing, trio exome or genome sequencing - analysing the child and both parents together - is the single highest-yield test for unexplained neuroregression, identifying a diagnostic variant in a substantial fraction of cases and often surfacing a treatable disorder that targeted testing had missed. A molecular diagnosis enables carrier testing, prenatal diagnosis, and preimplantation genetic testing for the wider family, and it carries prognosis. [2] [8]
Management — Resuscitation
When regression presents as acute or subacute decompensation - vomiting, altered consciousness, metabolic acidosis, or hyperammonaemia - the response is the emergency stop-the-regression protocol, which aims to halt catabolism and clear the toxin while the diagnostic workup proceeds. Stop all protein and any potential toxin (galactose, fructose) intake. Give intravenous 10% dextrose at maintenance-plus rate to suppress catabolism and provide an alternative fuel, with insulin if needed to control hyperglycaemia and drive anabolism. Treat metabolic acidosis with bicarbonate if severe. Manage hyperammonaemia with nitrogen-scavenger drugs and, if severe, haemofiltration or dialysis, because ammonia itself is neurotoxic. [6] [13]
While the workup proceeds, empirical cofactor therapy is warranted whenever a cofactor-responsive disorder is plausible, because these treatments are safe, cheap, and may transform the outcome. Biotin for biotinidase deficiency, thiamine for thiamine-responsive pyruvate dehydrogenase and maple syrup urine disease, folinic acid for cerebral folate deficiency and folate-receptor defects, hydroxocobalamin for cobalamin-responsive disorders and methylmalonic acidaemia, levodopa for aromatic L-amino-acid decarboxylase deficiency, and coenzyme Q10 for selected mitochondrial disorders are all reasonable while the diagnosis is confirmed. The principle is that a few days of empirical cofactor therapy is a small price to avoid missing a treatable cause. [4] [5]
Seizures, raised intracranial pressure, and encephalopathy are managed in parallel, with care to avoid worsening the metabolic defect: avoid prolonged fasting before procedures, choose anaesthetic and anticonvulsant agents that do not impair mitochondrial function, and keep the child metabolically safe. The recognition that a late-onset small-molecule disorder can decompensate under fasting, intercurrent illness, or surgery - and that this decompensation can be the first presentation of regression - is what makes the emergency protocol a core skill for the general paediatrician, not only the metabolic specialist. [6]
Management — Definitive & Stepwise
Definitive management means naming the disorder and matching it to the disease-modifying therapy that fits its pathway group and its central-nervous-system involvement. For the intoxicating small-molecule disorders, the backbone is a disease-specific diet that restricts the offending substrate and supplements deficient cofactors, with an emergency sick-day plan to prevent decompensation during illness. For GLUT1 deficiency, the ketogenic diet provides ketones as an alternative brain fuel and can control seizures, improve the movement disorder, and stabilise cognition. For selected storage disorders, substrate reduction therapy and pharmacological chaperones slow synthesis or rescue a misfolded enzyme, with the choice genotype-guided. [1] [12]
Enzyme replacement therapy is the disease-modifying backbone for non-neuronopathic viscera, reaching the liver, spleen, bone, and heart but not the brain. Haematopoietic stem cell transplant is reserved for neuronopathic disease and for cerebral X-linked adrenoleukodystrophy, because donor-derived microglia supply cross-correcting enzyme to the brain - and the window closes once the inflammatory or storage injury becomes fixed. Gene therapy and emerging mRNA approaches broaden the toolkit and extend treatment to disorders once considered untreatable, which is why early accurate diagnosis is more valuable now than ever. [3] [11]
The care plan is multidisciplinary and lifelong, built around the specialist metabolic and neurology service: dietetics for the restricted diets and feeding, neurology for seizures and movement disorder, developmental and educational support, orthopaedics and respiratory for complications, and palliative care involved early in the most severe neuronopathic forms - coexisting with disease-modifying treatment rather than replacing it. Once a molecular diagnosis is made, genetic counselling, carrier testing, prenatal diagnosis, and preimplantation genetic testing reshape the reproductive risk for the whole family, and this is part of management, not an afterthought. [5] [8]
Specific Subtypes & Scenarios
Leigh syndrome is the emblematic mitochondrial neurodegenerative disorder of childhood: a subacute regression with hypotonia, ataxia, dystonia, feeding and respiratory difficulty, and symmetric necrotic lesions in the basal ganglia and brainstem on MRI, often with a raised lactate. Its genetic heterogeneity is enormous - more than 75 monogenic causes across both mitochondrial and nuclear genomes - which is why a normal initial mitochondrial workup does not exclude it, and why trio exome is increasingly the diagnostic route. Cofactor therapy (coenzyme Q10, thiamine, riboflavin, biotin) helps a subset; the rest is supportive. [7]
X-linked adrenoleukodystrophy is a peroxisomal disorder of very-long-chain fatty-acid metabolism that affects boys. Most present in mid-childhood with behavioural change, school decline, new seizures, or adrenal insufficiency, and brain MRI shows a characteristic contrast-enhancing parieto-occipital white-matter lesion. Once the cerebral inflammatory form declares itself, progression is rapid and the transplant window is short - which is why any boy with unexplained regression and a white-matter lesion must have very-long-chain fatty acids measured urgently and be referred to a specialist service within days. Adrenal replacement therapy is given for adrenal insufficiency regardless of the neurological decision. [11]
GLUT1 deficiency is the treatable energy disorder every candidate must know. Defective glucose transport across the blood-brain barrier starves the brain of its preferred fuel and presents with epilepsy (often resistant to standard anticonvulsants), a movement disorder (paroxysmal exercise-induced dystonia, ataxia), and developmental regression, with a low cerebrospinal-fluid glucose and a low CSF-to-plasma glucose ratio on lumbar puncture. The ketogenic diet provides ketones as an alternative brain fuel and can control the seizures, improve the movement disorder, and stabilise cognition - a genuinely treatable cause of regression that is missed when the epilepsy is treated without a metabolic workup. [12]
PKAN and the wider neurodegeneration-with-brain-iron-accumulation group present with progressive dystonia, parkinsonism, dysarthria, and cognitive decline, with iron deposition in the globus pallidus - the eye-of-the-tiger sign on MRI in classic PKAN. The group is genetically heterogeneous, and some subtypes are treatable (for example, biotin-thiamine-responsive basal ganglia disease and some CoPLAN disorders respond to cofactors), which is why a diagnosis of NBIA is the start of a search for a treatable subtype, not the end of it. Symptom management with baclofen, botulinum toxin, and deep brain stimulation improves quality of life. [10]
Complications & Pitfalls
The most consequential complications are the consequences of delay: a forfeited transplant or enzyme window, irreversible brain injury, and the lost opportunity to offer the family accurate genetic counselling. Every day of delay matters for the disorders whose therapy works only early - cerebral X-ALD, infantile Krabbe, the neuronopathic storage disorders - and the single biggest contributor to delay is mislabelling. Calling a treatable IEM cerebral palsy, autism, or a behavioural problem is the cardinal avoidable error, because each label closes the diagnostic search and redirects the family away from the metabolic service. [1] [8]
Over-reliance on a normal initial metabolic screen is a second pitfall. Lactate can be normal between mitochondrial flares, urine organic acids can be normal outside a decompensation, and a disorder that is not on the newborn-bloodspot panel will not be caught by the panel. When the phenotype is suggestive, the answer is targeted assays and trio exome, not reassurance. A third pitfall is underestimating anaesthetic and fasting risk in a child with an undiagnosed IEM: prolonged fasting before a procedure, or certain anaesthetic agents, can trigger a metabolic decompensation that becomes the first presentation of the disorder, so any child under investigation should have a peri-procedure metabolic safety plan. [6] [13]
Prognosis & Disposition
Prognosis is determined by the specific disorder, the age and tempo of onset, the genotype, and - critically - the timing of treatment. The early-treated treatable IEM have a transformed prognosis: a child with GLUT1 deficiency started on a ketogenic diet early may have near-normal cognition; a child with a cofactor-responsive disorder may recover substantially; a boy with cerebral X-ALD transplanted early may halt the disease. The untreatable neuronopathic disorders - Tay-Sachs, the severe leukodystrophies, infantile neuronal ceroid lipofuscinosis - have a guarded outlook measured in years, and the task shifts to symptom control, developmental support, and palliative care. [1] [3]
Disposition is structured around the specialist metabolic and neurology service, with lifelong surveillance and a named coordinator. The general paediatrician holds the whole child, coordinates the multidisciplinary team, and owns the emergency sick-day plan and the peri-procedure metabolic safety plan. As the child grows, structured transition to adult metabolic and neurology services begins in early adolescence, with explicit handover of the diagnosis, the surveillance plan, and the reproductive counselling - because the patient who is a teenager now will be the parent who benefits from preimplantation genetic testing later. [4] [8]
Palliative care is involved early in the most severe neuronopathic forms, coexisting with disease-modifying treatment rather than replacing it: even when a therapy is being pursued, symptom control for seizures, dystonia, feeding difficulty, and respiratory compromise improves the child's quality of life and the family's capacity to cope. The conversation about goals of care is iterative, revisited as the disease declares its trajectory, and it is held with honesty and without abandoning the search for a treatable subset. [5]
Special Populations
The asymptomatic neonate flagged by an abnormal newborn-bloodspot screen is managed differently from the symptomatic regressing child. The screen is a probability, not a diagnosis: pseudodeficiency alleles, late-onset variants, and carrier states can all produce an abnormal screen in an infant who may never become unwell. Confirmatory testing - leucocyte enzyme assay and molecular testing of the relevant gene - is what distinguishes a child who needs treatment from one who needs surveillance, and it must happen within days, not weeks, so that a treatable window is not forfeited while uncertainty is resolved. [2] [3]
Consanguineous and founder populations carry a higher burden of autosomal recessive IEM, and the consultation reframes reproductive risk for the whole family: carrier testing, prenatal diagnosis, and preimplantation genetic testing become central to management once a molecular diagnosis is made. For Indigenous, migrant, refugee, and remote populations, equitable and culturally safe access to specialist metabolic services and expensive therapies is a real challenge, concentrated as those services are in tertiary centres; the principles are constant - recognise regression early, deploy the tiered workup, exclude the treatable IEM - but the pathway must be adapted to geography, language, and cultural safety, often with telehealth and a local coordinator. [8]
The technology-dependent and complex-chronic child, and the transition-age adolescent, need a plan that adapts across the lifespan. The adolescent who has lived with a metabolic diagnosis since childhood faces new questions - adherence to a restricted diet, transfer to adult services, reproductive decisions, and the risk of decompensation with new freedoms - and a structured transition, begun in early adolescence and completed before the eighteenth birthday, is the safeguard. [4]
Evidence, Guidelines & Regional Differences
The conceptual backbone of the field is the treatable-IEM literature: the systematic review of van Karnebeek and Stockler catalogued the treatable inborn errors causing intellectual disability, and the diagnostic-algorithm work and the 2021 review and digital app turned that catalogue into an actionable, tiered workup. The comprehensive-evaluation guidance from the American Academy of Pediatrics, the Saudubray clinical approach focused on treatable disease, and the disorder-specific consensus guidelines (PKAN, X-ALD, Leigh spectrum, the neuronal ceroid lipofuscinoses) frame a workup that converges across regions on the same principle: recognise regression, screen, target, and sequence. [1] [2] [8] [10]
In Australia and Aotearoa New Zealand, the newborn-bloodspot panel and funded access to exome, genome, enzyme therapies, and transplant services are governed by jurisdictional programmes that evolve over time. Specialist metabolic services are concentrated in tertiary paediatric centres, with outreach to regional and remote areas via telehealth and retrieval networks. State the local panel and the local funded pathway rather than assuming a universal list, and involve the regional metabolic service early. [3] [8]
The strength of evidence varies across the field. The evidence for early haematopoietic stem cell transplant in cerebral X-ALD and in selected neuronopathic storage disorders is mature enough to justify the urgency of diagnosis. The evidence for the ketogenic diet in GLUT1 deficiency is strong. The evidence for long-term outcomes of gene therapy and for the natural history of ultra-rare disorders is still maturing, and uncertainty changes counselling: a family should be told what is known, what is likely, and what is genuinely unknown, so that decisions are made on real information rather than false reassurance or undue pessimism. [11] [12]
Exam Pearls
Remember the golden rule that frames every consultation: never label a regressing child degenerative or palliative until the treatable inborn errors of metabolism have been excluded. The treatable subset spans every pathway group - diet and cofactor for the intoxicating and cofactor disorders, the ketogenic diet for GLUT1 deficiency, substrate reduction and chaperone for selected storage disorders, enzyme replacement for non-neuronopathic viscera, and haematopoietic stem cell transplant for the neuronopathic forms and cerebral X-ALD given early. The diagnostic rate-limiting step is naming the disorder early, and trio exome is now the single highest-yield test for unexplained neuroregression. [1] [2] [8]
Self-test: a four-year-old with progressive dystonia
A four-year-old presents with progressive dystonia, dysarthria, and declining gait over eight months. Brain MRI shows iron deposition in the globus pallidus with an eye-of-the-tiger sign. What is the most likely group, the diagnostic test, and the management principle? [10]
Answer: This is a neurodegeneration with brain iron accumulation, classically pantothenate kinase-associated neurodegeneration (PKAN), confirmed by PANK2 molecular testing. The management principle is that a diagnosis of NBIA is the start of a search for a treatable subtype - biotin-thiamine-responsive basal ganglia disease and several CoPLAN disorders respond to cofactors - alongside symptom management with baclofen, botulinum toxin, and deep brain stimulation. [10]
References
- [1]van Karnebeek CD, Stockler S. Treatable inborn errors of metabolism causing intellectual disability: a systematic literature review. Mol Genet Metab, 2012.PMID 22212131
- [2]van Karnebeek CD, Shevell M, Zschocke J, Moeschler JB, Stockler S. The metabolic evaluation of the child with an intellectual developmental disorder: diagnostic algorithm for identification of treatable causes and new digital resource. Mol Genet Metab, 2014.PMID 24518794
- [3]Hoytema van Konijnenburg EMM, Wortmann SB, Koelewijn MJ, Tseng LA, Houben R, Stockler-Ipsiroglu S, Ferreira CR, van Karnebeek CDM. Treatable inherited metabolic disorders causing intellectual disability: 2021 review and digital app. Orphanet J Rare Dis, 2021.PMID 33845862
- [4]Sedel F, Lyon-Caen O, Saudubray JM. Therapy insight: inborn errors of metabolism in adult neurology--a clinical approach focused on treatable diseases. Nat Clin Pract Neurol, 2007.PMID 17479075
- [5]Saudubray JM, Sedel F, Walter JH. Clinical approach to treatable inborn metabolic diseases: an introduction. J Inherit Metab Dis, 2006.PMID 16763886
- [6]Leonard JV, Morris AA. Diagnosis and early management of inborn errors of metabolism presenting around the time of birth. Acta Paediatr, 2006.PMID 16373289
- [7]Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann Neurol, 2016.PMID 26506407
- [8]Moeschler JB, Shevell M, Committee on Genetics. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics, 2014.PMID 25157020
- [9]Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology, 2012.PMID 22778232
- [10]Hogarth P, Kurian MA, Gregory A, Csanyi B, Zagustin T, Kmiec T, Wood P, Klucken A, Scalise N, Sofia F, Klopstock T, Zorzi G, Nardocci N, Hayflick SJ. Consensus clinical management guideline for pantothenate kinase-associated neurodegeneration (PKAN). Mol Genet Metab, 2017.PMID 28034613
- [11]Engelen M, Kemp S, de Visser M, van Geel BM, Wanders RJ, Aubourg P, Poll-The BT. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis, 2012.PMID 22889154
- [12]Pearson TS, Akman C, Hinton VJ, Engelstad K, De Vivo DC. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr Neurol Neurosci Rep, 2013.PMID 23443458
- [13]Saudubray JM, Garcia-Cazorla A. Inborn Errors of Metabolism Overview: Pathophysiology, Manifestations, Evaluation, and Management. Pediatr Clin North Am, 2018.PMID 29502909