Paeds · genetics-dysmorphology-and-metabolism
Lysosomal storage disorders
Also known as Lysosomal storage disorders · Lysosomal storage diseases · LSDs · Inborn errors of lysosomal metabolism · Sphingolipidoses
A fellowship approach to the lysosomal storage disorders: recognise the clinical phenotypes that trigger the search (developmental regression with organomegaly, coarse facies, or cardiomyopathy), group them by stored substrate, confirm with a layered enzyme-and-genotype workup, and match the disease-modifying therapy — enzyme replacement, substrate reduction, or haemopoietic stem cell transplant — to whether the central nervous system is involved.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A four-month-old who had been smiling and rolling stops meeting milestones, grows irritable, and is found at the six-month check to have a liver edge five centimetres below the costal margin and spastic limbs. Across the corridor, a newborn bloodspot screen flags low galactocerebrosidase activity, and the clock starts on whether a transplant can be arranged before the demyelination becomes irreversible. In both rooms the unifying question is the same: which lysosomal enzyme is missing, how fast is it injuring the brain, and what can be done before the damage is permanent. The fellowship task is to reason from phenotype to substrate to therapy without losing time. [3] [6]
L · Y · S · O · S · O · M · E
Overview & Definition
The lysosome is the cell's recycling organelle — an acidic compartment packed with hydrolases that break down macromolecules into building blocks the cell can reuse. A lysosomal storage disorder is what happens when one link in that degradative chain fails: the enzyme is absent or misfolded, its substrate cannot be cleaved, and undegraded material accumulates progressively inside the lysosome until the cell distends, malfunctions, and dies. The stored material, not the missing enzyme per se, drives the visible phenotype, which is why a Gaucher macrophage engorged with glucocerebroside looks and behaves differently from a neuron swollen with ganglioside. [1] [4]
Although each disorder maps to a different enzyme and substrate, the clinical consequences follow recurring themes because lysosomes exist in almost every cell. Macrophage-rich organs enlarge, giving the hepatosplenomegaly that is the single most useful physical sign. The central nervous system bears the brunt in the neuronopathic forms, where stored substrate triggers neuroinflammation, demyelination, and neuronal loss. Bone, connective tissue, heart valves, and airways suffer in the disorders that store glycosaminoglycans or glycogen. Recognising that the same mechanism produces different patterns in different tissues is the conceptual hinge of the whole topic. [1] [2]
The inheritance matters because it changes counselling and screening. Most lysosomal storage disorders are autosomal recessive, so a previously affected child reshapes the reproductive risk for every subsequent pregnancy and makes carrier testing and prenatal diagnosis central to management. A small but high-yield group is X-linked — Fabry, Hunter syndrome (MPS II), and Danon disease — where affected boys present with disease and carrier mothers may show attenuated features. Naming the inheritance at the first consultation frames every later conversation about siblings, future children, and the wider family. [2] [3]
Classification
Clinicians classify lysosomal storage disorders by the substrate that accumulates, because the substrate predicts the organ involvement and points to the first-line investigation. The figure below lays out the major groups alongside their deficient enzymes; the groups most likely to appear in an examination are the sphingolipidoses, the mucopolysaccharidoses, and Pompe disease. [1] [4]

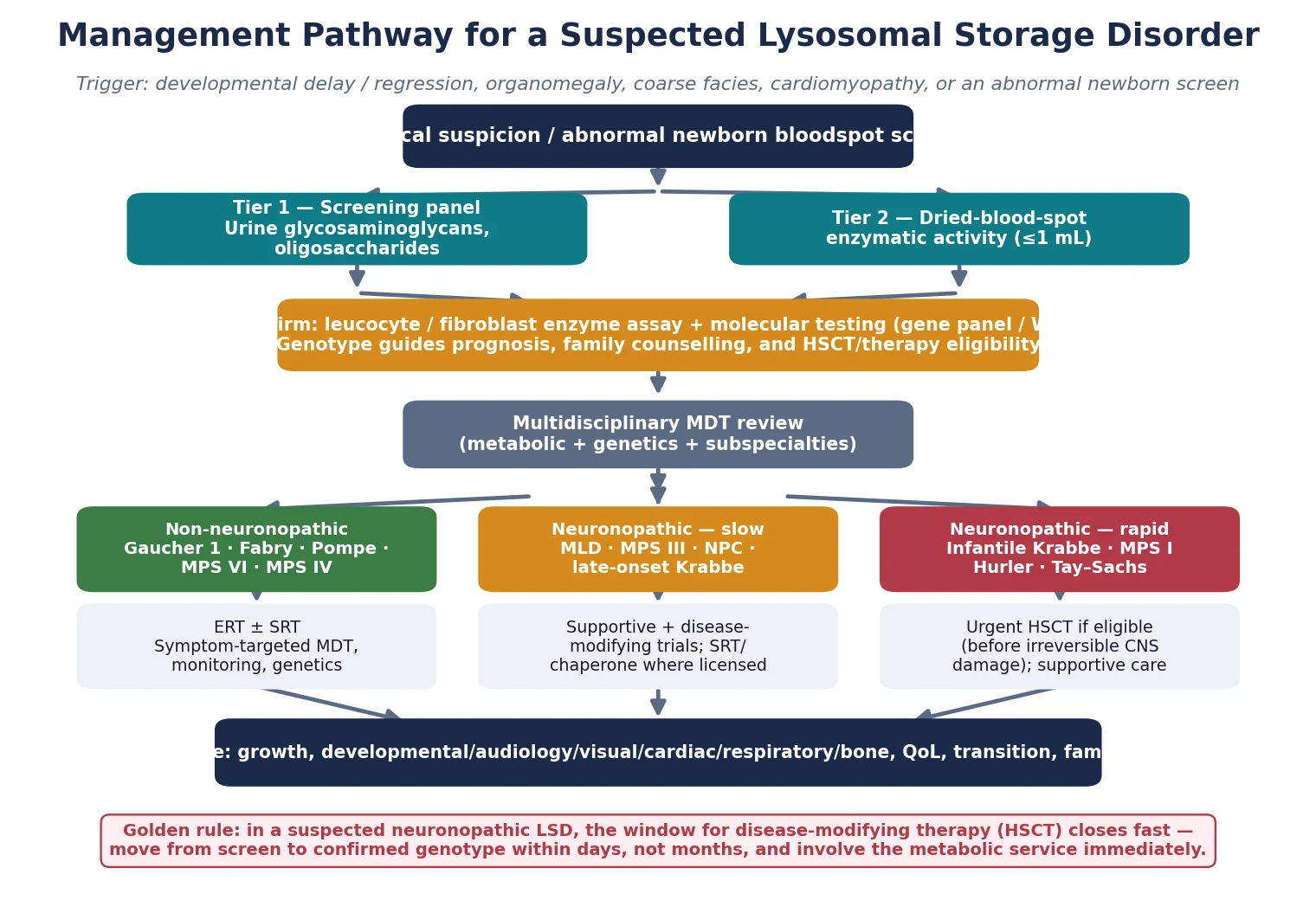
A second, more practical axis separates the disorders by whether the central nervous system is involved, because that distinction governs treatment choice. Non-neuronopathic disease — Gaucher type 1, Fabry, Pompe, MPS IV and VI — can be reached by intravenous enzyme replacement therapy, which does not cross the blood–brain barrier. Neuronopathic disease — infantile Krabbe, metachromatic leukodystrophy, Tay–Sachs, MPS III, and the severe Hurler phenotype of MPS I — needs a therapy that reaches the brain, and that usually means haemopoietic stem cell transplant given before the injury is fixed. Holding this binary in mind prevents the common error of offering intravenous enzyme to a child whose disease is primarily neurological. [2] [6]
Neuronopathic versus non-neuronopathic — the treatment-defining split
- Non-neuronopathic (ERT is effective): Gaucher type 1, Fabry, Pompe, MPS I Hurler–Scheie, MPS IV (Morquio), MPS VI (Maroteaux–Lamy). The viscera and skeleton bear the burden; the brain is spared.
- Neuronopathic (ERT cannot reach brain; consider HSCT or SRT): infantile Krabbe, metachromatic leukodystrophy, Tay–Sachs and Sandhoff, MPS III (Sanfilippo), MPS I Hurler, neuronopathic Gaucher (types 2 and 3).
- Mixed / progressive lipid traffic: Niemann–Pick type C — defective cholesterol transport, progressive neurodegeneration, and a treatment (miglustat) that may slow progression. [1] [2]
Epidemiology & Risk Factors
Individually each lysosomal storage disorder is rare, with prevalence figures between one in forty thousand and one in two hundred thousand, but collectively they are a significant burden in paediatric practice. Pooled estimates put the combined incidence at roughly one in five thousand to one in eight thousand live births, and several disorders are markedly more common in particular founder populations: Gaucher and Tay–Sachs in Ashkenazi Jewish families, metachromatic leukodystrophy in certain North African and Middle Eastern groups, and a cluster of sphingolipidoses among consanguineous communities. Consanguinity and a previously affected sibling are the two strongest risk factors and should lower the threshold for screening in any child with compatible features. [1] [2]
The epidemiology is shifting because of newborn screening. Krabbe disease, Pompe disease, and MPS I have been added to bloodspot panels in a growing number of regions, and that has changed the presenting population from symptomatic infants to asymptomatic neonates flagged by low enzyme activity on a dried blood spot. This presymptomatic detection is a double-edged benefit: it opens the transplant window for rapidly progressive Krabbe and the enzyme window for classic infantile Pompe, but it also surfaces infants with pseudodeficiency alleles or late-onset variants whose clinical course is uncertain and who may never become unwell. [6] [11]
Pathophysiology
The lysosome maintains an acidic interior near pH 4.5 that lets its hydrolases cleave complex macromolecules — glycosphingolipids, glycosaminoglycans, glycogen, and proteins — into reusable units. A pathogenic variant in the gene encoding one of these enzymes either abolishes its activity or, more often, causes misfolding so the enzyme is retained and degraded in the endoplasmic reticulum rather than trafficked to the lysosome. Either way the substrate cannot be processed, and it accumulates. The figure traces the cascade from gene change to multi-organ disease and overlays the therapeutic levers. [1] [2]
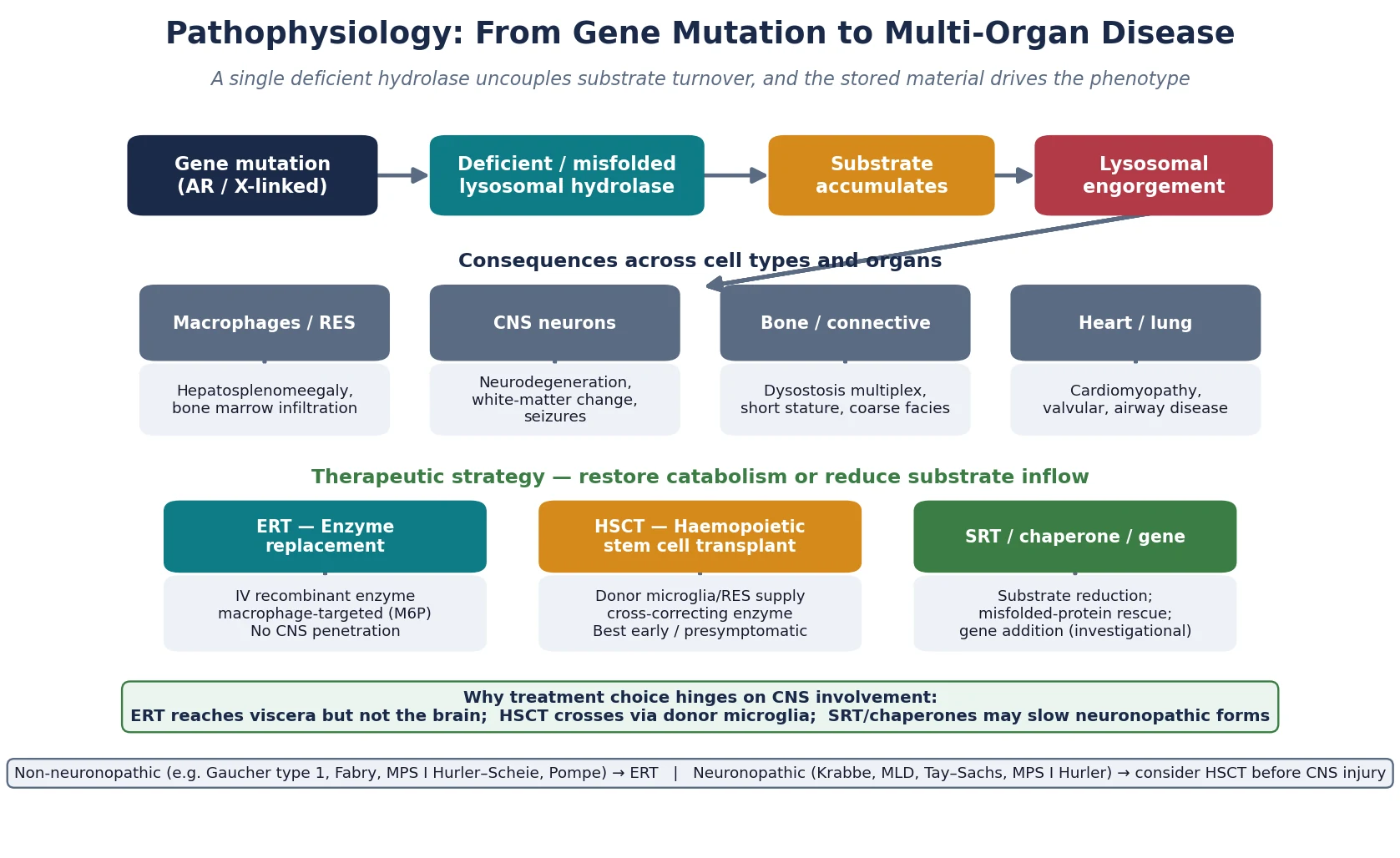
The organ phenotype follows where the substrate lands. In Gaucher disease, glucocerebroside accumulates in macrophages, producing the hepatosplenomegaly, cytopenias, bone infarction, and pulmonary disease that define type 1 disease, with neurological involvement reserved for types 2 and 3. In the gangliosidoses, ganglioside piles up in neurons, producing the relentless neurodegeneration, exaggerated startle, and cherry-red spot that mark Tay–Sachs. In Krabbe disease, psychosine — a toxic downstream metabolite of the blocked pathway — kills oligodendrocytes and drives the rapid demyelination that makes infantile Krabbe so devastating. Understanding that the storage material itself is often the injurious agent explains why these diseases progress even when the brain looks structurally intact early on. [4] [5]
Treatment is built on this same biology. Recombinant enzyme replacement therapy delivers a mannose-6-phosphate-tagged enzyme intravenously; the tag lets cells bind and internalise it, delivering functional enzyme to lysosomes in viscera — but the large molecule does not cross the blood–brain barrier. Haemopoietic stem cell transplant works differently: donor-derived microglia and macrophages engraft in the brain and viscera and secrete functional enzyme that neighbouring cells take up by cross-correction, which is why transplant can halt neuronopathic disease that enzyme replacement cannot reach. Substrate reduction therapy and pharmacological chaperones attack the problem from the other end, either slowing substrate synthesis or rescuing misfolded enzyme so it reaches the lysosome. [2] [10]
Clinical Presentation
The presentation depends on the enzyme, the substrate, and the age at onset, but a small number of patterns account for most encounters. The first is the infant or toddler with developmental regression — a child who acquired skills then loses them — accompanied by hepatosplenomegaly, seizures, hypotonia or spasticity, and sometimes a cherry-red macular spot. This neurodegenerative pattern points to the sphingolipidoses: Tay–Sachs and Sandhoff, Niemann–Pick, Gaucher types 2 and 3, and Krabbe. The pace is the clue: infantile Krabbe and type 2 Gaucher decline over weeks to months, whereas later-onset forms smoulder for years. [3] [4]
The second pattern is the hypertrophic-cardiomyopathy infant. A baby presents with poor feeding, sweating with feeds, respiratory distress, and a gallop, and echocardiography shows marked ventricular wall thickening with a short PR interval on electrocardiogram. Hypotonia and weak or absent reflexes distinguish Pompe disease (acid alpha-glucosidase deficiency) from the more common sarcomeric cardiomyopathies. The same infiltrative mechanism produces the concentric left-ventricular hypertrophy, arrhythmia, proteinuria, and neuropathic pain of Fabry disease, which may declare itself in childhood through acroparaesthesia, angiokeratomata, and anhidrosis in a boy, or in an affected adult through renal and cardiac failure. [7] [4]
The third pattern is the coarse-facies child with skeletal change. Coarsening of the facial features, corneal clouding, hepatosplenomegaly, herniae, joint stiffness, and the radiographic constellation of dysostosis multiplex point to a mucopolysaccharidosis. Hurler syndrome (MPS I) is the prototype: a child who seems normal at birth then develops coarse features, macroglossia, gibbus, claw hands, and progressive airway, cardiac-valve, and cognitive involvement. Hunter (MPS II, X-linked) resembles a milder Hurler without corneal clouding; Sanfilippo (MPS III) is distinguished by severe behavioural disturbance and sleep disruption with milder somatic change; Morquio (MPS IV) produces striking skeletal dysplasia with preservation of intellect. [8] [9]
Differential Diagnosis
When a lysosomal storage disorder is suspected, the differential depends on which presentation brought the child to attention. A regressing infant with organomegaly must be separated from other causes of developmental regression and neurodegeneration, including mitochondrial disorders, the neuronal ceroid lipofuscinoses, Rett syndrome (in girls, after a period of normal development), and acquired causes such as subacute sclerosing panencephalitis or lead encephalopathy. Mitochondrial disease often brings lactic acidosis, multi-organ failure, and a fluctuating course; the lipofuscinoses show visual failure and a characteristic electroencephalogram. [2] [3]
The cardiomyopathy infant raises a different list. Sarcomeric hypertrophic cardiomyopathy, Noonan syndrome and related RASopathies, glycogen storage diseases other than Pompe, and the infiltrative cardiomyopathy of Fabry or Danon all produce ventricular wall thickening in infancy. The discriminating features are the neuromuscular findings — severe hypotonia and weak reflexes point to Pompe — and the family history, since the X-linked disorders cluster in maternal males. Early measurement of creatine kinase (markedly raised in both Pompe and Danon) and a dried-blood-spot enzyme activity will redirect the workup within days. [7]
For the coarse-facies child, the differential includes the mucopolysaccharidoses, the oligosaccharidoses (mannosidosis, fucosidosis, sialidosis), mucolipidosis, and I-cell disease, all of which share dysostosis multiplex and varying degrees of intellectual impairment. Non-storage causes of coarse features — congenital hypothyroidism presenting late, and the rare congenital disorders of glycosylation — are excluded by thyroid function and transferrin isoelectric focusing. Urine glycosaminoglycan and oligosaccharide analysis is the single most useful first-line test here. [8] [9]
Clinical & Bedside Assessment
The bedside assessment is built around three questions. First, is the brain involved, and how fast is it declining — because this sets the urgency. Second, which viscera are affected, and how badly — because this sets the surveillance and the therapy targets. Third, is there a family history or a known affected sibling — because this sets the genetic counselling and the prenatal options for future pregnancies. A focused history and examination answer all three in a single consultation and direct the laboratory workup. [3] [6]
The neurological examination maps the pace and pattern of decline. An exaggerated startle response, loss of visual fixation, spasticity or rigidity, and loss of acquired motor and language skills mark the rapid neuronopathic sphingolipidoses. Irritability, feeding difficulty, rigid extended limbs, and unexplained low-grade fevers in an infant raise infantile Krabbe, where the window for transplant closes quickly. In Pompe, the picture is a paradoxical mix of marked hypotonia and weakness with brisk or preserved reflexes early on, and the heart is the dominant organ. Documenting a developmental trajectory — what the child could do three and six months ago — is more informative than any single examination finding. [5] [7]
The systemic examination records hepatosplenomegaly (measure the liver and spleen edges in centimetres below the costal margin), growth parameters, facial coarsening, corneal clouding, joint range of movement, herniae, and any cardiac murmur or gallop. Abdominal distension with a firm enlarged liver and spleen is the hallmark of macrophage storage and should be measured rather than described as "enlarged". A full cardiac and respiratory examination is essential: cardiomyopathy, valvular disease, and obstructive airway disease are treatable complications that determine anaesthetic risk and quality of life. [8] [9]
Investigations
The investigation strategy is layered and escalates from cheap screening tests to definitive enzyme and molecular confirmation. The first tier is urine metabolic screening — glycosaminoglycans and oligosaccharides — which is non-invasive, inexpensive, and points to the mucopolysaccharidoses and oligosaccharidoses. The second tier is enzyme activity measurement, now feasible on a dried blood spot or a small leucocyte sample for most of the common disorders, and this usually identifies the deficient enzyme. The third and confirmatory tier is molecular genetic testing — sequencing the relevant gene or a targeted lysosomal-disease gene panel — which confirms the diagnosis, defines the variant, and enables family testing, prenatal diagnosis, and genotype-based prognostication. [1] [11]
Newborn bloodspot screening has reshaped this pathway for the screened disorders — Krabbe, Pompe, and MPS I in many regions. A low enzyme activity on the bloodspot is a screening result, not a diagnosis, and must be confirmed urgently with leucocyte or fibroblast enzyme assay and molecular testing, because pseudodeficiency alleles, late-onset variants, and carrier states all produce abnormal screens in infants who may never develop disease. The confirmatory step is what distinguishes a child who needs a transplant from one who needs surveillance alone, and it must be completed within days for the rapidly progressive forms. [6] [11]
Supportive investigations quantify organ involvement and set the surveillance baseline. Echocardiography defines cardiomyopathy and valvular disease and is mandatory at diagnosis for Pompe, Fabry, the mucopolysaccharidoses, and Danon. Audiology, ophthalmology (including a dilated fundus examination for the cherry-red spot and corneal assessment), skeletal survey for dysostosis multiplex, and developmental assessment round out the baseline. Biochemistry tracks organ function — renal function and proteinuria in Fabry, liver function and haematology in Gaucher, creatine kinase in Pompe. The goal is to stage the disease precisely so that therapy and surveillance are matched to the individual child. [7] [9]
Management — Resuscitation
Most children with a lysosomal storage disorder do not present in collapse, but several acute scenarios demand prompt recognition. The infantile-Pompe heart fails quickly: a baby with a massively thickened myocardium may present in low-output cardiac failure or with a ventricular arrhythmia, and management is standard heart-failure therapy with careful fluid handling, beta-blockade avoided where obstruction predominates, and an urgent cardiology and metabolic referral, because enzyme replacement can reverse the cardiomyopathy if started early. Airway obstruction from glycosaminoglycan deposition in the mucopolysaccharidoses is an anaesthetic hazard and a cause of sleep-disordered breathing, so any MPS child needing sedation or intubation is managed by a team experienced in difficult airways. [7] [8]
The rapidly progressive neuronopathic disorders carry their own emergencies. Infantile Krabbe presents with irritability, feeding difficulty, rigidity, and unexplained fevers that can be mistaken for sepsis or colic; the recognition that this is a treatable neurodegenerative disorder, not a feeding problem, is the resuscitation-level decision, because every day of delay narrows the transplant window. Seizures, painful bone crises in Gaucher, and acute abdominal events from organomegaly all need symptom-directed management while the disease-modifying pathway is set in motion. In every case the resuscitation phase stabilises the child and secures the diagnosis in parallel. [5] [6]
Management — Definitive & Stepwise
The definitive management matches the therapy to the central-nervous-system burden. The figure lays out the decision pathway from suspicion to confirmed genotype to the treatment modality, and the guiding principle is that enzyme replacement therapy reaches the viscera but not the brain while haemopoietic stem cell transplant reaches both — but only if given early. [2] [6]
For non-neuronopathic disease, enzyme replacement therapy is the cornerstone. Intravenous recombinant enzyme every one to two weeks reduces organomegaly, improves haematology and biochemistry, and stabilises bone and lung disease in Gaucher type 1, Fabry, Pompe, and selected mucopolysaccharidoses (MPS I, II, IVA, VI). The enzyme does not cross the blood–brain barrier, so it does not halt neuronopathic decline, and infusion reactions, antibody formation, and the lifelong infusion burden are real limitations. Substrate reduction therapy offers an oral alternative for Gaucher (eliglustat, miglustat) and Fabry (migalastat where the genotype is amenable, and newer agents), and pharmacological chaperones can rescue misfolded enzyme in genotypes that respond. The choice is genotype-guided and made with the metabolic service. [2] [10]
For neuronopathic disease where a disease-modifying option exists, haemopoietic stem cell transplant is the established therapy. Transplant works because engrafted donor microglia and macrophages deliver functional enzyme to the brain and viscera by cross-correction. It is most effective in infantile Krabbe, the severe Hurler phenotype of MPS I, and a small number of other disorders, and its success is exquisitely time-sensitive: transplant given before neurological symptoms in early-infantile Krabbe and in Hurler preserves cognition and function, whereas transplant after injury adds toxicity without benefit. The decision to transplant carries significant morbidity and mortality and is made at a specialist centre with metabolic and transplant expertise, ideally before the child becomes symptomatic. [5] [6]
Enzyme replacement therapy — representative regimens (specialist-initiated, weight-based, confirm current dosing)
Symptom-directed multidisciplinary care runs in parallel with disease-modifying therapy and is often what determines quality of life. Cardiology manages cardiomyopathy, arrhythmia, and valvular disease; respiratory and ENT manage airway obstruction, sleep-disordered breathing, and recurrent infections, including consideration of tonsillectomy, continuous positive airway pressure, and tracheostomy in severe MPS; orthopaedics manages hip dysplasia, gibbus, and carpal tunnel; developmental and educational services support learning and communication; and palliative care is involved early in the most severe neuronopathic forms. Gene therapy and other investigational approaches are expanding and are accessed through clinical trials at specialist centres. [8] [9]
Specific Subtypes & Scenarios
Gaucher disease is the most common lysosomal storage disorder and the prototype for treatable non-neuronopathic disease. Type 1, which spares the brain, presents with hepatosplenomegaly, cytopenias, bone pain and infarction, and fatigue, and responds well to enzyme replacement or substrate reduction with eliglustat. The presence of the cherry-red spot, ophthalmoplegia, or neurodegeneration reclassifies the child as type 2 (acute neuronopathic, fatal in infancy) or type 3 (subacute, with progressive neurological disease), where treatment options are limited and the prognosis is guarded. Gaucher is the disorder most likely to appear as a long-case vignette, and the type 1 versus type 2 or 3 distinction is the central teaching point. [1] [2]
Pompe disease (glycogen storage disease type II) sits at the intersection of metabolic, cardiac, and neuromuscular medicine. Classic infantile-onset Pompe presents in the first months with severe hypotonia, cardiomegaly with a short PR interval, and respiratory insufficiency, and is rapidly fatal without enzyme replacement — which can rescue the cardiomyopathy and transform survival when started early. Late-onset Pompe presents at any age from childhood to adulthood with proximal myopathy and respiratory involvement but without cardiac disease. The creatine kinase is markedly raised, and the diagnosis is confirmed by acid alpha-glucosidase activity and GAA sequencing. [7]
Fabry disease is the great mimic of nephrology and cardiology clinics. An X-linked alpha-galactosidase A deficiency, it presents in childhood with acroparaesthesia (episodic burning pain in hands and feet), angiokeratomata, anhidrosis, and gastrointestinal symptoms, then progresses in adulthood to renal failure, hypertrophic cardiomyopathy, and stroke. Female carriers are not asymptomatic — many develop significant cardiac and renal disease — so treatment decisions are based on organ involvement, not on sex alone. Enzyme replacement or migalastat (for amenable variants) aims to prevent the irreversible organ damage that accumulates silently for decades. [4]
Krabbe disease is the emblematic treatable-if-early neuronopathic disorder. Infantile-onset Krabbe, caused by galactocerebrosidase deficiency, presents in the first months with irritability, feeding difficulty, rigidity, unexplained fevers, seizures, and rapid neurological regression, driven by psychosine-mediated oligodendrocyte death and demyelination. Untreated it is fatal within two years. Haemopoietic stem cell transplant given before the onset of symptoms — identified through newborn screening — can preserve cognitive function and modify the course, which is why Krabbe has been added to bloodspot panels and why a positive screen demands urgent confirmatory testing and specialist referral within days. [5] [6]
Complications & Pitfalls
The complications of lysosomal storage disorders are the complications of untreated or progressive storage, and they cluster by organ. The bones of a child with Gaucher or an MPS infarct and fracture, producing the painful bone crises and skeletal deformity that dominate quality of life. The airway narrows from glycosaminoglycan deposition, and the combination of macroglossia, short neck, and cervical spine instability makes anaesthesia genuinely dangerous in the mucopolysaccharidoses. The heart hypertrophies, fibroses, and fails in Pompe, Fabry, and Danon, and the valves thicken and regurgitate in the MPS disorders. The brain, once injured by storage, does not recover — which is why timing is the single most important determinant of outcome in the neuronopathic forms. [8] [9]
The most consequential pitfall is delay. Treating a neuronopathic disorder as a feeding problem, a developmental delay, or a behavioural issue for months forfeits the transplant window and converts a potentially modifiable disease into a fatal one. A second pitfall is misreading a screening result: a low enzyme activity on a newborn bloodspot reflects enzyme level, not clinical disease, and pseudodeficiency alleles, late-onset variants, and carriers all produce abnormal screens in children who may never become unwell. Confirmatory enzyme assay in leucocytes and molecular testing are essential before any irreversible decision such as transplant, and before reassuring or alarming a family. [6] [11]
A third pitfall is underestimating the anaesthetic and perioperative risk. Children with mucopolysaccharidoses have difficult airways, cervical spinal cord compression, and restrictive lung disease, and even routine procedures such as dental work or tonsillectomy carry a significant risk of airway loss or spinal cord injury. Any procedure should be planned with an anaesthetic team experienced in metabolic airways, with cervical spine imaging and a difficult-airway strategy in place. Finally, assuming that enzyme replacement therapy will halt neurological decline — when it cannot cross the blood–brain barrier — sets up both clinician and family for avoidable disappointment. [8]
Prognosis & Disposition
Prognosis is determined by the specific disorder, the age at onset, the genotype, and crucially the timing of treatment. Non-neuronopathic Gaucher type 1, treated with enzyme replacement or substrate reduction, now approaches a near-normal life expectancy with good quality of life, and is the model for what effective therapy can achieve. Classic infantile Pompe, uniformly fatal within the first year without treatment, is transformed by early enzyme replacement into a survivable chronic disease, albeit with residual myopathy and ongoing infusion burden. Fabry disease, treated early, delays or prevents the renal, cardiac, and cerebrovascular events that historically defined its adult course. [2] [7]
The neuronopathic disorders remain the most challenging. Infantile Krabbe transplanted before symptoms preserves cognition and substantially modifies survival, but transplant after symptom onset offers little neurological benefit and carries real transplant-related mortality. Hurler syndrome transplanted early preserves intellect but does not fully rescue the skeletal, airway, and valvular complications, so affected children need lifelong multidisciplinary care. Tay–Sachs, Sandhoff, and the neuronopathic Gaucher variants currently lack effective disease-modifying therapy, and management is supportive and palliative, with gene therapy and other investigational approaches the main hope for the future. Setting honest expectations with the family — grounded in the specific genotype and treatment response — is central to the disposition. [5] [6]
Disposition is to a specialist metabolic service with a structured transition to adult care, because these are lifelong diseases that cross every organ system and every life stage. The medical home coordinates surveillance — growth, development, audiology, ophthalmology, cardiology, respiratory, and bone health — and integrates the family's psychosocial, educational, and genetic-counselling needs. For the most severe neuronopathic forms, early involvement of paediatric palliative care alongside disease-directed therapy supports the child and the family through a progressive illness, and does not preclude continuing disease-modifying treatment where it is available. [1] [9]
Special Populations
Newborn screening has created a distinct population — the asymptomatic neonate with an abnormal screen — whose management differs from the symptomatic child. The metabolic service must distinguish true infantile disease, which demands urgent confirmatory testing and a rapid treatment decision, from late-onset variants and pseudodeficiency alleles that may never cause disease. Families need honest, calibrated counselling: not every abnormal screen means a sick child, but every abnormal screen demands a thorough, time-sensitive workup, because the cost of missing a treatable infantile disorder is irreversible neurological injury. [6] [11]
The conditions screened for lysosomal storage disease vary between jurisdictions and change over time, so the examining clinician should state the local panel rather than assume a universal list. In Australia and New Zealand, Krabbe, Pompe, mucopolysaccharidosis type I, and a small number of other disorders are being progressively added to bloodspot panels as evidence and treatment evolve. The principles are constant: a low enzyme activity is a screen, not a diagnosis; confirm with leucocyte enzyme assay and molecular testing within days; and refer urgently to the regional metabolic service for any screen positive for a rapidly progressive neuronopathic disorder. [6] [11]
Consanguineous communities and founder populations carry a higher burden of autosomal recessive lysosomal disease, and a previously affected sibling reshapes the reproductive risk for every subsequent pregnancy. In these families carrier testing, prenatal diagnosis by chorionic villus sampling or amniocentesis, and preimplantation genetic testing are central to management, and the metabolic and genetics services should be involved before the next pregnancy rather than after it. Indigenous, migrant, refugee, and remote populations face additional barriers — distance from specialist centres, language and cultural considerations, and inequitable access to expensive enzyme therapies — and the medical home must actively coordinate equitable, culturally safe care. [3]
Evidence, Guidelines & Regional Differences
The evidence base for treatment has grown substantially over the last two decades but remains uneven across the family of disorders. Enzyme replacement therapy for Gaucher type 1 has the most mature evidence, with decades of registry data showing reduced organomegaly, normalised haematology, and improved survival, and it is the benchmark against which newer therapies are judged. Pompe and Fabry enzyme therapies are supported by randomised trials showing biochemical and functional benefit, though residual muscle and renal disease persist. Haemopoietic stem cell transplant for infantile Krabbe and Hurler rests on cohort and registry evidence showing that pre-symptomatic transplant preserves cognition — the rationale that underpins newborn screening for these disorders. [2] [5]
Guidelines are disorder-specific and specialist-led. The Pompe diagnosis and management guideline sets the framework for enzyme replacement and multidisciplinary care; the Krabbe newborn-screening consensus defines the urgent confirmatory and treatment pathway; and management guidelines for the individual mucopolysaccharidoses address surveillance and therapy by subtype. [6] [7] Regional differences persist in what is screened, what therapies are funded, and how transplant is accessed, so the clinician should ground recommendations in local policy while applying the universal principles: screen early, confirm precisely, treat before the brain is injured, and support the family across the lifespan. [9] [11]
The frontier is gene therapy. Adeno-associated-virus-mediated gene addition is approved or in advanced trials for several disorders, offering the prospect of durable enzyme expression from a single treatment, and substrate reduction and chaperone therapies are expanding to new genotypes and disorders. These advances make early, accurate diagnosis more valuable than ever, because a disorder that was untreatable a decade ago may be modifiable today — provided it is found before irreversible injury. The fellowship answer therefore closes with an active posture: keep the lysosomal storage disorders on the differential of any unexplained regression, organomegaly, or cardiomyopathy, confirm fast, and refer early. [2] [10]
Exam Pearls
In a viva, lead with the clinical pattern, name the stored substrate and deficient enzyme, and then state the confirmatory test and the treatment modality — that four-step structure answers almost any lysosomal storage question. For a short case, the cherry-red spot, hepatosplenomegaly, cardiomyopathy, and dysostosis multiplex are the findings that earn marks; for a long case, the management plan that matches the therapy to the central-nervous-system burden and sets out surveillance and genetic counselling is what distinguishes a pass from a commendation. Remember the X-linked disorders — Fabry, Hunter, and Danon — because the inheritance changes the family counselling and the screening of at-risk males. [1] [4]
The single most testable principle is that enzyme replacement therapy does not cross the blood–brain barrier, so it cannot halt neuronopathic decline, whereas haemopoietic stem cell transplant can — but only before the brain is injured. Couple that with the recognition that newborn screening for Krabbe, Pompe, and MPS I exists precisely to find these children pre-symptomatically, and you have the framework that ties the whole topic together. Every lysosomal storage disorder question ultimately reduces to: which enzyme, which substrate, is the brain involved, and what is the window for the therapy that reaches it. [2] [6]
References
- [1]Platt FM, d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primers, 2018.PMID 30275469
- [2]Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med, 2015.PMID 25587658
- [3]Staretz-Chacham O, Lang TC, LaMarca ME, Krasnewich D, Sidransky E. Lysosomal storage disorders in the newborn. Pediatrics, 2009.PMID 19336380
- [4]Boustany RM. Lysosomal storage diseases--the horizon expands. Nat Rev Neurol, 2013.PMID 23938739
- [5]Wenger DA, Rafi MA, Luzi P. Krabbe disease: One hundred years from the bedside to the bench to the clinic. J Neurosci Res, 2016.PMID 27638583
- [6]Kwon JM, Matern D, Kurtzberg J, Wrabetz L, Gelb M, Foss AH, et al. Consensus guidelines for newborn screening, diagnosis and treatment of infantile Krabbe disease. Orphanet J Rare Dis, 2018.PMID 29391017
- [7]Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, et al. Pompe disease diagnosis and management guideline. Genet Med, 2006.PMID 16702877
- [8]Muenzer J. Overview of the mucopolysaccharidoses. Rheumatology (Oxford), 2011.PMID 22210669
- [9]Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics, 2007.PMID 17671068
- [10]Parenti G, Moracci M, Fecarotta S, Monti M, Gregorio SD, Meroni G, et al. Pharmacological chaperone therapy for lysosomal storage diseases. Future Med Chem, 2014.PMID 25068986
- [11]Matern D, Gavrilov D, Oglesbee D, Raymond K, Rinaldo P, Tortorelli S. Newborn screening for lysosomal storage disorders. Semin Perinatol, 2015.PMID 25891428