Paeds · genetics-dysmorphology-and-metabolism
Marfan syndrome and heritable connective-tissue disorders
Also known as Marfan syndrome · MFS · Marfanoid habitus · Loeys-Dietz syndrome · Ehlers-Danlos syndrome · Vascular Ehlers-Danlos syndrome · Congenital contractural arachnodactyly · Beals syndrome · Heritable thoracic aortic disease
A fellowship approach to Marfan syndrome and the heritable connective-tissue disorders: recognise the marfanoid child, confirm Marfan with the revised Ghent nosology and FBN1 sequencing rather than a karyotype, image the aortic root from diagnosis and protect it for life with beta-blockade or losartan and elective valve-sparing root repair, and separate Marfan from Loeys-Dietz syndrome, vascular and hypermobile Ehlers-Danlos, Beals syndrome and homocystinuria by gene and by the direction the lens has fallen.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The candidate who wins the marks holds three layers in view at once. The first is the child in front of you: their stature, their limbs, their lens, their heart, and the aortic root that quietly dilates until it tears. The second is the molecule: a fibrillin-1 defect that breaks the elastic-fibre scaffold and, just as importantly, releases the brake on transforming growth factor-beta (TGF-beta), the signalling discovery that reframed the whole disease. The third is the family: because Marfan and its relatives are autosomal dominant, every first-degree relative carries a one-in-two chance and deserves assessment and counselling. [1] [3]
Overview & Definition
Marfan syndrome is an autosomal-dominant disorder of fibrillin-1, a large glycoprotein that is the building block of the microfibrils scaffolding elastic fibres throughout the body. When fibrillin-1 is defective the load-bearing tissues — the aortic wall, the suspensory ligament of the lens, the dura, the lungs and the growing skeleton — lose their structural integrity in a predictable pattern. Marfan is the commonest of the heritable connective-tissue disorders and the one a general paediatrician is most likely to meet, affecting roughly one in 3,000 to 5,000 people of all populations. [1] [2]
Around the core diagnosis sits a family of related conditions that share a marfanoid habitus or a connective-tissue mechanism but differ sharply in their gene, their lens, and the behaviour of their arteries. Loeys-Dietz syndrome is driven by signalling defects in the TGF-beta pathway (TGFBR1 or TGFBR2, and the related SMAD3, TGFB2 and TGFB3 genes) and produces aggressive, early arterial aneurysms and dissections alongside a cleft palate, bifid uvula and hypertelorism. Vascular Ehlers-Danlos syndrome is a type-III collagen defect (COL3A1) that causes fragile arteries, spontaneous bowel rupture and the gravest prognosis in the family. Congenital contractural arachnodactyly (Beals syndrome) is an FBN2 disorder that mimics Marfan but adds congenital joint contractures and crumpled ears. [6] [8] [10]
The trap the fellowship candidate must avoid is the tall child labelled "Marfan" on habitus alone. The lens that has fallen downward and inward belongs to homocystinuria, a treatable autosomal-recessive enzyme defect whose thrombotic and cognitive course is entirely different. Separating these conditions is not a naming exercise — it sets the surveillance schedule, the surgical thresholds, the reproductive counselling and, in homocystinuria, the diet and vitamin therapy that change the outcome. [9] [2]
Classification
The classification that governs practice is the revised Ghent nosology of 2010, which moved the centre of gravity of diagnosis from the old systemic score to the two cardinal features: the aortic root aneurysm and the lens. In a child without a family history, aortic-root dilation plus ectopia lentis is enough to call Marfan syndrome outright, because that pairing is almost pathognomonic. Where the lens is normal, root dilation combined with a supportive systemic score of seven or more points, or with a pathogenic FBN1 variant, closes the diagnosis. The older systemic score becomes a supporting tool rather than the gateway. [2]
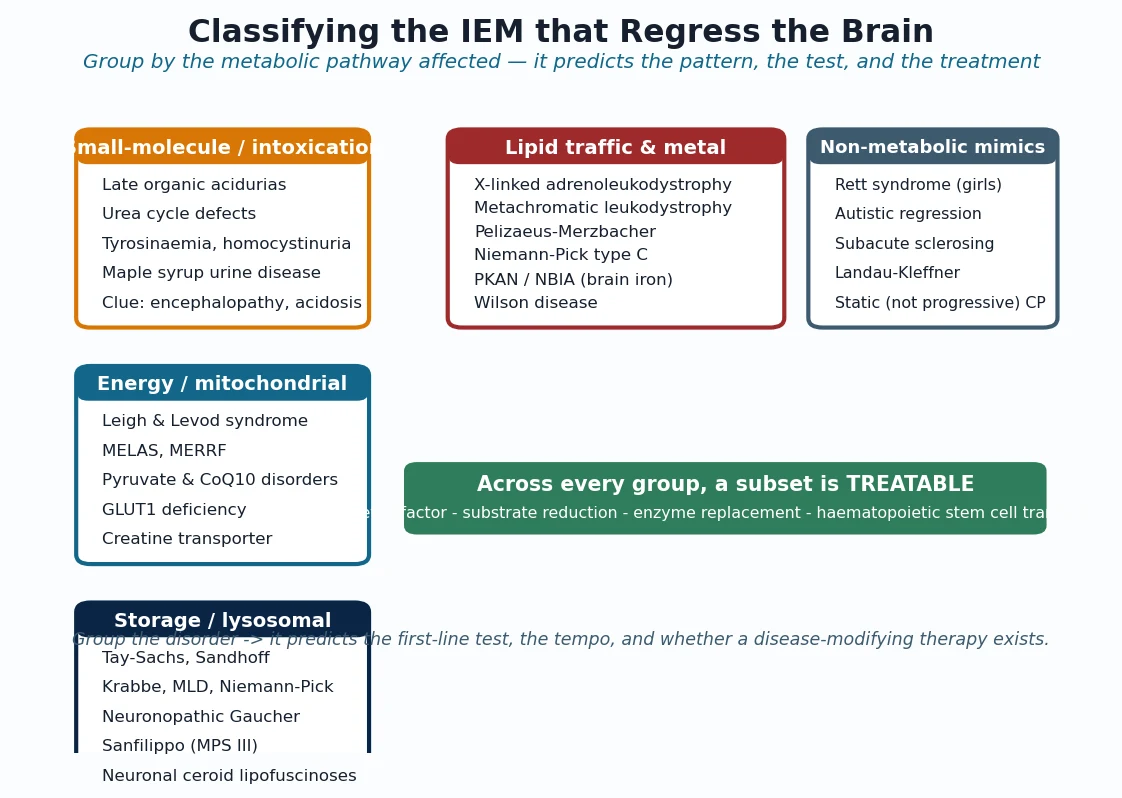
The systemic score rewards the features a clinician can elicit at the bedside, and its highest-yield items carry the most weight. The combination of a positive wrist and thumb sign is worth three points, pectus carinatum and hindfoot deformity each score two, and spontaneous pneumothorax, dural ectasia and protuberant acetabulae each add two more. A score of seven or more supports the diagnosis once the root is dilated, and it is especially useful in the young child whose phenotype is still emerging. [2]
Revised Ghent cardinal pairs \u2014 the lens and the root
The family of related disorders is classified by gene rather than by gestalt, because the management flows from the gene. Loeys-Dietz sits beside Marfan in the TGF-beta signalling pathway, vascular Ehlers-Danlos sits in the collagen family, and Beals syndrome sits beside Marfan in the fibrillin family (FBN2). Recognising the right member of the family is what decides whether the arteries will tear at a small diameter, whether surgery is safe, and whether the children should be tested. [6] [8]
Epidemiology & Risk Factors
Marfan syndrome affects roughly one in 3,000 to 5,000 people and shows no major ethnic predilection. It is autosomal dominant with complete penetrance but striking variable expressivity, so two relatives carrying the same variant can look entirely different. About a quarter of cases arise from a de novo variant, which means a negative family history never excludes the diagnosis and the parents of a sporadic case should be examined and offered testing to detect mosaicism. [1]
The single most important risk factor is a confirmed affected first-degree relative, which is why a three-generation pedigree is part of the first assessment. The preventable complications — dissection, retinal detachment, severe scoliosis, pneumothorax — cluster in those whose root has not been measured and whose beta-blockade is not started. Loeys-Dietz is far rarer than Marfan, perhaps one in 100,000, but it dissects at smaller diameters and younger ages, and vascular Ehlers-Danlos carries a median survival in the fifth decade without diagnosis and avoidance of vascular injury. [6] [7] [8]
Population and access factors shape detection as much as biology. In remote and Indigenous communities, and in migrant, refugee and asylum-seeking families, the combination of an absent family history, reduced access to echocardiography and lower rates of cascade testing means that the root is measured late and the aneurysm presents at a larger size, which is the gap that surveillance is designed to close. [11]
Pathophysiology
The fibrillin-1 defect injures the body through two arms, and both matter. The structural arm is the one taught first: fibrillin-1 assembles into the beaded microfibrils that scaffold elastin, and without intact microfibrils the elastic fibres of the aortic media fragment and weaken. The media then dilates under systolic pressure at its weakest point, the sinuses of Valsalva, producing the root aneurysm that defines the disease. The same weakness explains dural ectasia, spontaneous pneumothorax and the over-compliant joints. [1] [3]
The signalling arm is the discovery that reframed the disease. Fibrillin-1 normally binds and stores latent TGF-beta in the extracellular matrix, holding it in reserve; when fibrillin is defective, free TGF-beta accumulates and over-signals its receptors. Neptune and colleagues showed in 2003 that fibrillin-deficient mice developed a Marfan-like lung and vascular disease, and that blocking TGF-beta rescued them — evidence that the phenotype is not only a weak wall but an over-driven growth and remodelling signal. This second arm is why bone over-grows, why mitral valve tissue is redundant, and why a drug that antagonises the TGF-beta axis, losartan, was a rational therapy. [3]
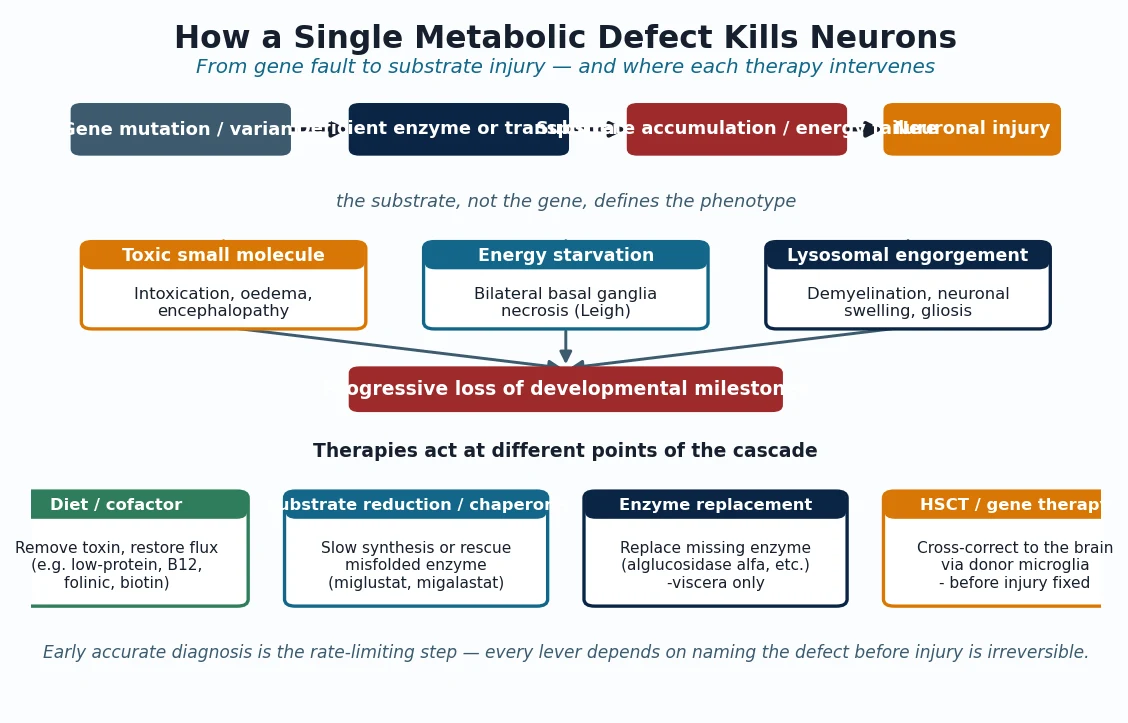
Loeys-Dietz syndrome is the signalling arm writ large. Its causative genes encode the TGF-beta receptors themselves (TGFBR1 and TGFBR2) or downstream and ligand partners (SMAD3, TGFB2, TGFB3), so the same over-signalling that is a secondary consequence in Marfan is the primary defect here. That explains why Loeys-Dietz dissects earlier and at smaller diameters, and why its craniofacial features (bifid uvula, hypertelorism, craniosynostosis) are more florid: the developmental TGF-beta signal is grossly amplified. [6] [7]
Vascular Ehlers-Danlos sits in a different mechanism entirely. Type-III collagen is the structural collagen of blood vessels, bowel and uterus, and its defect produces tissues so friable that the arteries and viscera can rupture with minimal provocation. The principle unifies the family: each condition removes a different load-bearing molecule, and the management follows the molecule, because the tissue that fails predicts the catastrophe that must be prevented. [8]
Clinical Presentation
Marfan syndrome declares itself in an age-dependent sequence. The infant may simply be long, with a long fingers and toes and a systolic click of mitral valve prolapse. The child accumulates the musculoskeletal phenotype — arm span greater than height, a reduced upper-to-lower-segment ratio, pectus excavatum or carinatum, scoliosis, pes planus with hindfoot deformity, joint hypermobility, a high-arched palate and dental crowding. The lens, which is held by fibrillin-rich zonules, subluxes upward and outward, presenting as visual blur, strabismus or a finding at routine screening. [1] [2]
The complications that change the trajectory emerge as the child grows. The aortic root, normal in early childhood, most often begins to dilate in adolescence and young adulthood, and it is here that the lethal risk of acute type-A dissection rises. Spontaneous pneumothorax reflects the weak pulmonary connective tissue. Apical blebs and dural ectasia produce back pain and headache. Retinal detachment and glaucoma threaten vision once the lens has moved. In a previously undiagnosed young person, the first manifestation can be sudden tearing chest or back pain, and that is the disaster the lifetime of surveillance is built to prevent. [1] [11]

The other members of the family present in characteristic ways. Loeys-Dietz adds a bifid uvula or cleft palate, hypertelorism, craniosynostosis and arterial tortuosity on imaging, and dissects at a young age. Vascular Ehlers-Danlos presents with easy bruising, translucent skin visible veins, acrogeric hands, and arterial, intestinal or uterine rupture. Beals syndrome presents as a marfanoid infant with congenital contractures of the large joints and crumpled ears. Homocystinuria presents as a marfanoid child with intellectual disability, malar flush, osteoporosis and a lens that has fallen downward and inward. [6] [8] [9] [10]
Differential Diagnosis
The differential of the marfanoid child turns first on the lens, because the direction it has fallen is the classic discriminator. In Marfan the zonules rupture superiorly and the lens dislocates upward and superotemporal. In homocystinuria the inferior zonules fail first and the lens sinks downward and inferonasal. The marfanoid child with intellectual disability, a malar flush or any thrombosis must have a plasma total homocysteine measured before the diagnosis of Marfan is committed, because homocystinuria is autosomal recessive, is treated with pyridoxine, folate and vitamin B12 and a methionine-restricted diet, and carries a thromboembolic — not aneurysmal — risk. [9] [2]
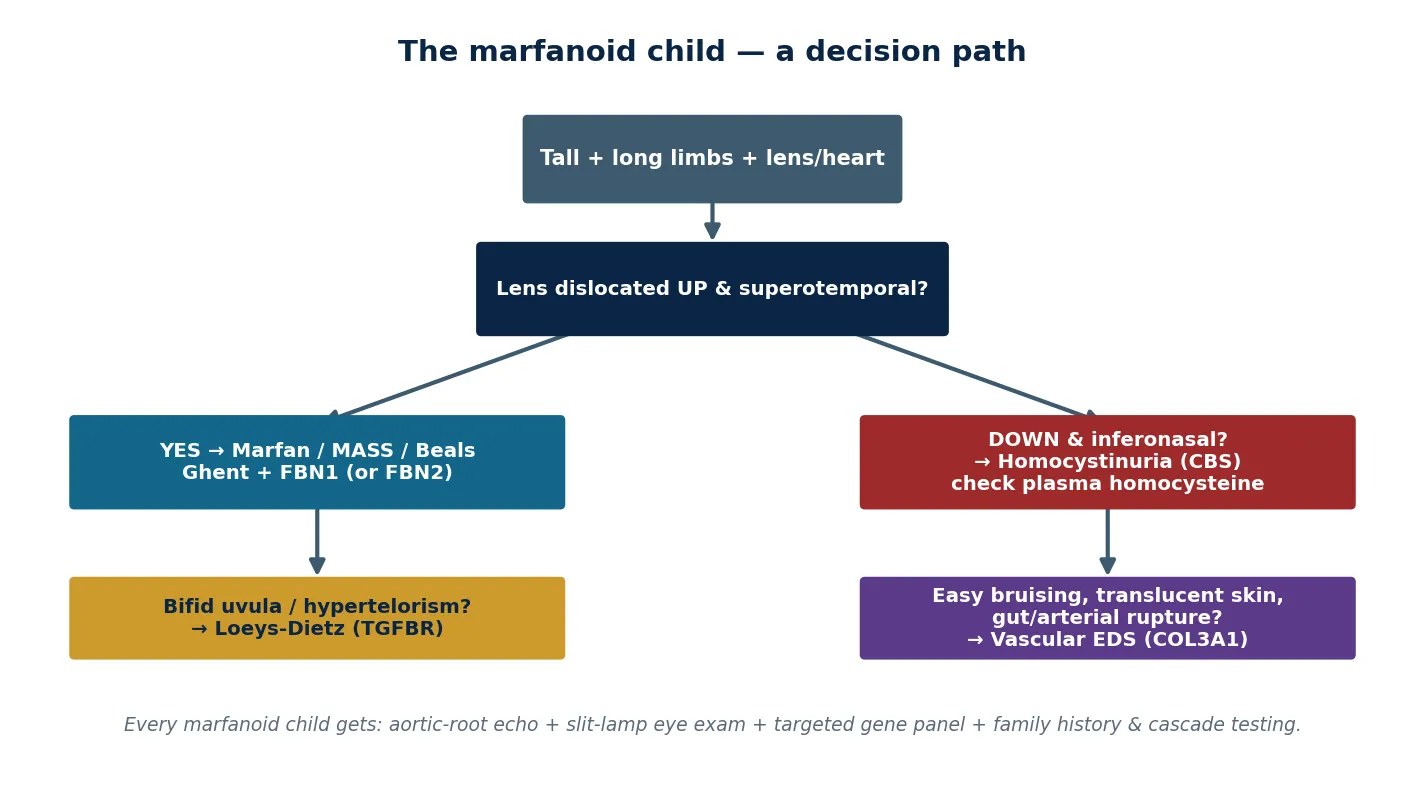
The vascular and craniofacial features then separate the dangerous relatives. Bifid uvula or cleft palate with hypertelorism and arterial tortuosity points to Loeys-Dietz, where dissection occurs at smaller diameters and the whole arterial tree must be imaged. Easy bruising, translucent skin and a history of bowel or arterial rupture point to vascular Ehlers-Danlos, where invasive procedures are themselves dangerous and arteriography is avoided. A marfanoid infant with congenital contractures and crumpled ears points to Beals syndrome (FBN2). [6] [7] [8] [10]
The remaining differentials are the tall-child and hypermobility presentations that are not Marfan at all. Familial (constitutional) tall stature and familial joint hypermobility are common and benign, and hypermobile Ehlers-Danlos syndrome is a clinical diagnosis of joint hypermobility with chronic pain and dysautonomia but no consistent aortic dilation. The trap in clinic is labelling a tall child as hypermobile EDS without looking at the aortic root; the trap in the exam is missing the one marfanoid child whose lens has fallen down. [8] [2]
Clinical & Bedside Assessment
The bedside assessment of a marfanoid child is a structured examination of the skeleton, the eyes, the heart and the family. Measure height, weight and arm span, and calculate the upper-to-lower segment ratio; plot them against standard and, where available, Marfan-specific growth charts. Examine the chest wall for pectus carinatum or excavatum, the spine for scoliosis, the feet for hindfoot deformity and plain pes planus, and the palate for a high arch. Perform the wrist and thumb signs — the highest-yield manoeuvres — and test the elbows for reduced extension. [2] [1]
The two examinations that cannot be deferred are the slit-lamp eye examination and the echocardiogram. The slit lamp finds the upward and superotemporal subluxation that closes the diagnosis and sets the ophthalmology plan; the echocardiogram measures the aortic root at the sinuses of Valsalva, expressed as a Z-score in children, and finds the mitral valve prolapse that is the most common cardiac lesion. Measure blood pressure at every visit, because hypertension accelerates the root and, in Loeys-Dietz, may reflect renal artery tortuosity. [1] [11]
The family history is non-negotiable. Ask explicitly about sudden death, dissection or aortic surgery, tall stature, lens surgery, and unexplained early arthritis or pneumothorax in relatives across three generations, and examine both parents where possible. A subtle Marfan phenotype in a parent confirms the diagnosis and unmasks an affected family whose other members need cascade testing. In remote and Indigenous communities and in migrant families, the family history may be incomplete, and the threshold to offer genetic testing should fall rather than rise. [1] [11]
Investigations
The diagnosis of Marfan is clinical, made by the revised Ghent nosology, and molecular testing with FBN1 sequencing confirms it. Sequencing and deletion/duplication testing of FBN1 finds a pathogenic variant in the large majority of clinically affected adults and is essential when the phenotype is incomplete, when the result will guide reproductive counselling, or when a related disorder such as Loeys-Dietz is in the differential. A broader heritable-connective-tissue-disorder gene panel is now the rational first test when the clinical picture is ambiguous, because it covers FBN1, FBN2, TGFBR1/2, SMAD3, TGFB2/3, COL3A1 and the other relevant genes in a single assay. [1] [2]
The cardiac investigation is the echocardiogram, and the measurement that matters is the aortic root at the sinuses of Valsalva, expressed as a Z-score that corrects for body size in children. Annual echocardiography from diagnosis is the standard, with more frequent imaging when the root is enlarging or approaches the operative threshold, and a baseline whole-aorta assessment as the child grows. In Loeys-Dietz the imaging extends to a whole-body magnetic resonance angiogram to map arterial tortuosity and aneurysms beyond the root, because dissection can occur anywhere. [11] [7]

The ophthalmological investigation is the slit-lamp examination with dilation, which defines the lens position, the refraction and the retina. Where a marfanoid child has a lens that has fallen downward, a plasma total homocysteine (with methylmalonic acid where needed) excludes homocystinuria. Magnetic resonance imaging of the lumbosacral spine is reserved for the child with symptoms of dural ectasia. Routine brain imaging is not part of Marfan surveillance. [9] [1]
Management — Resuscitation
Acute resuscitation in the heritable connective-tissue disorders is uncommon but decisive when it occurs. The first scenario is acute type-A aortic dissection, presenting as sudden tearing chest or back pain in a young person. The response is immediate referral to a cardiothoracic centre, intravenous blood-pressure control with a beta-blocker first to reduce the rate of pressure rise, analgesia, cross-sectional imaging and surgical repair. The second is spontaneous pneumothorax, managed with aspiration or small-bore drainage and an awareness that recurrence is common and pleurodesis is often needed. The third is retinal detachment in a child with ectopia lentis, which needs same-day ophthalmology referral. [11] [1]
In Loeys-Dietz and vascular Ehlers-Danlos the acute events are grimmer. Arterial dissection or rupture can occur at smaller diameters and in unusual locations, and the threshold for imaging any new pain is lower. Vascular Ehlers-Danlos carries the additional hazard that invasive procedures — arterial lines, arteriography, even surgery — can themselves cause fatal arterial injury, so diagnosis by COL3A1 testing and non-invasive imaging (CT angiography with careful technique) is preferred, and any procedure is planned with a specialist centre. [7] [8]
Management — Definitive & Stepwise
Definitive management of Marfan is a four-pillar plan for life: surveillance, lifestyle modification, medical therapy, and elective surgery, with cascade testing of the family threaded through. Surveillance is the annual echocardiogram measuring the root, the annual ophthalmology review, and periodic assessment of growth, spine, puberty and development through a specialist connective-tissue-disorder service. Lifestyle modifies the activities that raise blood pressure and shear stress on the aorta: competitive and contact sport, heavy resistance training and any Valsalva-heavy exertion are avoided, while moderate aerobic activity is encouraged. [1] [11]
Medical therapy begins with a beta-blocker, which reduces the rate of aortic growth and the risk of dissection by lowering heart rate and the rate of pressure rise. The angiotensin-II receptor blocker losartan, rational because angiotensin-II signalling drives TGF-beta, is used alone or added to a beta-blocker. The Pediatric Heart Network trial (Lacro et al., NEJM 2014) compared atenolol with losartan in children and young adults with Marfan and found the two drugs similar in slowing aortic-root enlargement — losartan was not superior, but it was an acceptable alternative or addition, and its long-term follow-up confirmed a comparable effect. [4] [5]
Elective surgery is what prevents the lethal event. The valve-sparing aortic-root replacement (the David procedure or related operations) replaces the dilated root while preserving the native aortic valve, sparing the child lifelong anticoagulation. The adult operative threshold is roughly 4.5 to 5.0 cm of root diameter, and in children the decision is individualised on indexed diameter, Z-score and rate of growth. Timing the operation before dissection is the whole point of the surveillance programme. Mitral valve repair is offered for severe prolapse with regurgitation. [11] [1]
The plan closes with the family. A first-degree relative of a confirmed case has a one-in-two chance of carrying the variant and deserves targeted testing once the pathogenic FBN1 variant in the family is known, with cardiology and ophthalmology review of any affected relative. For the young woman with Marfan who wants to conceive, pre-pregnancy counselling is mandatory: an aortic root greater than 4.0 cm carries a high risk of pregnancy-related dissection, and elective root repair before conception, beta-blockade through pregnancy, and a planned mode of delivery with a specialist team are the standard of care. [1] [11]
Specific Subtypes & Scenarios
Loeys-Dietz syndrome demands more aggressive surveillance than Marfan because dissection occurs at smaller diameters and younger ages and anywhere in the arterial tree. Management adds a baseline whole-body magnetic resonance angiogram to map tortuosity and aneurysms, an intervention threshold that is lower than in Marfan, and a craniofacial and cervical-spine assessment for craniosynostosis and instability. The bifid uvula or cleft palate is repaired as needed, and allergic and inflammatory disease is common and treated actively. [6] [7]
Vascular Ehlers-Dietz syndrome — vascular Ehlers-Danlos — is managed by diagnosis, avoidance and contingency. Diagnosis is by COL3A1 sequencing, and the family is cascade-tested. Arteriography is avoided because the punctured artery can rupture; cross-sectional imaging with careful technique is preferred. Surgery is reserved for life-threatening rupture and is high-risk because the tissues do not hold sutures. Bowel surveillance is cautious, pregnancy is high-risk for uterine rupture, and a MedicAlert and a specialist centre are part of the plan. [8]
Hypermobile Ehlers-Danlos syndrome is the commonest member of the EDS family and is managed clinically, because no gene has yet been identified. The plan is joint protection and graded physiotherapy, management of chronic pain, recognition and treatment of postural tachycardia and gastrointestinal dysmotility, and psychological support. A baseline echocardiogram excludes aortic dilation, but routine aortic surveillance is not required in the absence of dilation or a positive family history. [8]
Homocystinuria is the mimic whose management changes everything. Once confirmed by elevated plasma total homocysteine, it is treated with pyridoxine (vitamin B6), folate and vitamin B12 supplementation, a methionine-restricted diet with cystine supplementation, and thromboprophylaxis around surgery and dehydration. The lens that has fallen downward may need surgical removal, and the thrombotic risk, not the aneurysm, is what shortens life. Recognising homocystinuria converts a terminal cardiac story into a treatable metabolic one. [9]
Complications & Pitfalls
The dominant complication is acute aortic dissection and rupture, and it is preventable in the child who is diagnosed, surveilled, treated and repaired at the threshold. The complication is more dangerous in Loeys-Dietz, where dissection occurs at smaller diameters and younger ages, and grimmest in vascular Ehlers-Danlos, where arterial rupture can occur without warning. The ocular complications — retinal detachment, glaucoma and amblyopia from an unrecognised ectopia lentis — are under-diagnosed and rob vision in children whose root is well managed. [1] [6] [8]
The procedural pitfalls are specific to the disorder. In vascular Ehlers-Danlos, arteriography and arterial lines can cause fatal arterial injury, and surgery is reserved for life-threatening events. In Marfan, pregnancy-related dissection is the classic pitfall in a young woman whose root was thought "borderline." Dural ectasia causes back and leg pain that is mistaken for musculoskeletal causes, and spontaneous pneumothorax recurs if pleurodesis is not considered. [8] [1]
The pitfalls that cost marks in the exam are few and repeatable. Ordering a karyotype instead of FBN1 sequencing, measuring the aortic root without a Z-score in a child, omitting the slit-lamp examination, labelling a tall child as hypermobile EDS without imaging the root, and forgetting homocystinuria in the marfanoid child with intellectual disability — each is a classic and avoidable error. [2] [9]
Prognosis & Disposition
The life expectancy of a person with Marfan syndrome who is diagnosed early and managed with surveillance, beta-blockade or losartan and timely elective root repair now approaches that of the general population. Prognosis is shaped by the size and growth rate of the root, the responsible gene, adherence to medical therapy and the timing of surgery. Loeys-Dietz and vascular Ehlers-Danlos carry a more guarded prognosis because of their more aggressive vascular behaviour. [1] [6] [8]
The disposition is shared, structured, lifelong care coordinated through a specialist connective-tissue-disorder service, with clinical genetics, paediatric cardiology and cardiothoracic surgery, ophthalmology, orthopaedics and a named coordinator at the centre. Social factors — mainstream schooling with appropriate sport modification, family support, and strengths-based framing around the person rather than the diagnosis — shape quality of life as much as the cardiac management. [1] [11]
Special Populations
Pregnancy is the defining special population for Marfan syndrome, because the haemodynamic load of gestation magnifies the risk of aortic dissection; a root greater than 4.0 cm is a red flag, and pre-pregnancy valve-sparing root repair, beta-blockade through pregnancy and a planned delivery with a specialist team are the standard of care. In remote and Indigenous communities, and in migrant, refugee and asylum-seeking families, the combination of an absent family history, reduced access to echocardiography and lower rates of cascade testing means the root is measured late and the aneurysm presents larger, which is the gap that surveillance and telehealth are designed to close. Adolescents and young adults need a structured handover of the surveillance schedule to adult care, reproductive counselling, and honest guidance on sport and occupation. [1] [11]
In Australia and New Zealand the international Ghent and ACC/AHA frameworks are applied through clinical genetics services for multigene panel testing, paediatric and adolescent cardiology for structured aortic surveillance, and telehealth to extend the schedule into rural and remote communities. Transition to adult care is increasingly structured through young-adult connective-tissue-disorder clinics, and reproductive counselling before pregnancy is a recognised ANZ standard, with elective root repair offered before conception where the root is dilated. [11]
Evidence, Guidelines & Regional Differences
The framing references are the revised Ghent nosology (Loeys et al., J Med Genet 2010), which reset the diagnostic logic around the root and the lens, and the canonical FBN1 GeneReviews entry. The 2003 Neptune paper reframed the mechanism by proving the TGF-beta arm in the fibrillin-deficient mouse, and the 2005 Loeys paper extended the concept to the TGF-beta-receptor disorders that bear his name. [2] [3] [6]
The key trial a candidate must be able to discuss is the Pediatric Heart Network trial of atenolol versus losartan (Lacro et al., NEJM 2014), which randomised children and young adults with Marfan to atenolol or losartan and found the two drugs similar in reducing the rate of aortic-root enlargement. Losartan was not superior to a beta-blocker, and the long-term follow-up confirmed a comparable effect; both remain acceptable first-line or combination therapy, and the trial is a model of how a signalling hypothesis was tested in a randomised paediatric study. [4] [5]
The operative thresholds and surveillance intervals are set by the 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease, which provides the diameter and Z-score-based recommendations applied in ANZ cardiology practice with regional adjustment for access. The Ehlers-Danlos syndromes are classified by the 2017 international classification, which named thirteen subtypes and fixed vascular EDS as the dangerous, COL3A1-driven member. [11] [8]
Exam Pearls
The five fellowship anchor points are these: the root kills; the lens goes up; fibrillin loss drives both a weak matrix and runaway TGF-beta; beta-blockade or losartan and elective repair are the defence; and cascade the family. The classic pairings reward the candidate who knows them cold: Marfan and FBN1 with an upward lens; homocystinuria and CBS with a downward lens; Loeys-Dietz and TGFBR with a bifid uvula; vascular EDS and COL3A1 with easy bruising. [2] [9]
The wrist-and-thumb sign is worth three systemic-score points, and a score of seven or more supports the diagnosis once the root is dilated. In a child the aortic root must be expressed as a Z-score, and the adult diameter threshold of roughly 4.5 to 5.0 cm does not translate directly to a small child. The most common cardiac lesion is mitral valve prolapse, not the aneurysm, and the most common cause of early death is the dissection that surveillance and elective repair prevent. [2] [11]
References
- [1]Adam MP, Cao Y, Colman M, Dice J, Hall BD, Gray K, et al. FBN1-related Marfan syndrome. GeneReviews, 1993.PMID 20301510
- [2]Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet, 2010.PMID 20591885
- [3]Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet, 2003.PMID 12598898
- [4]Lacro RV, Dietz HC, Sleeper LA, Yetman AT, Bradley TJ, Colan SD, et al. Atenolol versus losartan in children and young adults with Marfan's syndrome. N Engl J Med, 2014.PMID 25405392
- [5]Robertson DM, Wei H, Pearson GD, Yankelevitz L, Gersony WM, Hahn RT, et al. Pediatric Heart Network trial of losartan vs. atenolol in children and young adults with Marfan syndrome: impact on prespecified subgroups. Pediatr Cardiol, 2023.PMID 35902413
- [6]Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet, 2005.PMID 15731757
- [7]MacCarrick G, Black JH, Bowdin S, El-Hamamsy I, Frischmeyer-Guerrerio PA, Guerrerio AL, et al. Loeys-Dietz syndrome: a primer for diagnosis and management. Genet Med, 2014.PMID 24577266
- [8]Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet, 2017.PMID 28306229
- [9]Sacharow SJ, Picker JD, Levy HL. Homocystinuria due to Cystathionine Beta-Synthase Deficiency. GeneReviews, 1993.PMID 20301697
- [10]Adam MP, Clauser KS, Viskochil DH, Meck JM, Graham JM Congenital contractural arachnodactyly. GeneReviews, 1993.PMID 20301560
- [11]Isselbacher EM, Preventza O, Hamilton Black I, Augoustides JG, Beck AW, Bolen MA, et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: a report of the American Heart Association. Circulation, 2022.PMID 36322642