Paeds · genetics-dysmorphology-and-metabolism
Klinefelter syndrome and sex chromosome aneuploidy
Also known as Klinefelter syndrome · 47,XXY · Triple X syndrome · 47,XXX · XYY syndrome · 47,XYY · Jacobs syndrome · Sex chromosome trisomy · Sex chromosome aneuploidy
A fellowship approach to sex chromosome aneuploidies: recognise that Klinefelter syndrome (47,XXY) is the commonest chromosomal aneuploidy in humans yet more than half of cases are never diagnosed, understand the extra-X dosage model that links the neurodevelopmental phenotype to the gonadal trajectory, and apply a lifespan surveillance framework covering testosterone replacement, bone health, metabolic and cardiovascular risk, fertility preservation, and psychosocial advocacy.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A tall, shy fourteen-year-old boy is referred by his school counsellor because he is struggling academically, socially withdrawn, and has not started puberty while his peers have. On examination his testicular volume is 4 mL bilaterally — small and firm — and he has mild gynaecomastia. The same boy may have been flagged years earlier by his speech pathologist for a language disorder, or he may present only in his thirties at a fertility clinic with azoospermia. The fellowship task is to recognise that this is a sex chromosome aneuploidy — the commonest chromosomal disorder in humans — and to lay out a surveillance and management framework that follows the person through childhood, adolescence, and adulthood. [1] [2]
K.L.I.N.E.F.E.L.T.E.R. — the ten domains
Overview & Definition
Sex chromosome aneuploidies are chromosomal disorders caused by the presence of one or more additional sex chromosomes — or, in the case of Turner syndrome, the absence of one — and they are distinguished from autosomal aneuploidies by a paradox: they are the commonest chromosomal disorders in humans, yet the mildest in phenotype. Klinefelter syndrome, the prototypical and commonest form, is the phenotype produced by a 47,XXY karyotype: one or more extra X chromosomes in a male. It was first described by Harry Klinefelter in 1942 as a syndrome of small testes, gynaecomastia, and azoospermia, and the chromosomal basis was established by Jacobs and Strong in 1959, making it the first human chromosomal disorder to be mapped to a specific karyotype. [2] [1]
The clinical challenge of sex chromosome aneuploidies is that the phenotype is mild, variable, and overlaps with the general population. A boy with 47,XXY may have language delay, tall stature, and learning difficulties — features shared with many children in any classroom — and the diagnosis is easily missed. An estimated fifty to seventy-five per cent of males with Klinefelter syndrome are never diagnosed in life, and the diagnosis rate for 47,XXX and 47,XYY is even lower, because those phenotypes are subtler still. This is the fundamental teaching point: sex chromosome aneuploidies are common but invisible, and the clinician's task is to recognise the pattern across the lifespan rather than to wait for a neonatal diagnosis that, for most, never comes. [1] [5]
The distinction between sex chromosome aneuploidies and autosomal trisomies is mechanistic and phenotypic. An extra chromosome 21 — as in Down syndrome — produces a severe, recognisable, multisystem phenotype because the gene dosage of an entire autosome is disrupted. An extra sex chromosome produces a milder phenotype because of the X-inactivation mechanism: the additional X is largely inactivated, and the residual effect comes from escape genes — genes that evade inactivation and are therefore overexpressed — and from the overall dosage of XIST RNA and the subtle disruption of the inactivation machinery. The Y chromosome, by contrast, carries few genes, and its presence in 47,XYY adds mainly the SRY effect and the neuroanatomical contribution of Y-linked gene expression. [3] [6]
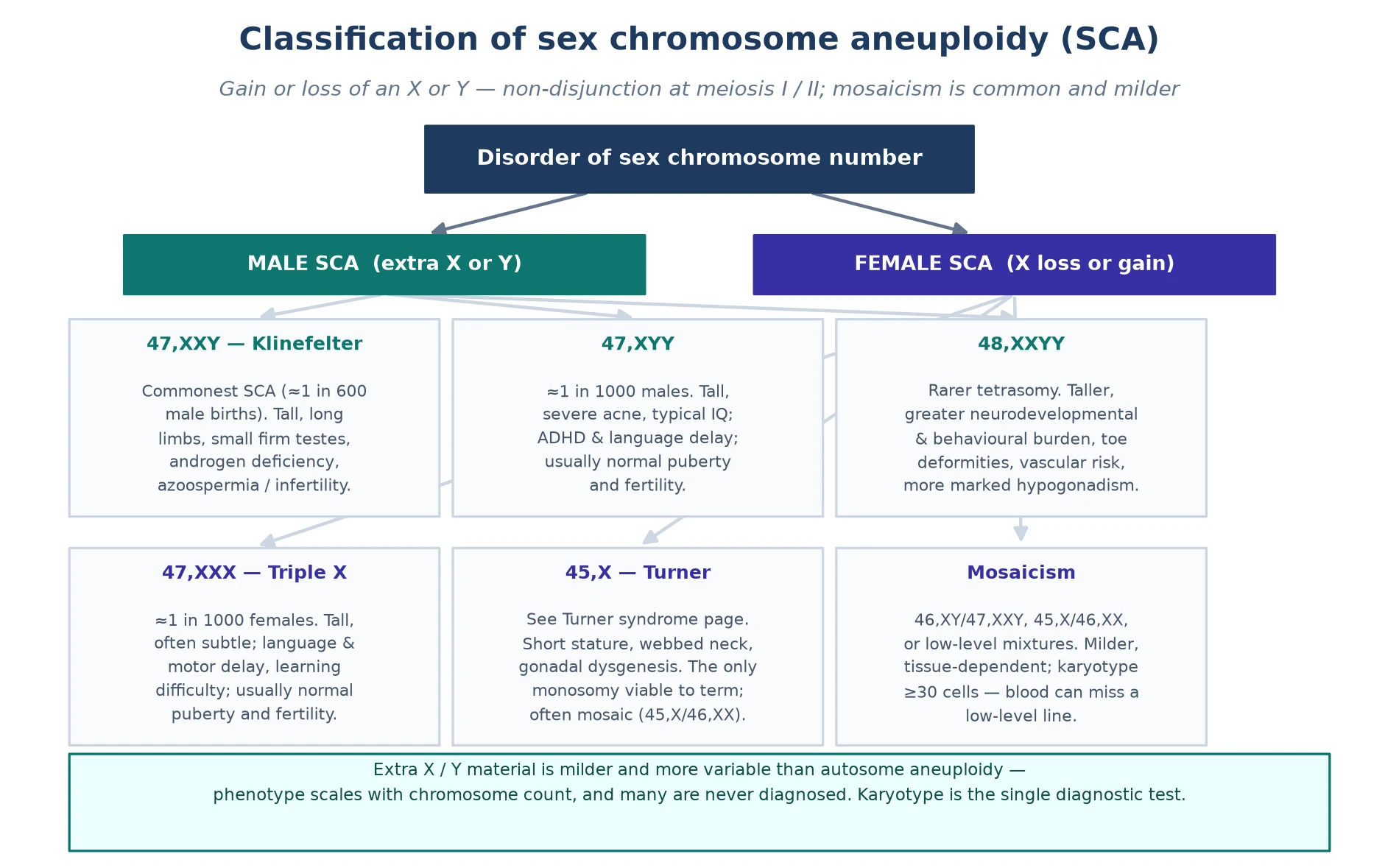
Classification
Sex chromosome aneuploidies are classified by the karyotype, and each karyotype carries a distinctive phenotype and prognosis. The three common sex chromosome trisomies — 47,XXY, 47,XXX, and 47,XYY — share the feature of an additional sex chromosome with a predominantly neurodevelopmental phenotype, while 45,X (Turner syndrome) is the monosomy counterpart that is clinically distinct and covered on its own page. [1] [6]
Klinefelter syndrome (47,XXY) accounts for the majority of sex chromosome aneuploidy diagnoses and is the prototype. It occurs in approximately one in six hundred male births, making it the commonest chromosomal aneuploidy in humans. The karyotype is typically non-mosaic 47,XXY, but approximately ten per cent of cases are mosaic (46,XY/47,XXY), and higher-grade variants — 48,XXXY, 49,XXXXY — carry a progressively more severe phenotype that includes intellectual disability, more pronounced dysmorphism, and a greater cardiac and skeletal burden. The extra-X dosage model explains the gradient: each additional X chromosome adds more overexpressed escape genes, producing a more severe neurodevelopmental and physical phenotype. [1] [3]
Triple X syndrome (47,XXX) occurs in approximately one in one thousand female births and is even less likely to be diagnosed than Klinefelter syndrome, because the phenotype is subtler. Affected girls and women are typically tall, with mild learning difficulties — particularly in language and executive function — motor clumsiness, and an increased risk of anxiety and social difficulties. Pubertal development and fertility are usually normal, and many women with 47,XXX are never diagnosed. The postnatal-diagnosis literature shows a more severe phenotype than the prenatal-diagnosis literature, reflecting ascertainment bias: girls diagnosed because of symptoms are more affected than those identified prenatally and followed prospectively. [7] [5]
XYY syndrome (47,XYY) occurs in approximately one in one thousand male births and shares the tall-stature and neurodevelopmental phenotype with 47,XXY — language delay, learning difficulties, and increased risk of attention-deficit and behavioural challenges — but without gonadal failure, testosterone deficiency, or infertility. Pubertal development and fertility are typically normal. The historical association with criminality, based on prison screening studies with profound ascertainment bias, has been comprehensively refuted by prospective cohort studies, and the modern framing emphasises that the vast majority of males with 47,XYY lead typical lives. [6] [5]
The reason a karyotype is the diagnostic test rather than a chromosomal microarray alone is twofold. First, microarray cannot reliably detect low-level mosaicism — a mosaic 46,XY/47,XXY line may be present at a level below the array's detection threshold, and it matters because mosaicism carries a milder phenotype, a greater likelihood of spontaneous fertility, and a different surveillance threshold. Second, the karyotype shows the mechanism — whether the sex chromosome complement is a simple trisomy, a mosaic, or a structurally abnormal chromosome — and the mechanism frames the genetic counselling, even though the recurrence risk for a de novo sex chromosome aneuploidy is low. [1] [2]
Epidemiology & Risk Factors
Sex chromosome aneuploidies are the commonest group of chromosomal disorders in humans. Klinefelter syndrome has a prevalence of approximately one in six hundred male births — a figure remarkably consistent across populations — making it more common than Down syndrome. Triple X syndrome and XYY syndrome each occur in approximately one in one thousand births, and together the sex chromosome trisomies account for a substantial proportion of all chromosomal disorders. Yet despite this high prevalence, the population of diagnosed individuals is small, because the diagnosis rate is low. [1] [5]
The single most important epidemiological fact is the under-diagnosis rate. An estimated fifty to seventy-five per cent of males with Klinefelter syndrome are never diagnosed in life, and the figures for 47,XXX and 47,XYY are even higher — some studies suggest that fewer than ten per cent of cases are ever identified. The reasons are the mild and variable phenotype, the overlap with the general population, and the absence of a newborn screening programme. This under-diagnosis has two consequences: the published phenotype, drawn from diagnosed cases, is biased towards the more severe end, and the clinician must maintain a low threshold to test when the pattern is present. [1] [3]
Advanced maternal age is a risk factor for sex chromosome aneuploidy, as it is for autosomal trisomies, because meiotic nondisjunction becomes more likely as oocytes age. However, the maternal-age effect for sex chromosome aneuploidies is smaller than for autosomal trisomies, partly because a proportion of cases arise from paternal meiotic nondisjunction — particularly for 47,XXY, where approximately half of cases are of paternal origin. This means that counselling about recurrence risk in a future pregnancy is generally reassuring: the recurrence risk after one child with a de novo sex chromosome aneuploidy is low, close to the population baseline. [1] [2]
The risk factors for individual complications — separate from the aneuploidy itself — are the risk factors the surveillance framework is built to modify. A man with untreated testosterone deficiency is at risk of osteoporosis and fracture; a man with metabolic syndrome is at risk of type 2 diabetes and cardiovascular disease; a man with venous stasis and a thrombophilia is at risk of thromboembolism. Identifying these risks early, and modifying them with testosterone replacement, bone protection, metabolic management, and lifestyle intervention, is the epidemiology that matters at the bedside. [3] [4]
Pathophysiology
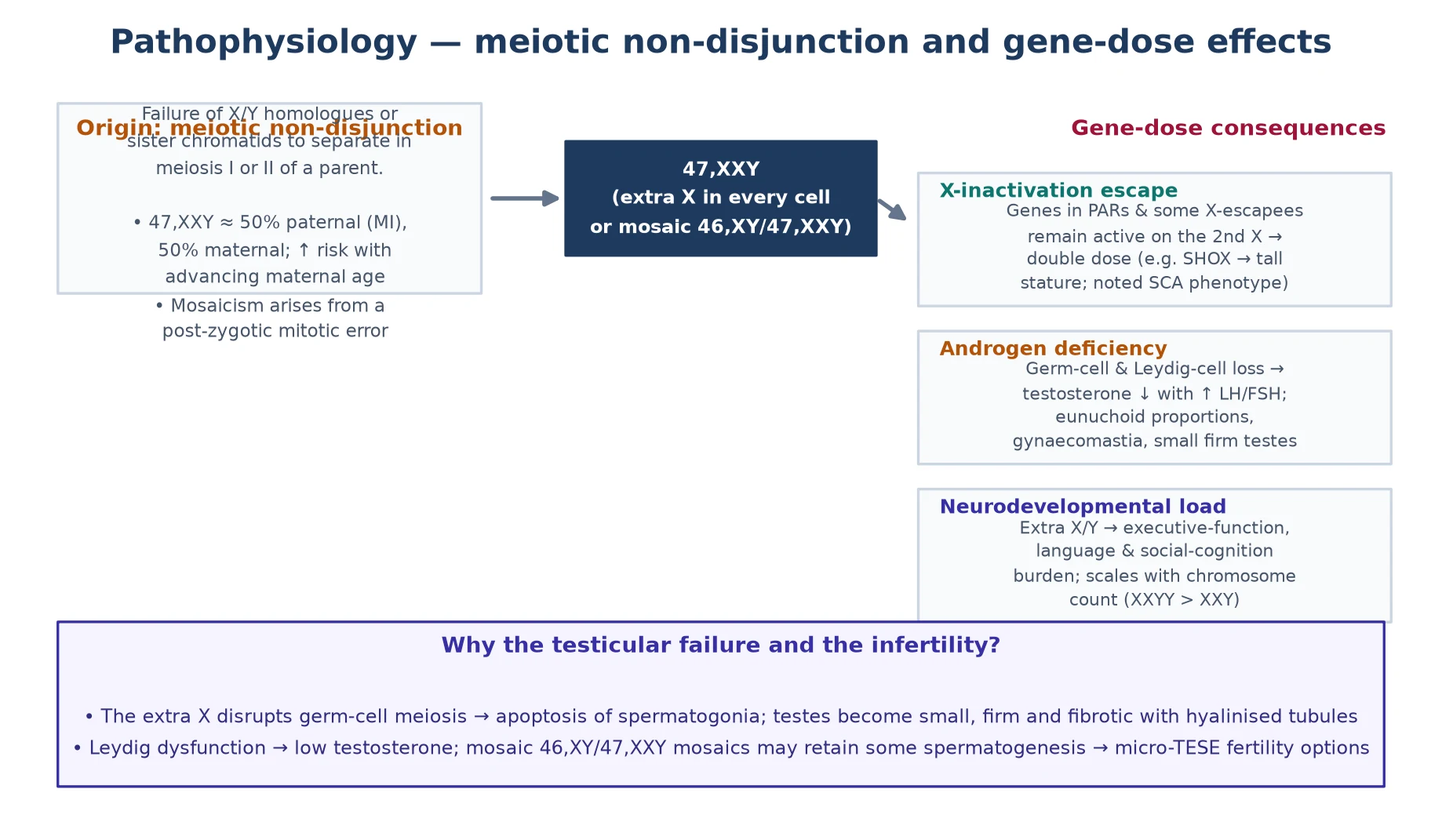
The pathophysiology of sex chromosome aneuploidies is a gene-dosage problem shaped by the X-inactivation mechanism. In Klinefelter syndrome, the extra X chromosome is largely inactivated — as it is in typical females, where one X is randomly inactivated in each cell — but the inactivation is incomplete. A subset of genes, known as escape genes, evade X-inactivation and are therefore expressed from both X chromosomes in 47,XXY cells, producing a gene-dosage excess that drives the phenotype. [2] [3]
The escape genes are the molecular link between the karyotype and the clinical features. Genes in the pseudoautosomal regions of the X and Y chromosomes — SHOX, the short-stature homeobox gene, is the best characterised — are normally expressed from both sex chromosomes, and their overexpression in 47,XXY and 47,XYY contributes to the tall stature. Genes involved in neural development and language that escape inactivation contribute to the learning and language difficulties. The overall effect is subtle but pervasive: the gene-dosage excess does not produce a severe malformation syndrome, as an extra autosome does, but it shapes neurodevelopment, endocrine function, and metabolism in ways that accumulate across the lifespan. [3] [6]
The gonadal pathophysiology in Klinefelter syndrome is distinctive and progressive. At birth, the testes may appear normal or near-normal, and germ cell numbers are present. Over childhood and early puberty, however, the seminiferous tubules undergo progressive hyalinisation and fibrosis, the germ cells are lost, and the Sertoli cell function declines — reflected in falling inhibin B and anti-Müllerian hormone (AMH) levels. Leydig cell dysfunction follows, producing a rise in luteinising hormone (LH) and follicle-stimulating hormone (FSH) and a fall in testosterone, and the testes become small and firm. The result is primary gonadal failure: the biochemical signature is high LH and FSH with low or low-normal testosterone, and the clinical signature is small testes, gynaecomastia, and, in adulthood, infertility with azoospermia. [1] [2]
The neurobiological basis of the cognitive phenotype is being mapped by neuroimaging. Boys and men with 47,XXY show reduced total brain volume, with disproportionate reduction in temporal and frontal grey matter — regions involved in language processing and executive function — and alterations in white matter integrity. These structural differences correlate with the language-based learning difficulties and executive function challenges that are the core of the neurodevelopmental phenotype. The extra X chromosome in females with 47,XXX produces a similar but often milder neurodevelopmental profile, and the extra Y in 47,XYY produces a motor and language phenotype that overlaps with 47,XXY. [6] [5]
Clinical Presentation
The clinical presentation of a sex chromosome aneuploidy depends entirely on the age at which the person is seen and the karyotype involved, and a fellowship answer earns depth by handling each life stage separately. The diagnosis may be made prenatally, in childhood through neurodevelopmental assessment, in adolescence through pubertal evaluation, or in adulthood through fertility or metabolic investigation — or, for the majority, never at all. [1] [5]
In the prenatal period, a sex chromosome aneuploidy is most often identified incidentally — on a cell-free DNA screen performed for autosomal trisomy risk, or on a karyotype performed for an unrelated indication such as advanced maternal age or an abnormal ultrasound finding. The counselling challenge is substantial: the cell-free DNA screen for sex chromosome aneuploidies has a higher false-positive rate and a higher no-call rate than for autosomal trisomies, and the phenotype described in the postnatal literature is biased towards the more severe end because it is drawn from diagnosed cases. The prospective eXtraordinarY babies study is addressing this bias by following prenatally diagnosed children, and its early data confirm that the phenotype is milder and more variable than the case-report literature suggests. [5] [7]
In childhood, the presentation centres on neurodevelopment. Language delay is often the first sign — a boy who is slow to talk and whose speech is difficult to understand — followed by learning difficulties, particularly in reading, spelling, and written expression, and by motor clumsiness and hypotonia. Tall stature may be noted, with long limbs and a eunuchoid body build, and clinodactyly of the fifth finger and a single palmar crease may be present but are non-specific. Behavioural features include shyness, anxiety, social difficulties, and an increased rate of autism and attention-deficit traits. The features overlap substantially with the general population, which is why the diagnosis is so often missed in childhood. [1] [6]
In adolescence, the presentation is dominated by puberty and the genital examination. Small, firm testes — a testicular volume less than 6 mL by mid-puberty, or testes disproportionately small for the Tanner stage — is the clinical hallmark of Klinefelter syndrome and the finding that should prompt a karyotype. Gynaecomastia, delayed or incomplete puberty, and a eunuchoid habitus may accompany it. Biochemical primary gonadal failure — elevated LH and FSH with low or low-normal testosterone — confirms the endocrine component. For 47,XXX and 47,XYY, pubertal development is typically normal, and the diagnosis is more likely to be made through the neurodevelopmental pathway than through endocrine presentation. [1] [2]
In adulthood, Klinefelter syndrome may present for the first time at a fertility clinic with azoospermia and small testes, or to a physician with osteoporosis, metabolic syndrome, type 2 diabetes, cardiovascular disease, or venous thromboembolism. The vignette of a tall man with osteoporotic fracture, or a young man with an unexplained deep vein thrombosis, should prompt consideration of Klinefelter syndrome alongside the standard differential. Mental health presentations — depression, anxiety, fatigue — are common and may reflect testosterone deficiency, psychosocial factors, or both. The principle is that the diagnosis may surface at any age, and the clinician who recognises the pattern earns the diagnosis. [3] [4]
Differential Diagnosis
The differential diagnosis of a suspected sex chromosome aneuploidy splits into two questions: what else causes the neurodevelopmental phenotype, and what else causes the endocrine phenotype. The discriminating skill is to know when to send the karyotype and when to pursue an alternative diagnosis. [1] [6]
The neurodevelopmental differential — language delay, learning difficulties, tall stature, and behavioural features — is broad because these features overlap with the general population. Fragile X syndrome is the most important genetic differential of developmental delay with learning difficulties in a boy, and it shares the social and language phenotype; the distinguishing features are the family history of intellectual disability in an X-linked pattern, the characteristic facial features (long face, large ears, prominent jaw), and macro-orchidism in postpubertal males, which contrasts with the small testes of Klinefelter syndrome. Noonan syndrome shares the learning difficulties and may share a cardiac phenotype, but it produces short stature rather than tall stature, and the karyotype is normal. [1]
The endocrine differential of hypogonadism in an adolescent male includes constitutional delay of growth and puberty, the commonest cause of delayed puberty in boys. Constitutional delay is distinguished by a positive family history, bone age delay consistent with the height age, and a normal karyotype; the testes are small because the boy is prepubertal, not because they are failing, and they will grow as puberty begins. Kallmann syndrome combines hypogonadotrophic hypogonadism with anosmia, and it is distinguished from Klinefelter syndrome by the low LH and FSH (hypogonadotrophic, not hypergonadotrophic) and the anosmia. A pituitary tumour may produce secondary hypogonadism and is excluded by prolactin and imaging. [1] [2]
Tall stature with eunuchoid proportions may raise Marfan syndrome or homocystinuria, both of which produce long limbs, but neither produces small testes or primary gonadal failure. Prior testicular damage — from undescended testes, torsion, mumps orchitis, chemotherapy, or anabolic steroid use — may produce small testes and gonadal failure in a 46,XY male, and the history and the karyotype separate these from Klinefelter syndrome. The principle is that the combination of small firm testes, gynaecomastia, tall stature, and learning difficulties in a boy is sufficiently distinctive that the karyotype is diagnostic. [2] [3]
The trap that costs marks is confirming the diagnosis with a chromosomal microarray but no karyotype. Microarray may identify the sex chromosome complement, but it cannot reliably detect low-level mosaicism, and it cannot show the mechanism in the way a karyotype can. When the clinical diagnosis is a sex chromosome aneuploidy, request a karyotype — it is the diagnostic test, and the information it provides about mosaicism frames the counselling and the management. [1]
| Feature | Klinefelter (47,XXY) | Constitutional delay | Kallmann syndrome |
|---|---|---|---|
| Testosterone | Low or low-normal | Low (prepubertal) | Low |
| LH / FSH | High (hypergonadotrophic) | Low (prepubertal) | Low (hypogonadotrophic) |
| Testes | Small, firm, below expected volume | Small (prepubertal, will grow) | Small (prepubertal) |
| Stature | Tall, eunuchoid | Short for age, delayed bone age | Normal |
| Karyotype | 47,XXY | 46,XY (normal) | 46,XY (normal) |
| Other features | Gynaecomastia, learning difficulties | Family history of late puberty | Anosmia, midline defects |
Clinical & Bedside Assessment
The bedside assessment of a person with a suspected or confirmed sex chromosome aneuploidy is structured around the lifespan framework rather than a single complaint, because the point of the visit is to run the surveillance net as well as to address the presenting problem. A complete assessment covers the neurodevelopmental, endocrine, metabolic, bone, and psychosocial domains at every relevant visit. [1] [4]
The history explores developmental milestones, language development, school performance, and behaviour — because the neurodevelopmental phenotype is the feature that shapes the person's life most. Ask about puberty onset and progression, gynaecomastia, and any fertility concerns. Explore symptoms of testosterone deficiency — fatigue, low mood, reduced libido, poor concentration — in adolescents and adults. A careful growth history, plotted on standard charts with awareness of the tall-stature trajectory, frames the physical assessment. Ask about family history of learning difficulty, delayed puberty, or infertility, and about the prenatal diagnosis pathway if the diagnosis was made antenatally. [1] [6]
Examination begins with growth parameters — height, weight, and body proportions — because tall stature with eunuchoid proportions (arm span exceeding height, upper-to-lower segment ratio reduced) is a hallmark of an extra sex chromosome. The genital examination is non-negotiable in an adolescent male: measure testicular volume with a Prader orchidometer, because a volume below 6 mL by mid-puberty, or testes disproportionately small for the Tanner stage, is the clinical finding that should prompt a karyotype. Assess for gynaecomastia — true glandular tissue, not just adipose — and Tanner staging. [1] [2]
Neurodevelopmental and psychosocial assessment is essential at every visit. Screen for language and literacy difficulties with age-appropriate tools, assess executive function, and screen for anxiety, depression, and autism traits. The social dimension matters: people with sex chromosome aneuploidies may have social vulnerability — difficulty reading social cues, risk of exploitation — and a careful assessment of the person's social network, educational placement, and employment is part of the clinical picture. Assessment is strengths-based: the person's abilities — which often include strong verbal reasoning when language is supported, good long-term memory, and creative thinking — are named alongside the challenges. [4] [5]
Assess development and learning with standardised tools appropriate to the person's age, and interpret the result against the expected profile of the specific aneuploidy. A fellowship answer makes clear that learning difficulties in sex chromosome aneuploidies are expected and are not, by themselves, a sign of a new problem — but a regression, a plateau, or a change in behaviour demands investigation for a complication such as testosterone deficiency, depression, thyroid dysfunction, or an unrelated neurological event. [3] [4]
Investigations
The investigation plan for a sex chromosome aneuploidy combines diagnostic confirmation with lifespan surveillance, and it is a schedule rather than a one-off work-up. The first test is the karyotype — to confirm the diagnosis, to identify mosaicism, and to frame the genetic counselling — followed by endocrine, metabolic, bone, and psychosocial assessment. [1] [2]
Diagnostic confirmation is by karyotype, and a chromosomal microarray may be added but never replaces the karyotype. The karyotype distinguishes a non-mosaic 47,XXY from a mosaic 46,XY/47,XXY, and it identifies higher-grade variants such as 48,XXXY and 49,XXXXY that carry a more severe phenotype and a different management pathway. When the diagnosis is made prenatally on cell-free DNA, a confirmatory karyotype — by amniocentesis or, postnatally, by peripheral blood — is mandatory, because the false-positive rate for sex chromosome aneuploidies on cell-free DNA is substantially higher than for autosomal trisomies. [1] [5]
Endocrine investigation in confirmed Klinefelter syndrome centres on the gonadal axis. Measure testosterone (total, ideally morning fasting), LH, FSH, oestradiol, inhibin B, and AMH. The biochemical signature of primary gonadal failure is elevated LH and FSH — often markedly elevated — with low or low-normal testosterone and low inhibin B. The pattern evolves over puberty: in early puberty, LH and FSH may rise before testosterone falls, and the full picture may not be evident until mid-to-late puberty. Thyroid function should be checked, because autoimmune thyroid disease is over-represented. [1] [4]
Metabolic and cardiovascular investigation is part of the baseline and ongoing schedule. A fasting lipid panel, HbA1c or fasting glucose, liver function tests, and blood pressure measurement screen for the metabolic syndrome that is over-represented in Klinefelter syndrome. A coagulation screen is prudent, given the elevated venous thromboembolism risk, particularly before starting testosterone replacement. Bone density assessment by DEXA is recommended at baseline in adults and at the point of diagnosis in adolescents, because untreated testosterone deficiency drives osteoporosis, and the DEXA provides the baseline against which the response to testosterone replacement and bone protection is measured. [3] [4]
Management — Resuscitation
Resuscitation in the context of a sex chromosome aneuploidy is unusual, because the diagnosis is most often elective and the presentation chronic. Two acute scenarios, however, may bring a person with Klinefelter syndrome to an emergency department, and both demand recognition of the underlying condition. [1] [3]
The first is venous thromboembolism. Men with Klinefelter syndrome have a substantially elevated risk of deep vein thrombosis and pulmonary embolism — estimates range from five to ten times the population baseline — driven by a combination of hypogonadism-related haemoconcentration, altered coagulation factor synthesis, and, in some, the effect of exogenous testosterone on erythropoiesis and platelet reactivity. A young man presenting with an unexplained thromboembolism warrants consideration of Klinefelter syndrome alongside the standard thrombophilia screen, and the testosterone replacement regimen should be reviewed — a high haematocrit may signal over-replacement and the need for dose reduction. Standard anticoagulation is the treatment, with the duration guided by whether the thrombosis was provoked. [3]
The second is an osteoporotic fracture in a young adult male. Untreated testosterone deficiency in Klinefelter syndrome drives accelerated bone loss, and a fragility fracture — a vertebral compression fracture, a wrist fracture from a low-energy fall — may be the presenting event. Management is analgesia, fracture stabilisation, and investigation of the underlying cause, including a testosterone level and a karyotype if the phenotype is suggestive. Testosterone replacement, with or without a bisphosphonate depending on the DEXA T-score, is the bone-protection strategy. [4]
Severe symptomatic hypogonadism at presentation — profound fatigue, depression, and sexual dysfunction in a man with untreated Klinefelter syndrome — may be disabling enough to constitute an urgent management priority, though it is not a resuscitation scenario in the traditional sense. The principle is that untreated testosterone deficiency has measurable, progressive harms — bone loss, metabolic deterioration, cardiovascular risk, and impaired quality of life — and that testosterone replacement, titrated to clinical and biochemical targets, reverses many of these harms. [1] [4]
Management — Definitive & Stepwise
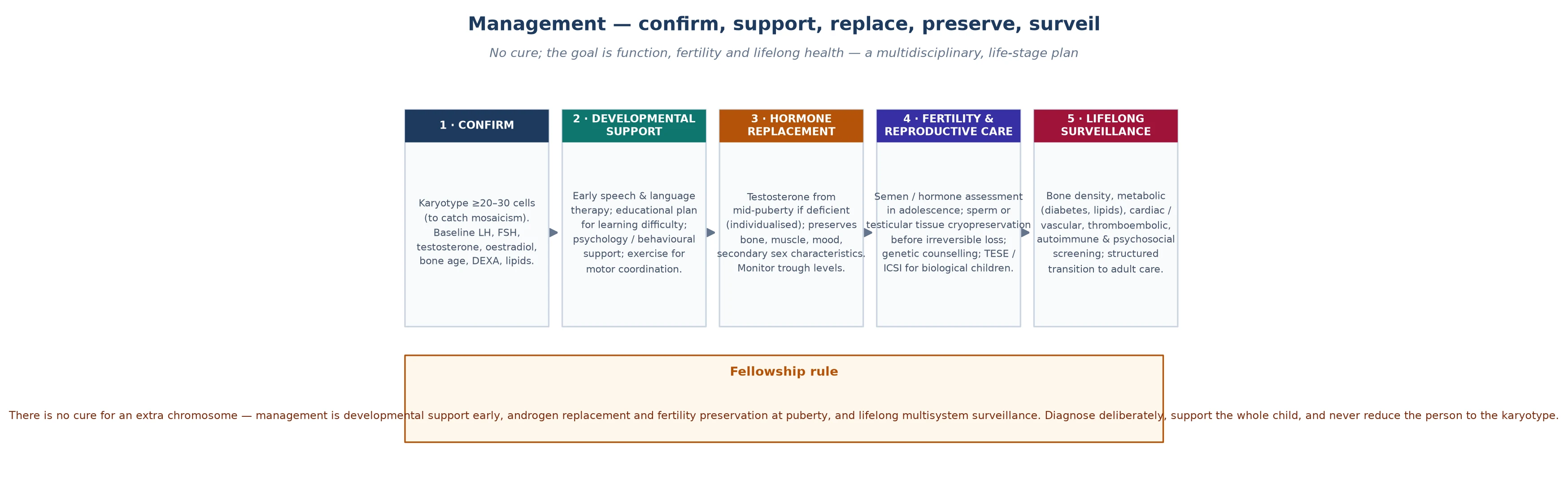
Definitive management of sex chromosome aneuploidies is a lifespan surveillance framework, and it is the core of fellowship-level competence. The framework moves with the person from the prenatal counselling conversation through childhood neurodevelopmental support, pubertal monitoring, adolescent transition, and adult endocrine and metabolic management. Each stage carries non-negotiable items that prevent specific, predictable harm. [1] [4]
In the prenatal period, the management is counselling. When a sex chromosome aneuploidy is identified on cell-free DNA screening or amniocentesis, the counselling must be balanced, honest, and strengths-based. The key messages are that the phenotype is mild and variable, that the published literature is biased towards the more severe end, that many people with sex chromosome aneuploidies are never diagnosed and live typical lives, and that the available surveillance and treatment — particularly testosterone replacement in Klinefelter syndrome — can modify the outcome. The counselling should be non-directive, and the family should be connected to a genetic counsellor and, where available, to a support organisation. [5] [7]
In childhood, the management is neurodevelopmental. Early intervention for language delay — speech pathology from the toddler years — is the single most impactful intervention, because the language-based learning difficulties are the core of the phenotype and they compound if unsupported. Educational support, including individualised learning plans and classroom accommodations for attention and executive function challenges, is essential through the school years. Motor therapy for clumsiness and hypotonia, behavioural support for anxiety and social difficulties, and autism and ADHD screening where the phenotype suggests it, complete the childhood framework. [5] [6]
In adolescence, the management is endocrine. When primary gonadal failure is confirmed biochemically — rising LH and FSH with a testosterone that fails to rise into the pubertal range — testosterone replacement is initiated. The timing and the regimen are tailored to the individual: pubertal induction may begin at around twelve to fourteen years with low-dose testosterone, escalating to adult replacement doses over two to three years, guided by clinical response (Tanner staging, virilisation, growth, wellbeing) and biochemical targets (morning testosterone in the normal range). The route — topical gel, intramuscular injection, or transdermal patch — is chosen for adherence and acceptability. Gynaecomastia may require surgical reduction if it persists despite testosterone adequacy. [1] [4]
In adulthood, the framework adds metabolic and cardiovascular risk management (lipid optimisation, glycaemic control, blood pressure), bone density surveillance (DEXA every two to five years, with bisphosphonate if osteoporosis is confirmed), fertility counselling (micro-TESE for sperm retrieval, with assisted reproduction), and psychosocial support. The transition to adult care should be planned, documented, and begun in late adolescence, with a structured handover that preserves endocrine, bone, metabolic, and psychosocial continuity. The framework does not end at twenty-one; it changes shape. [4] [3]
Specific Subtypes & Scenarios
Each sex chromosome aneuploidy carries a distinctive management profile, and a fellowship answer earns depth by handling them individually rather than as a single block. The surveillance items overlap, but the management is karyotype-specific. [1] [5]
The Klinefelter syndrome scenario centres on testosterone replacement, bone health, metabolic risk, and fertility. Testosterone replacement is the mainstay of adult management, titrated to clinical and biochemical targets, with monitoring for erythrocytosis (haematocrit), prostate health, and cardiovascular risk. Bone density surveillance by DEXA, with bisphosphonate when osteoporosis is confirmed, protects the skeleton. Fertility counselling is central: although azoospermia is the rule, micro-TESE (microsurgical testicular sperm extraction) can retrieve sperm in approximately forty to fifty per cent of men with Klinefelter syndrome, enabling intracytoplasmic sperm injection (ICSI) and biological fatherhood. The counselling should begin in adolescence, because the prospects for successful sperm retrieval decline with age, and semen cryopreservation or testicular tissue cryopreservation may be discussed. [2] [4]
The Triple X syndrome scenario is predominantly neurodevelopmental. Girls and women with 47,XXX benefit from early language and literacy intervention, educational support, and psychosocial support, but hormonal treatment is typically not required — pubertal development and fertility are usually normal. The management is monitoring for the neurodevelopmental and mental health features — anxiety, social difficulties, and, in some, executive function challenges — and providing support tailored to the individual. The postnatal-diagnosis phenotype is more severe than the prenatal-diagnosis phenotype, reflecting ascertainment bias, and the counselling of a prenatally diagnosed family should be cautious and optimistic. [7] [5]
The XYY syndrome scenario is similarly neurodevelopmental. Boys and men with 47,XYY share the language delay, learning difficulties, and behavioural features of the other sex chromosome trisomies, but without gonadal failure, testosterone deficiency, or infertility. Management is educational and psychosocial: early language intervention, classroom support, and behavioural monitoring. The historical association with criminality — based on prison screening studies in the 1960s and 1970s — has been comprehensively refuted by prospective cohorts showing that the vast majority of males with 47,XYY lead typical, productive lives, and the counselling must explicitly address and correct this misconception. [6]
The mosaicism scenario (46,XY/47,XXY) carries a milder and less predictable phenotype than non-mosaic 47,XXY. Gonadal function may be partially preserved, testosterone deficiency may be absent or mild, and spontaneous fertility is more likely. The management is tailored to the individual's endocrine and neurodevelopmental profile rather than to the full Klinefelter framework, with a lower threshold for monitoring and a higher threshold for testosterone replacement than in non-mosaic disease. The karyotype is the test that identifies mosaicism and sets the management pathway. [1]
The higher-grade variant scenario (48,XXXY, 49,XXXXY) is clinically distinct. These karyotypes carry a more severe phenotype — intellectual disability, more pronounced dysmorphism, cardiac defects (particularly in 49,XXXXY), skeletal anomalies, and more severe gonadal failure — and they follow a different management pathway with earlier and more intensive multidisciplinary involvement. The principle is that each additional X chromosome adds gene-dosage burden, and the phenotype scales accordingly. [1] [3]
Why micro-TESE has changed the fertility counselling in Klinefelter syndrome
For decades, the teaching was that Klinefelter syndrome means infertility, full stop. This is no longer accurate. Microsurgical testicular sperm extraction (micro-TESE), performed by an experienced urological surgeon, can retrieve sperm from the testicular tissue of approximately forty to fifty per cent of men with non-mosaic 47,XXY, and the retrieved sperm can be used for intracytoplasmic sperm injection (ICSI) to achieve biological fatherhood. The success rate declines with age, which is why fertility counselling should begin in adolescence and why some centres offer testicular tissue cryopreservation at the time of diagnostic biopsy. The message to the young man with Klinefelter syndrome is no longer "you will never have biological children" but "there is a realistic possibility, and here is the pathway." [2] [4]
Complications & Pitfalls
The complications of sex chromosome aneuploidies are the harms left undetected, and the pitfalls are the assumptions that lead clinicians to miss them. A fellowship answer handles both, because the harm is rarely the karyotype itself — it is the preventable complication that the surveillance framework was designed to catch. [1] [3]
Metabolic and cardiovascular complications are a major contributor to the morbidity and mortality of Klinefelter syndrome. Men with KS have an elevated risk of type 2 diabetes, metabolic syndrome, and cardiovascular disease, driven by a combination of testosterone deficiency, altered body composition (increased fat mass, reduced muscle mass), and possibly direct metabolic effects of the extra X chromosome. Testosterone replacement improves body composition and insulin sensitivity, and metabolic surveillance — lipid panel, HbA1c, blood pressure — is part of the annual schedule. [3]
Venous thromboembolism is a distinctive and high-yield complication. Men with Klinefelter syndrome have a five-to-ten-fold elevated risk of deep vein thrombosis and pulmonary embolism, driven by hypogonadism-related haemoconcentration, altered coagulation factor synthesis, and, in some, the effect of exogenous testosterone on erythropoiesis. Monitoring the haematocrit during testosterone replacement — and reducing the dose if it exceeds 0.54 — is the safeguard. A young man presenting with a thromboembolism warrants consideration of Klinefelter syndrome alongside the standard thrombophilia screen. [3]
Bone complications — osteoporosis and fragility fracture — follow from untreated testosterone deficiency. Testosterone is essential for bone formation and maintenance, and its deficiency drives accelerated bone loss that begins in adolescence if replacement is delayed. DEXA surveillance from the point of diagnosis, with bisphosphonate when osteoporosis is confirmed, is the bone-protection strategy. The principle is that bone health is a treatable complication if caught early, and an irreversible harm if missed. [4]
Neurodevelopmental and psychosocial complications run through the lifespan. Untreated learning difficulties compound across the school years, reducing educational attainment, employment prospects, and self-esteem. Anxiety, depression, and social vulnerability are over-represented, and an autism or ADHD phenotype may coexist and require its own management. The pitfall is attributing all of the person's difficulties to the karyotype — a form of diagnostic overshadowing — and missing a treatable comorbidity such as testosterone deficiency, thyroid dysfunction, depression, or a specific learning difficulty that would benefit from targeted intervention. [5] [6]
Prognosis & Disposition
The prognosis of sex chromosome aneuploidies has improved with modern surveillance and treatment, and the lifespan of a person with Klinefelter syndrome who receives adequate testosterone replacement and metabolic management is now close to the population average. Cohort studies from Denmark, which has near-complete national registry data, suggest a reduction in life expectancy of approximately two to five years, driven largely by cardiovascular and metabolic disease and by complications of untreated hypogonadism. With early diagnosis and structured management, this gap narrows. [1] [3]
The determinants of prognosis are the age at diagnosis, the adequacy of testosterone replacement, the quality of neurodevelopmental support, and the management of metabolic and bone risk. A boy diagnosed in childhood who receives early language intervention, educational support, timely testosterone replacement, bone surveillance, and psychosocial advocacy will follow a trajectory close to his potential. A man diagnosed late, with untreated hypogonadism, osteoporosis, and metabolic syndrome, will have accumulated preventable harm. The prognosis is therefore not fixed by the karyotype; it is shaped by the quality of the surveillance. [4] [1]
Quality of life is shaped as much by social and educational factors as by medical ones. Educational attainment, employment, relationships, and social inclusion are the outcomes that matter to the person, and the fellowship answer frames prognosis in those terms rather than in deficit language alone. People with sex chromosome aneuploidies work, form relationships, and contribute to their communities, and the disposition plan should support that trajectory. The neurodiversity-affirming framing — recognising the person's strengths and differences rather than pathologising them — is increasingly the standard. [5]
Disposition for a general paediatrician is shared, structured care. The paediatrician owns the surveillance schedule, the coordination of subspecialty input, the developmental and educational support, and the transition to adult care. Endocrinology, speech pathology, psychology, fertility services, and genetic counselling each contribute at the relevant point, and a named coordinator prevents the fragmentation that is the enemy of a checklist-based schedule. Early referral to a structured multidisciplinary sex chromosome aneuploidy clinic, where one exists, supports the person and the family from the point of diagnosis. [4] [1]
Special Populations
Sex chromosome aneuploidies interact with the person's social, cultural, and developmental context, and the same karyotype behaves differently across populations. Access, adherence, and late presentation all shape outcome, and a fellowship answer recognises that the surveillance framework is only as good as the person's ability to engage with it. [1] [5]
Families with limited health literacy or language barriers face the challenge of understanding a diagnosis whose implications are complex and variable. The counselling conversation — particularly after a prenatal diagnosis — must be delivered in the family's language, with an interpreter if needed, and with written and visual materials that explain the phenotype, the surveillance, and the support pathway in plain language. The risk of miscommunication is high, because the phenotype is mild and the literature is biased, and a balanced, non-directive approach that respects the family's values is essential. [7]
Migrant, refugee, and asylum-seeking families may have had no prenatal screening, may arrive with an unrecognised diagnosis, and may face barriers to engaging with a complex multidisciplinary schedule. A careful developmental history, confirmation of the diagnosis by karyotype, an interpreter-mediated explanation, and a written schedule in the family's language are the foundations. The neurodevelopmental phenotype may be attributed to language disruption or trauma rather than to the karyotype, and the discriminating clinician holds both possibilities in view. [1]
Socioeconomically disadvantaged families carry the burden that adherence to a multi-specialist schedule is harder, and the limiting step is often attendance rather than the medicine. Structuring the schedule around a single coordinated visit, providing written and visual materials, and linking the family to a support organisation and to transport and appointment support all improve engagement. The aim is to fit the surveillance to the family's reality rather than the reverse. [4]
The neurodiversity-affirming population — people with sex chromosome aneuploidies and their advocates, and the broader neurodiversity community — is reshaping the conversation. The traditional framing of sex chromosome aneuploidies as disorders to be treated is giving way to a framing of difference and variation, with an emphasis on the person's strengths, on removing barriers, and on providing support rather than cure. This is particularly relevant to the prenatal counselling conversation, where the question of whether to continue a pregnancy is framed by the information the family receives, and where a balanced, strengths-based approach is ethically mandatory. [5] [6]
Evidence, Guidelines & Regional Differences
The evidence base for sex chromosome aneuploidies rests on clinical reviews, prospective cohort studies, and an evolving set of clinical practice guidelines, supplemented by the emerging data from the eXtraordinarY babies study that is redefining the prenatal-diagnosis phenotype. The field is hampered by the under-diagnosis rate — which means that the published literature is drawn from the more severe, diagnosed end of the spectrum — and by the historical legacy of ascertainment bias in the 47,XYY literature. [1] [3]
The Groth et al. clinical review in the Journal of Clinical Endocrinology and Metabolism (2013) remains the framing reference for Klinefelter syndrome, organising the genetics, the comorbidity map, and the lifespan trajectory. The Lanfranco et al. review in the Lancet (2004) is the landmark earlier review from the Nieschlag group, and the Kanakis and Nieschlag review in Metabolism (2018) extends the framing beyond hypogonadism to the multisystem phenotype. Together these documents define a structured, evidence-based approach that generalises across settings. [1] [2] [3]
The eXtraordinarY babies study (Tartaglia et al., 2020) is a landmark prospective study of prenatally diagnosed children with sex chromosome trisomies, and its early data are transforming the counselling conversation. By following children identified prenatally — rather than retrospectively from diagnosed cases — the study is documenting a milder and more variable phenotype than the case-report literature, confirming that the majority of children with sex chromosome trisomies have relatively subtle neurodevelopmental differences and good overall health. This is the evidence that underpins the modern, balanced, strengths-based counselling approach. [5] [7]
The TESTO trial (Davis et al., 2025) is informing the controversy over early testosterone therapy in infants and children with 47,XXY. The question is whether early, brief testosterone supplementation in infancy improves neurodevelopmental outcomes, and the trial data are being interpreted alongside the broader principle that testosterone replacement should be guided by biochemical evidence of gonadal failure rather than by the karyotype alone. The controversy is unresolved, and the fellowship answer should acknowledge both sides while emphasising the evidence-based standard of pubertal monitoring and replacement when gonadal failure is confirmed. [9]
Regional differences are practical rather than scientific. Australia and New Zealand apply the international surveillance frameworks with access to karyotyping through public genetics services, paediatric and adult endocrinology, and multidisciplinary sex chromosome aneuploidy clinics in the major centres. There is no newborn screening programme for sex chromosome aneuploidies in ANZ or internationally, reflecting the controversy over medicalising a condition whose phenotype overlaps with the general population. Telehealth and outreach extend the surveillance framework into rural and remote communities. The Turner syndrome clinical practice guidelines (Gravholt et al., 2024) provide the parallel framework for the 45,X monosomy counterpart. [10] [4]
In Australia and New Zealand, karyotyping is accessible through public genetics services, and paediatric and adult endocrinology services manage testosterone replacement, bone surveillance, and metabolic monitoring. Multidisciplinary sex chromosome aneuploidy clinics are established in the major paediatric centres, coordinating endocrinology, speech pathology, psychology, and genetic counselling. There is no newborn screening programme for sex chromosome aneuploidies, and the diagnosis rate remains low, with most cases identified through the neurodevelopmental, pubertal, or fertility pathways. Transition to adult care is increasingly structured, and the neurodiversity-affirming approach is gaining ground in the counselling and management conversation. [1] [4]
Exam Pearls
A fellowship candidate answering on sex chromosome aneuploidies should land five anchor points and avoid four classic traps. The anchors are the framework examiners listen for; the traps are where candidates lose easy marks. [1] [3]
Anchor one: the commonest chromosomal aneuploidy in humans. Klinefelter syndrome, at one in six hundred males, is the commonest chromosomal disorder in humans — more common than Down syndrome — yet more than half of cases are never diagnosed. The teaching point is that the phenotype is mild, variable, and overlaps with the general population, and the clinician must maintain a low threshold to test when the pattern is present. [1]
Anchor two: the karyotype is the diagnostic test, not the microarray. Confirm the diagnosis with a karyotype, because it identifies mosaicism — which changes the management — and it shows the mechanism that frames the counselling. A microarray alone cannot reliably detect low-level mosaicism. [2]
Anchor three: testosterone replacement is the mainstay of Klinefelter management. From puberty onwards, when gonadal failure is confirmed biochemically, testosterone replacement protects the bones, improves body composition and metabolism, supports mood and wellbeing, and is titrated to clinical and biochemical targets. The dose is monitored for haematocrit — because testosterone replacement drives erythropoiesis and the thromboembolism risk is real. [4]
Anchor four: micro-TESE makes fertility possible. Infertility is no longer an absolute. Microsurgical testicular sperm extraction can retrieve sperm in approximately forty to fifty per cent of men with Klinefelter syndrome, enabling biological fatherhood through ICSI. The counselling should begin in adolescence, and the prospects decline with age. [2]
Anchor five: the neurodevelopmental phenotype is the feature that shapes a person's life. Language delay, learning difficulties, executive dysfunction, and social vulnerability are the core of the sex chromosome aneuploidy phenotype, and early intervention — speech pathology from the toddler years, educational support, psychosocial advocacy — is the most impactful management. Speak in neurodiversity-affirming, strengths-based language, and frame the person by their abilities, not their karyotype. [5] [6]
The four traps to avoid are confirming the diagnosis with a microarray but no karyotype, telling a young man he is infertile without discussing micro-TESE, attributing all difficulties to the karyotype (diagnostic overshadowing), and counselling a prenatal diagnosis using the biased postnatal literature. Avoid these and the rest of the answer falls into place. [1] [3]
References
- [1]Groth KA, Skakkebæk A, Høst C, Gravholt CH, Bojesen A. Clinical review: Klinefelter syndrome--a clinical update. J Clin Endocrinol Metab, 2013.PMID 23118429
- [2]Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E. Klinefelter's syndrome. Lancet, 2004.PMID 15262106
- [3]Kanakis GA, Nieschlag E. Klinefelter syndrome: more than hypogonadism. Metabolism, 2018.PMID 29382506
- [4]Gies I, Unuane D, Velkeniers B, De Schepper J. Management of Klinefelter syndrome during transition. Eur J Endocrinol, 2014.PMID 24801585
- [5]Tartaglia N, Ayari N, Howell S, D'Epagnier C, Zeitler P. Early neurodevelopmental and medical profile in children with sex chromosome trisomies: Background for the prospective eXtraordinarY babies study to identify early risk factors and targets for intervention. Am J Med Genet C Semin Med Genet, 2020.PMID 32506668
- [6]Ross JL, Roeltgen DP, Stefanatos G, et al. An extra X or Y chromosome: contrasting the cognitive and motor phenotypes in childhood in boys with 47,XYY syndrome or 47,XXY Klinefelter syndrome. Dev Disabil Res Rev, 2009.PMID 20014371
- [7]Wigby K, D'Epagnier C, Howell S, et al. Expanding the phenotype of Triple X syndrome: A comparison of prenatal versus postnatal diagnosis. Am J Med Genet A, 2016.PMID 27644018
- [8]Howell S, Hintz SR, Davis SM, et al. Eosinophilic esophagitis in individuals with sex chromosome aneuploidies: Clinical presentations and management implications. Mol Genet Genomic Med, 2021.PMID 34738344
- [9]Davis SM, Pyle L, Ross JL, et al. Testosterone Effects on Short-term Physical, Hormonal, and Neurodevelopmental Outcomes (TESTO) in Infants With 47,XXY. J Clin Endocrinol Metab, 2025.PMID 40177735
- [10]Gravholt CH, Viuff MH, Brun S, Stochholm K, Wallentin M. Clinical practice guidelines for the care of girls and women with Turner syndrome. Eur J Endocrinol, 2024.PMID 38748847