Paeds · genetics-dysmorphology-and-metabolism
Mitochondrial disease
Also known as Mitochondrial disease · Mitochondrial cytopathy · Mitochondrial encephalomyopathy · OXPHOS disorder · Respiratory chain disease · MELAS · Leigh syndrome
A fellowship approach to mitochondrial disease: recognise the energy-failure phenotype across high-demand tissues, hold the dual-genome logic (maternal mtDNA versus Mendelian nDNA) and the heteroplasmy-threshold principle that explains variability, investigate in tiers from lactate to dual-genome sequencing to selective muscle biochemistry, and manage with supportive multidisciplinary care plus the hard pharmacological constraints — above all the absolute avoidance of valproate in POLG-related disease.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A two-year-old is admitted with refractory seizures, vomiting, and drowsiness after a febrile illness. The lactate is high, the liver is enlarging, and the development that the parents were proud of a month ago has stalled. Across the ward, a teenager presents with painless loss of central vision, and in clinic a mother and her son both have diabetes and deafness. The fellowship task in mitochondrial disease is not to memorise a single syndrome but to recognise the energy-failure signature — the way a defect in ATP supply produces symptoms that cross conventional specialty boundaries — and to act on the few decisions that genuinely change outcome: confirm the genetic defect, avoid the mitochondrial toxins, and support the failing systems. [2] [7]
M.I.T.O.C.H.O.N.D.R.I.A. — the high-yield framework
Overview & Definition
Mitochondrial diseases are a group of genetically determined disorders in which the oxidative phosphorylation system fails to meet cellular energy demand, producing a characteristic pattern of dysfunction in tissues with the highest ATP requirement. They are among the most common inherited metabolic disorders, and they are also among the most protean — a single molecular defect can present in infancy with catastrophic encephalopathy or in adulthood with isolated deafness, and everything in between. The reason for this variability is biological, not arbitrary, and understanding it is the foundation of the fellowship answer. [2] [1]
The unit of function is the respiratory chain: five multi-subunit enzyme complexes (I–V) embedded in the inner mitochondrial membrane that use electrons harvested from food to pump protons, generating the electrochemical gradient that drives ATP synthase. Only thirteen of the roughly ninety subunits of this machinery are encoded by mitochondrial DNA; the remainder, and all of the assembly, maintenance, translation and import factors, are encoded by nuclear genes. A defect in either genome — and in over a thousand nuclear genes by current count — can disable the chain, which is why the clinical and genetic landscape is so large. [2] [3]
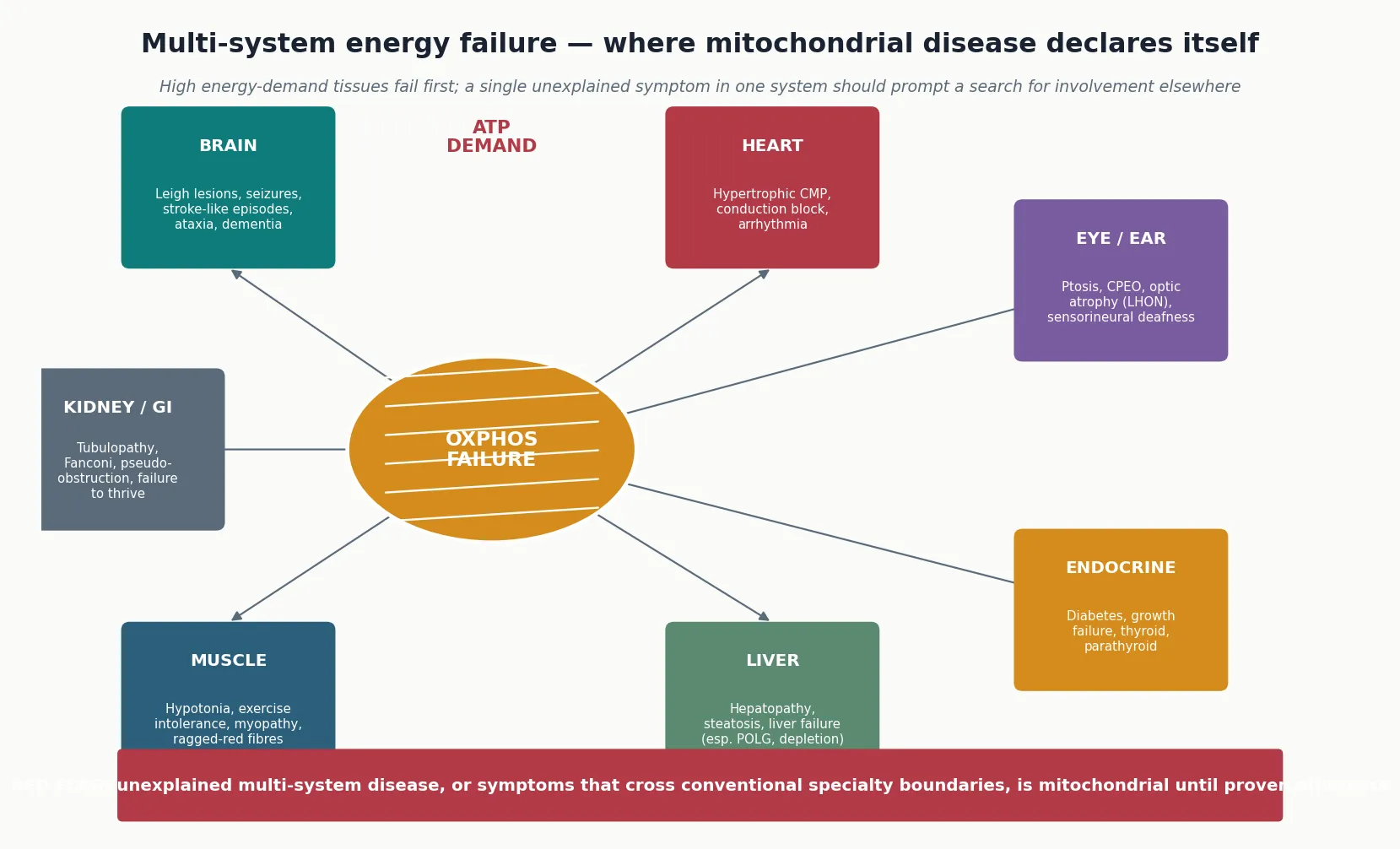
Because the phenotype reflects inadequate energy supply, it tracks the tissues that consume the most energy. The central nervous system, skeletal muscle, cardiac muscle, liver, renal tubules, pancreatic beta cells, endocrine glands, retina and cochlea are all high-demand tissues, and mitochondrial disease declares itself across them in combinations that no single-organ diagnosis explains. The bedside clue that unifies these combinations is the energy-failure signature: symptoms that cross specialty boundaries, fluctuate with metabolic stress, and are out of proportion to the apparent trigger. [2] [9]
Classification
Mitochondrial disease is classified in two complementary frames — by the genome involved and by the clinical syndrome — and the fellowship candidate must use both, because the genome sets the inheritance pattern and the counselling, while the syndrome anchors recognition. The genetic frame divides defects into those of mitochondrial DNA and those of nuclear DNA, and the distinction is the most important single fact for the family. [2] [1]
Mitochondrial DNA defects are maternally inherited — every child inherits all of their mitochondria from the mother and none from the father — and they carry the additional property of heteroplasmy, meaning that a cell contains a mixture of normal and mutant genomes. Three categories of mtDNA defect are recognised: point mutations (such as m.3243A>G in MELAS, m.8344A>G in MERRF, and m.11778G>A in Leber hereditary optic neuropathy), single large-scale deletions (Kearns-Sayre syndrome and sporadic chronic progressive external ophthalmoplegia), and mtDNA depletion, in which the copy number of mtDNA falls because of a defect in a nuclear maintenance gene. The clinical effect depends on the mutant load and the tissue distribution. [4] [9]
Nuclear DNA defects follow Mendelian inheritance, most often autosomal recessive, and they are the cause of most paediatric mitochondrial disease. They include defects of the respiratory-chain subunits and assembly factors themselves (the complex I deficiency that underlies many cases of Leigh syndrome), defects of mtDNA maintenance and replication (POLG, TWNK, and the nucleotide salvage enzymes whose loss causes mtDNA depletion syndromes), defects of mitochondrial translation and protein import, and defects of coenzyme Q10 biosynthesis. Because nuclear defects can affect any component of the mitochondrial machinery, their phenotypes are broad, and many children do not fit a named syndrome. [6] [7]
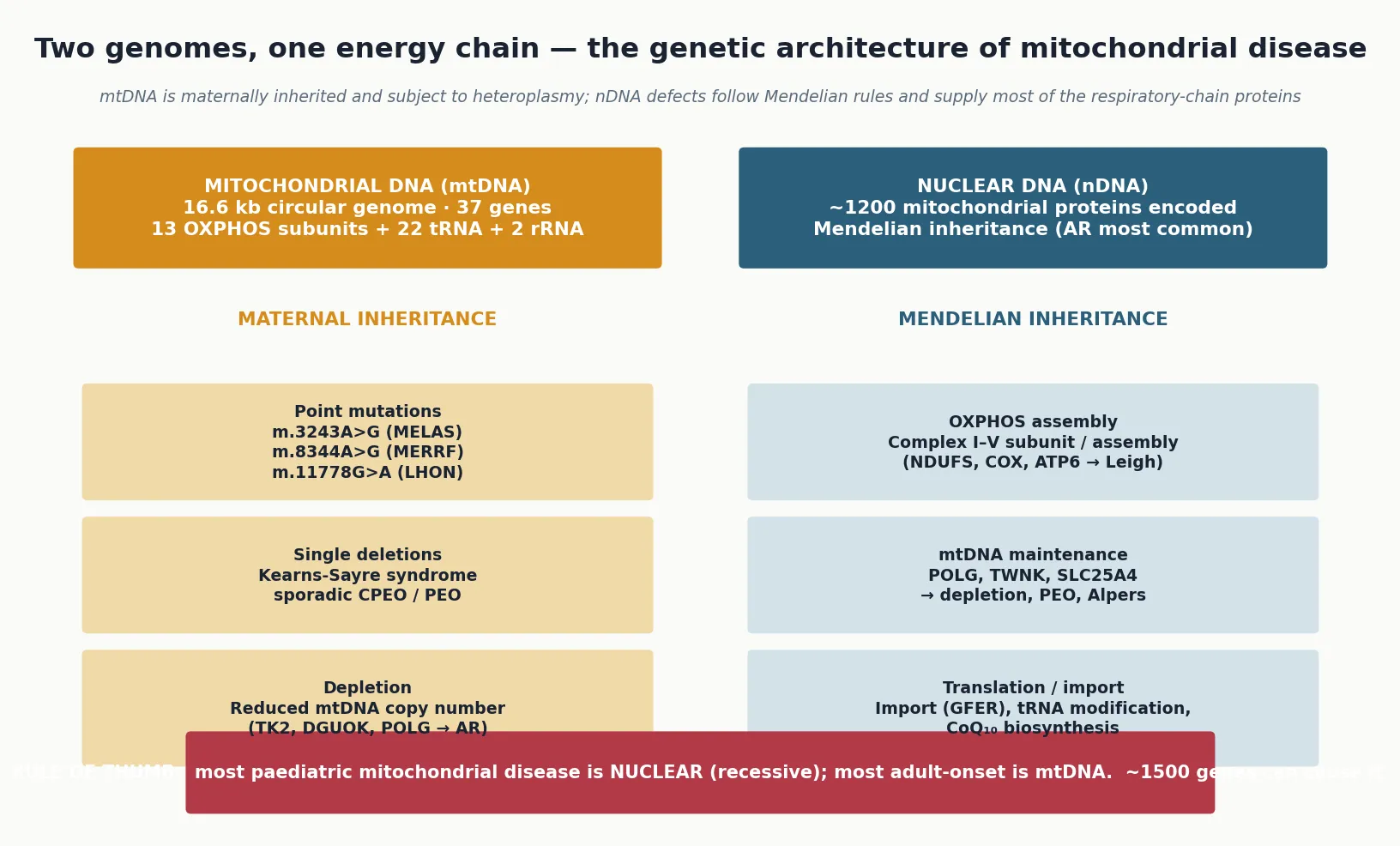
The clinical-syndrome frame organises the classical eponymous entities that anchor exam answers: MELAS, MERRF, Leigh syndrome, Kearns-Sayre syndrome, Leber hereditary optic neuropathy, NARP, Pearson syndrome, and Alpers-Huttenlocher syndrome. These syndromes are useful because they link a recognisable phenotype to a characteristic genetic lesion, but the fellowship answer must also state their limitation — there is substantial overlap between them, many patients have atypical or mixed features, and a significant proportion of children with confirmed mitochondrial disease do not fit any named syndrome at all. The syndrome is the starting point for recognition, not the boundary of the disease. [2] [3]
Epidemiology & Risk Factors
Mitochondrial disease is more common than was once believed. The point prevalence of clinically manifest mitochondrial disease is around one in 4300 individuals, and when pathogenic mtDNA mutations are counted regardless of whether they cause disease, the carrier rate is approximately one in 200 in the general population. Together, mtDNA and nuclear DNA mutations make mitochondrial disease one of the commonest inherited metabolic disorders, and the prevalence is likely to rise further as genomic testing identifies milder and atypical cases that would previously have gone undiagnosed. [1] [2]
The age and pattern of presentation differ between the two genomes. Nuclear DNA defects, which are usually recessive and often severe, present most often in infancy and early childhood with encephalopathy, hypotonia, liver failure, or cardiomyopathy. Mitochondrial DNA defects are more evenly spread across the lifespan, because heteroplasmy and the threshold effect allow some mutant genomes to remain silent for decades before a triggering event or age-related accumulation pushes the load over threshold. This age split is why the paediatric population is dominated by nuclear disease and the adult population by mtDNA disease. [1] [6]
The risk factors for presentation and decompensation, distinct from the genetic lesion itself, are the factors that the clinician can modify. Intercurrent infection, fever, fasting, surgery and anaesthesia are the classic precipitants of metabolic decompensation, because each raises energy demand or disrupts substrate supply at exactly the moment when the failing respiratory chain is least able to compensate. Recognising these precipitants is the epidemiology that matters at the bedside — they are the events that convert a compensated mitochondrial defect into a life-threatening crisis, and aggressive early management of them is one of the few interventions that genuinely changes outcome. [2] [12]
Pathophysiology
The pathophysiology of mitochondrial disease is a problem of energy supply. Oxidative phosphorylation converts the energy of nutrients into ATP through the electron-transport chain, and a defect anywhere in the chain — in a structural subunit, an assembly factor, a maintenance or replication enzyme, a translation factor, or a coenzyme Q10 biosynthetic enzyme — reduces the cell's capacity to generate ATP. When demand exceeds the reduced supply, the cell fails, and because demand is highest in brain, muscle, heart, liver and kidney, these are the tissues that fail first and most severely. [2] [3]
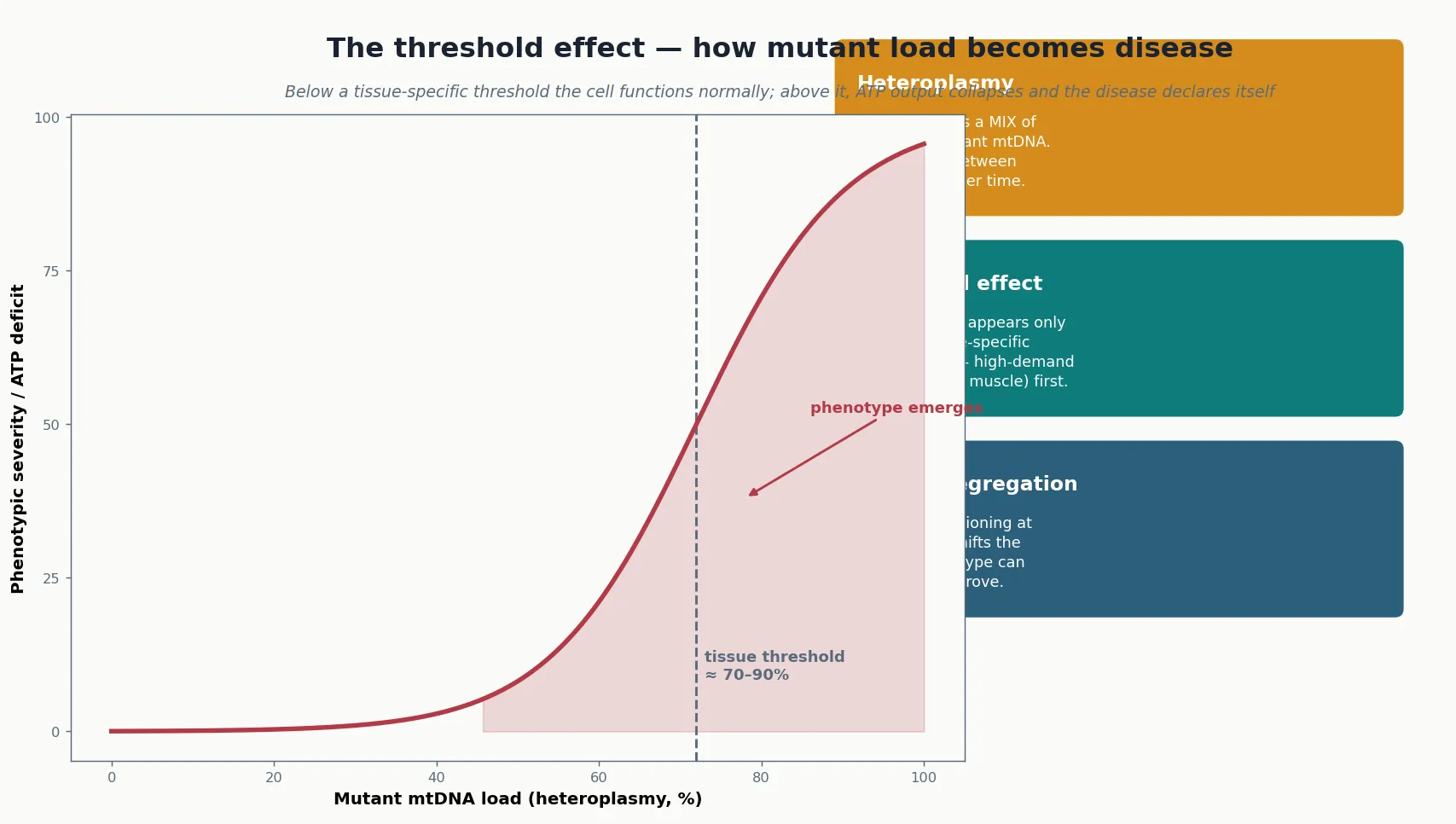
For mitochondrial DNA defects, two additional principles govern how the molecular defect becomes disease. The first is heteroplasmy: each cell contains hundreds to thousands of mtDNA copies, and in a patient with an mtDNA mutation the cell carries a mixture of normal and mutant genomes. The second is the threshold effect: a cell functions normally until the proportion of mutant genomes exceeds a tissue-specific level, typically in the range of sixty to ninety per cent, at which point ATP output collapses and the disease declares itself. Together these principles explain why two members of the same family, carrying the same mutation, can be asymptomatic or severely affected — their heteroplasmy loads differ. [4] [9]
Mitotic segregation adds a further layer of variability. As cells divide, the normal and mutant genomes are distributed unevenly to daughter cells, so the mutant load in a given tissue can drift upward or downward over time. This drift explains the age-related emergence of disease in some patients, the shift in phenotype as a child grows, and the observation that a blood heteroplasmy measurement may not reflect the load in muscle or brain. The practical consequence is that a single, low blood heteroplasmy does not exclude disease — the relevant tissue may carry a much higher load. [1] [3]
Beyond ATP deficiency, the failing respiratory chain generates increased reactive oxygen species and, through reverse electron transport and impaired NADH re-oxidation, drives lactic acidosis. The accumulation of lactate and other metabolites contributes to the metabolic encephalopathy, the stroke-like episodes of MELAS, and the tissue injury seen on neuroimaging. In POLG-related disease, the failure to replicate mtDNA in high-turnover tissues — liver and brain — produces the catastrophic hepatocerebral syndrome of Alpers, in which valproate is catastrophically hepatotoxic because it further depletes the already failing mtDNA copy number and disrupts beta-oxidation. [7] [8]
Clinical Presentation
The clinical presentation of mitochondrial disease is as broad as the energy-demand tissues it affects, and the fellowship answer earns depth by organising it around a set of presenting patterns rather than a single vignette. The unifying theme is multi-system energy failure: symptoms that cross conventional specialty boundaries, that may fluctuate with metabolic stress, and that are frequently out of proportion to the apparent trigger. [2] [3]
In the neonate and infant, mitochondrial disease presents with hypotonia, feeding difficulty, failure to thrive, encephalopathy, seizures, liver dysfunction, or cardiomyopathy — often in combination. Leigh syndrome is the archetypal presentation of infancy: a subacute, relapsing encephalopathy with hypotonia, psychomotor regression, ataxia, dystonia, brain-stem and bulbar dysfunction, and characteristic bilateral, symmetric lesions in the basal ganglia, thalamus and brain stem on MRI. The mtDNA depletion syndromes present in infancy with severe hepatopathy, myopathy, or encephalopathy, depending on the nuclear gene involved. [6] [13]
In the older child and adolescent, the presentation shifts toward discrete syndromes. MELAS presents with stroke-like episodes — focal neurological deficits that do not conform to vascular territories, classically posterior, with seizures and encephalopathy — together with short stature, migraine, deafness and diabetes. MERRF presents with myoclonus, generalised epilepsy, ataxia and ragged-red fibres. Kearns-Sayre syndrome presents with the progressive triad of external ophthalmoplegia and ptosis, pigmentary retinopathy, and onset before age twenty, with added cardiac conduction block, cerebellar ataxia and raised CSF protein. Leber hereditary optic neuropathy presents in late adolescence, predominantly in males, with painless subacute central visual loss. [4] [9] [11]
The isolated-organ presentations are easy to miss because they masquerade as a single-system disease. Mitochondrial diabetes — often matrilineal, slowly progressive, and non-insulin-dependent at onset — is the clue when diabetes coexists with deafness in a child or family. Sensorineural hearing loss, hypertrophic cardiomyopathy, renal tubulopathy (Fanconi syndrome), ptosis and ophthalmoplegia, optic atrophy, and gastrointestinal dysmotility may each be the presenting feature. The discriminating skill is to look for a second affected system, to take a careful maternal family history, and to measure a lactate whenever the pattern is atypical or multi-system. [10] [3]
Differential Diagnosis
The differential diagnosis of mitochondrial disease depends on the presenting pattern, and the fellowship candidate should frame it around three questions: what else causes a Leigh-like neurodegenerative picture, what else causes lactic acidosis, and what else causes a multi-system phenotype. Most of the time the clinical and genetic context separates these, but the discriminating skill is to know when the mitochondrial door stays open. [2] [6]
A Leigh-like presentation — bilateral basal-ganglia and brain-stem signal change with regression — raises a defined group of metabolic mimics. Organic acidaemias (propionic and methylmalonic acidaemia) can produce basal-ganglia injury and acidosis. Biotin-thiamine-responsive basal-ganglia disease, pyridoxine-dependent epilepsy, and glucose transporter type 1 deficiency share features of seizures and movement disorder and are treatable, so they must be excluded or confirmed early because the response to supplementation is dramatic. Wilson disease, intoxication-type encephalopathy, and infection (particularly in the immunocompromised) complete the acute differential. [6] [8]
Elevated lactate itself has a broad differential, and distinguishing primary mitochondrial lactic acidosis from secondary causes is a core skill. Sepsis, hypoxia, shock, and seizure activity all raise lactate through tissue hypoperfusion and anaerobic metabolism, and a high lactate in a sick child is not diagnostic of mitochondrial disease. Thiamine deficiency, liver failure (impaired lactate clearance), and certain drugs and toxins also raise lactate. The discriminating features are persistence of the elevation once the acute illness resolves, an elevated lactate-to-pyruvate ratio suggesting a pyruvate-metabolism defect, and concordance with a compatible neurological phenotype. [2] [3]
The multi-system phenotype raises the lysosomal storage disorders (which share hepatosplenomegaly, neurodegeneration and coarse features), peroxisomal disorders (with characteristic dysmorphism and sensory neural deafness), congenital disorders of glycosylation, and syndromic causes of diabetes and deafness such as Wolfram syndrome (DIDMOAD). A chromosomal microarray and a broad metabolic screen are the discriminating investigations, and the fellowship answer makes the point that a negative single test never closes the door on mitochondrial disease — the diagnosis is built from the convergence of phenotype, metabolites, imaging and genetics. [3] [10]
| Feature | Leigh (mitochondrial) | Organic acidaemia | GLUT1 deficiency | Biotin-thiamine-responsive |
|---|---|---|---|---|
| Imaging | Basal ganglia + brain stem | Globus pallidus (MMA), putamen | Usually normal | Basal ganglia, caudate |
| Lactate | Often elevated | May be elevated | Low CSF glucose | Variable |
| Key test | Respiratory chain / genetics | Organic acids, acylcarnitines | CSF:plasma glucose ratio | SLC19A3 / trial of biotin+thiamine |
| Treatment response | Supportive | Diet, carnitine | Ketogenic diet | Biotin + thiamine — dramatic |
Clinical & Bedside Assessment
The bedside assessment of a child with suspected mitochondrial disease is built around the search for multi-system involvement, because a single-organ diagnosis rarely captures the full burden of disease and the recognition of a second affected system is often what converts suspicion into a diagnosis. Begin with growth — short stature and failure to thrive are common — and take a meticulous history that probes every system rather than the presenting complaint alone. [3] [2]
The history explores, system by system, the energy-failure phenotype. Ask about neurological symptoms (developmental delay or regression, seizures, ataxia, movement disorder, migraine, stroke-like episodes), muscle symptoms (exercise intolerance, weakness, myalgia), cardiac symptoms (palpitations, syncope, heart failure), gastrointestinal symptoms (feeding difficulty, vomiting, dysmotility, constipation, liver disease), endocrine symptoms (diabetes, growth failure, thyroid dysfunction), and sensory symptoms (visual loss, ptosis, ophthalmoplegia, deafness). A maternal family history of diabetes and deafness, of unexplained visual loss in young males, or of infantile deaths is highly suggestive of an mtDNA defect. [1] [10]
Examination is multi-system and looks specifically for the stigmata of energy failure. Neurologically, assess tone, power, reflexes, coordination, eye movements (ptosis and ophthalmoplegia), fundoscopy (optic atrophy, pigmentary retinopathy) and hearing. Cardiac examination listens for the murmur and gallop of hypertrophic cardiomyopathy and screens for arrhythmia, recognising that conduction disease in Kearns-Sayre syndrome can cause sudden death. Abdominal examination checks for hepatosplenomegaly. Skin, stature and dysmorphism complete the survey, and the whole examination is interpreted as a search for the second affected system that confirms the multi-system pattern. [9] [13]
Assess development with a standardised tool and interpret change over time, because regression — the loss of previously acquired skills — is a cardinal red flag. A child with mitochondrial disease may plateau during intercurrent illness and recover incompletely, so serial assessment is more informative than a single snapshot. The fellowship answer frames the examination as a deliberate hunt for multi-system involvement, because finding a second affected system is often the step that justifies the metabolic work-up. [2] [3]
Investigations
The investigation of suspected mitochondrial disease proceeds in tiers, from a compatible phenotype and a first-line metabolic screen to dual-genome genetic testing, with muscle biochemistry reserved for cases in which genetics is negative or equivocal. The era of genome-first diagnosis has transformed the work-up, but the tiered structure remains because no single test is sensitive or specific enough to stand alone. [3] [2]
Tier one is the clinical phenotype plus first-line blood and urine tests. A blood lactate, drawn free-flowing and with a paired pyruvate to calculate the lactate-to-pyruvate ratio, is the entry-point test, but its limitations must be understood: a normal blood lactate does not exclude mitochondrial disease, because sensitivity is only around fifty per cent and the relevant tissue may not be blood. A creatine kinase (which may be normal or mildly elevated), blood gas, full blood count, liver function, glucose, ammonia, and urinary organic acids complete the first-line metabolic screen. Audiometry and ophthalmology review screen for the sensory involvement that frequently accompanies mitochondrial disease. [2] [3]
Tier two is neuroimaging and cerebrospinal fluid analysis. Brain MRI is essential: Leigh syndrome shows bilateral, symmetric T2 hyperintensity in the basal ganglia (especially the putamen), thalamus and brain stem, while MELAS shows stroke-like lesions that do not conform to vascular territories and classically involve the posterior cortex. Magnetic resonance spectroscopy may demonstrate a lactate peak at 1.3 ppm, which is highly suggestive when present. A CSF lactate is more sensitive than a blood lactate and should be measured whenever a lumbar puncture is performed for another reason, recognising that it is the metabolic signature of the central nervous system. [6] [5]
Tier three is genomic testing, and it is now the diagnostic mainstay. Because both genomes can be implicated, both must be tested — an mtDNA analysis and a nuclear gene panel, whole-exome or whole-genome sequencing together. For mtDNA point mutations, testing blood is the starting point, but urine epithelial cells and muscle carry a higher heteroplasmy load and should be tested when blood is negative or when the variant load needs quantification for counselling. For nuclear defects, a gene panel or exome captures the large and growing list of responsible genes, and trio sequencing (testing both parents) aids variant interpretation. [1] [3]
Tier four is tissue biochemistry, and it is increasingly selective. Muscle biopsy, once the diagnostic gold standard, is now reserved for cases in which genetic testing is negative, equivocal, or identifies a variant of uncertain significance. When performed, it provides ragged-red fibres on modified Gomori trichrome, COX-negative and SDH-positive fibres on sequential histochemistry, and respiratory-chain enzymology that quantifies the activity of complexes I–V. The combination of histological and enzymatic abnormalities can confirm a respiratory-chain defect even when the gene is unknown. [3] [2]

Management — Resuscitation
Resuscitation in a child with known or suspected mitochondrial disease means recognising and arresting metabolic decompensation, because the child decompensates on a reversible precipitant and the precipitant is often more treatable than the underlying defect. The common scenario is a febrile, fasting, or post-operative child who develops encephalopathy, worsening acidosis, seizures, or liver dysfunction. The principles are the same across syndromes: identify and treat the trigger, restore energy substrate, correct acid-base disturbance, and escalate to intensive care for encephalopathy, status epilepticus, or refractory acidosis. [2] [12]
The first intervention is to stop the catabolism. Fever, infection and fasting drive energy demand and deplete substrate, so aggressive antipyresis, early antibiotic or antiviral therapy for the suspected infection, and the prompt provision of glucose are the foundations. Intravenous fluids should contain glucose at an appropriate concentration, with careful monitoring to avoid both hypoglycaemia and the hyperglycaemia that can worsen cerebral injury. Avoid prolonged fasting for procedures — a child with mitochondrial disease should receive intravenous glucose during any period of enforced fasting. [12] [2]
Metabolic acidosis is managed with fluid resuscitation and, when severe, cautious bicarbonate, recognising that over-correction can be harmful. In MELAS stroke-like episodes, intravenous L-arginine given early reduces the severity and duration of the episode by stabilising endothelial nitric oxide production and improving cerebral perfusion, and it should be started promptly in any patient with known MELAS who presents with a stroke-like episode. Lactic acidosis refractory to standard measures is an indication for intensive-care involvement and, in severe cases, consideration of haemofiltration. [5] [4]
Status epilepticus in mitochondrial disease is common and dangerous, and the choice of anticonvulsant matters. Levetiracetam and lamotrigine are preferred first-line agents because they are well tolerated and do not impair mitochondrial function. Valproate must be avoided until POLG-related disease has been excluded, because of the risk of fatal hepatotoxicity; if it has already been given, it should be stopped and liver function monitored closely. Phenobarbitone and benzodiazepines are used for acute control. Refractory status epilepticus mandates PICU admission for continuous infusions. [12] [8]
Management — Definitive & Stepwise
Definitive management of mitochondrial disease is multidisciplinary, supportive, and symptom-led, because no disease-modifying therapy exists for the great majority of mitochondrial disorders. The fellowship answer frames management as a structured programme built on four pillars: avoidance of mitochondrial toxins, a cofactor and antioxidant 'mito cocktail', system-based organ support, and aggressive prevention and management of metabolic decompensation. Each pillar is modest in isolation, but together they shape the trajectory. [2] [3]
Avoidance of mitochondrial toxins is the pillar with the clearest evidence of harm prevention. Valproate is contraindicated in POLG-related disease and should be avoided in any patient with suspected mitochondrial disease until the genetic diagnosis is secure. Metformin carries a risk of lactic acidosis and is generally avoided in mitochondrial diabetes, where insulin is preferred. Aminoglycosides cause ototoxicity in carriers of the m.A1555G mtDNA variant and should be used with caution or avoided where possible. Certain drugs — statins (myopathy risk), tetracyclines and linezolid (inhibitors of the mitochondrial ribosome), and prolonged propofol infusion — warrant caution. The principle is to review every prescription against the mitochondrial safety profile. [12] [7]
The mito cocktail is a combination of cofactors and antioxidants given empirically to support residual respiratory-chain function and limit oxidative damage. Co-enzyme Q10 (ubiquinone or its analogue idebenone) is the cornerstone, particularly in primary or secondary CoQ10 deficiency; riboflavin (vitamin B2) supports complex I and flavoprotein enzymes and is especially useful in riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency; L-carnitine corrects secondary carnitine deficiency; and a range of antioxidants — vitamin C, vitamin E, alpha-lipoic acid — limit reactive-oxygen-species damage. L-arginine or citrulline is used both acutely and as maintenance in MELAS. Evidence for the cocktail is largely observational, but it is well tolerated and widely used. [2] [4]
System-based organ support addresses each affected system directly. Seizures are managed with mitochondrial-safe anticonvulsants. Cardiac surveillance with echocardiography and ECG detects cardiomyopathy and conduction disease — in Kearns-Sayre syndrome, prophylactic pacing for conduction block prevents sudden death. Endocrine management provides insulin for diabetes and hormone replacement for pituitary, thyroid or parathyroid failure. Sensorineural hearing loss is addressed with hearing aids or cochlear implantation. Nutrition — often via gastrostomy — prevents fasting and supports growth, and a structured emergency plan for illness is provided to every family. [9] [13]

Specific Subtypes & Scenarios
Each named mitochondrial syndrome carries a distinctive teaching point, and the fellowship answer earns depth by handling them individually. The surveillance principles generalise, but the management and the counselling are syndrome-specific, and several carry decisions that change outcome. [2] [3]

MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes), most often caused by the m.3243A>G point mutation in MT-TL1, presents from childhood with stroke-like episodes — focal deficits that do not respect vascular territories — together with seizures, encephalopathy, short stature, migraine, sensorineural deafness and diabetes. The stroke-like episodes are thought to arise from a combination of metabolic failure and impaired angiogenesis in the cortex, and early intravenous L-arginine reduces their severity and recurrence. The m.3243A>G mutation is the single commonest pathogenic mtDNA mutation and underlies a spectrum from severe MELAS through to isolated diabetes and deafness, depending on heteroplasmy. [4] [5]
Leigh syndrome is the archetypal paediatric mitochondrial encephalopathy, presenting in infancy with psychomotor regression, hypotonia, ataxia, dystonia, bulbar and respiratory dysfunction, and bilateral symmetric basal-ganglia and brain-stem lesions on MRI. It is genetically heterogeneous — caused by defects of both mtDNA and over seventy-five nuclear genes — which reflects the fact that any component of the respiratory chain, when defective, can produce the Leigh phenotype. Prognosis is generally poor, with death often in early childhood from respiratory failure, and management is supportive with aggressive treatment of intercurrent illness. [6] [8]
POLG-related disorders and Alpers-Huttenlocher syndrome deserve special attention because of the valproate hazard. POLG encodes the catalytic subunit of the mitochondrial DNA polymerase, and its defects produce a spectrum from adult-onset progressive external ophthalmoplegia and ataxia through to the catastrophic childhood hepatocerebral syndrome of Alpers — refractory seizures (often occipital epilepsia partialis continua), developmental regression and liver failure. The liver failure is frequently precipitated or worsened by valproate, which is why every child with unexplained encephalopathy and seizures must be assumed to have POLG disease until excluded, and why levetiracetam is the anticonvulsant of first choice. [7] [12]
Kearns-Sayre syndrome and chronic progressive external ophthalmoplegia are caused by single large-scale mtDNA deletions. Kearns-Sayre syndrome is defined by the triad of onset before age twenty, progressive external ophthalmoplegia, and pigmentary retinopathy, with added cardiac conduction block, cerebellar ataxia, raised CSF protein, and endocrine dysfunction. The conduction block can progress to complete heart block and sudden death, so serial ECG and prophylactic pacing are essential — the cardiac surveillance is the intervention that most reliably prevents death in this syndrome. [9] [2]
Why L-arginine works in MELAS stroke-like episodes
The stroke-like episodes of MELAS were long thought to be ischaemic, but they do not conform to vascular territories and the mechanism is now understood to involve impaired nitric-oxide-mediated cerebral vasodilation, driven by reduced citrulline and arginine recycling. Intravenous L-arginine, given early in an episode, restores nitric-oxide availability and cerebral perfusion, reducing the severity and duration of the deficit. Oral L-arginine or citrulline is used as maintenance to reduce recurrence. This is one of the few targeted, mechanism-based treatments in mitochondrial medicine. [5] [4]
Leber hereditary optic neuropathy causes painless, subacute, bilateral central visual loss, most often in adolescent or young-adult males carrying one of three primary mtDNA point mutations (m.11778G>A, m.3460G>A, m.14484T>C). The visual loss is due to selective degeneration of retinal ganglion cells. Idebenone, a CoQ10 analogue, is the first approved treatment and improves visual outcome in a proportion of patients when started early, and it represents a milestone as the first licensed therapy for a mitochondrial disorder. [11] [2]
Pearson syndrome and NARP complete the high-yield set. Pearson syndrome, caused by a single large-scale mtDNA deletion, presents in infancy with transfusion-dependent sideroblastic anaemia, pancytopenia and exocrine pancreatic failure; infants who survive the marrow failure evolve into Kearns-Sayre syndrome as the deletion-rich haemopoietic line is replaced. NARP (neuropathy, ataxia and retinitis pigmentosa), caused by the m.8993T>G or T>C mutation in MT-ATP6, sits on a severity continuum with Leigh syndrome — a high heteroplasmy load converts the NARP phenotype into Leigh. [2] [6]
Complications & Pitfalls
The complications of mitochondrial disease are the consequences of untreated organ involvement and of avoidable harm, and the pitfalls are the assumptions that lead clinicians to miss or worsen the disease. A fellowship answer handles both, because the harm is rarely the genetic defect itself — it is the preventable complication. [2] [3]
Neurological complications dominate. Refractory epilepsy, progressive encephalopathy, stroke-like episodes and neurodegeneration drive much of the morbidity. The pitfall is treating the seizures without considering the underlying cause — reaching for valproate in a child with undiagnosed POLG disease — and the safeguard is to assume POLG disease in any young child with unexplained encephalopathic epilepsy and to use levetiracetam first. Another pitfall is attributing regression or new neurological signs to a static encephalopathy, when they signal decompensation on a reversible precipitant. [7] [8]
Cardiac complications include hypertrophic cardiomyopathy and, critically, conduction disease. In Kearns-Sayre syndrome the conduction block can progress silently to complete heart block and sudden death, and the pitfall is performing a single normal ECG and discharging the patient — serial ECG and a low threshold for prophylactic pacing are the safeguards. mtDNA depletion syndromes and complex I deficiency can cause severe infantile cardiomyopathy, and echocardiographic surveillance is part of every mitochondrial work-up. [13] [9]
Hepatic and metabolic complications are most dangerous in the mtDNA depletion syndromes and in POLG-related disease, where liver failure can be rapid and is worsened by valproate. Lactic acidosis can become refractory during decompensation. The pitfall common to both is failing to recognise that a sick mitochondrial child is decompensating on a reversible trigger, and the safeguard is aggressive early treatment of infection, fever and fasting with glucose and fluids. [7] [13]
Anaesthetic complications deserve emphasis because every child with mitochondrial disease will encounter anaesthesia. Volatile agents, propofol infusion, and prolonged fasting all carry risk; succinylcholine is avoided in myopathy. The safeguard is a planned, multidisciplinary anaesthetic that provides energy substrate throughout, avoids mitochondrial toxins, and monitors for acidosis and arrhythmia. The pitfall is treating a mitochondrial child as a routine anaesthetic candidate. [2] [3]
Prognosis & Disposition
The prognosis of mitochondrial disease is set by the genotype, the phenotype and the age at onset, and it ranges from a near-normal lifespan in mild or tissue-restricted disease to death in infancy in severe encephalopathic or hepatocerebral presentations. The fellowship answer does not collapse this range into a single figure but frames prognosis around its determinants: the severity and pace of the disease, the organs involved, and the quality of supportive care. [2] [6]
Leigh syndrome with infantile onset and brain-stem involvement carries a poor prognosis, with death often in early childhood from respiratory failure. The mtDNA depletion syndromes with hepatic or encephalopathic presentation are similarly severe and often fatal in infancy or early childhood. POLG-related Alpers syndrome is frequently fatal once liver failure develops. In contrast, Leber hereditary optic neuropathy carries a relatively preserved lifespan with visual disability, chronic progressive external ophthalmoplegia is compatible with long survival, and many patients with milder mtDNA mutations or riboflavin-responsive nuclear defects follow a chronic, stable course. [6] [7]
The determinants that the clinician can modify are the prevention of decompensation and the early treatment of organ complications. A child whose intercurrent illnesses are managed aggressively, whose cardiac conduction is monitored and paced, whose seizures are controlled with mitochondrial-safe drugs, and whose nutrition is maintained will follow a trajectory closer to the best achievable for their genotype. The prognosis is therefore not fixed by the mutation alone — it is shaped by the quality of the supportive care and by the avoidance of the drugs and circumstances that worsen mitochondrial function. [3] [12]
Disposition for a general paediatrician is shared, structured care with a metabolic or neurology centre. The metabolic team owns the diagnostic work-up, the emergency illness plan and the genetic counselling; the paediatrician owns the coordination of subspecialty input, developmental support, and the family-facing emergency plan. A written, portable illness plan — detailing the need for glucose during fasting, the anticonvulsant choices, and the drugs to avoid — is the single most useful disposition document, because decompensation often presents to emergency departments unfamiliar with the child. [2] [3]
Special Populations
Mitochondrial disease interacts with the child's genetic, social and geographic context, and the same molecular defect behaves differently across populations. Access to specialist metabolic services, the ability to engage with a complex schedule, and the genetic counselling implications of maternal inheritance all shape outcome, and the fellowship answer recognises that the management plan is only as good as the family's ability to act on it. [2] [3]
Maternal inheritance and genetic counselling define a population in their own right. When an mtDNA mutation is identified, the mother's heteroplasmy should be measured, because it predicts — imperfectly — the risk to subsequent children, and siblings may carry a clinically significant load. The counselling is complex and probabilistic: a mother with a low load may still transmit a high load to a child, because of the mitochondrial bottleneck, and no test can predict an individual child's outcome with certainty. Nuclear defects carry standard Mendelian recurrence risks and lend themselves to prenatal and preimplantation genetic diagnosis. The fellowship answer makes clear that reproductive counselling is integral to management, not an afterthought. [1] [4]
Indigenous children, particularly in Australia and New Zealand, may face a higher background burden of the infections that precipitate decompensation, alongside reduced access to specialist metabolic services in remote communities. These factors intensify the need for a robust, portable illness plan, for a low threshold to provide glucose and fluids during intercurrent illness, and for telehealth and outreach that extend the metabolic team's expertise into communities a clinic-based model would miss. The margin for delay is smaller when the baseline infection burden is higher. [2] [3]
Rural and remote families carry the burden that the nearest emergency department may be unfamiliar with mitochondrial disease, and the limiting step is often communication rather than medicine. A written emergency plan, a medic-alert identifier listing the drugs to avoid, and a direct line to the metabolic team are the safeguards. Telehealth consultation during acute illness can guide local teams through the first critical hours, and the plan should be designed to be executed by a clinician who has never met the child. [3] [12]
Migrant, refugee and asylum-seeking families may arrive with an unrecognised diagnosis, no prior metabolic work-up, and language and trauma barriers to engaging with a complex schedule. A careful reconstruction of the family history — specifically the maternal history of diabetes, deafness and unexplained childhood deaths — confirmation of the diagnosis, an interpreter-mediated explanation, and a written illness plan in the family's language are the foundations. Vaccination status should be confirmed and completed, because vaccine-preventable infection is a precipitant of decompensation. [2] [10]
Evidence, Guidelines & Regional Differences
The evidence base for mitochondrial disease rests on prevalence studies, consensus diagnostic and management statements, and an expanding corpus of genotype-phenotype descriptions, supplemented by emerging trial evidence for a small number of targeted therapies. The field has moved from a muscle-biopsy-based diagnosis to a genome-first diagnosis within a generation, and the guidelines have evolved with it. [1] [3]
The Gorman prevalence study in Annals of Neurology remains the framing epidemiology reference, documenting that pathogenic mtDNA mutations are carried by approximately one in two hundred individuals and that manifest disease affects around one in forty-three hundred — a prevalence that places mitochondrial disease among the commonest inherited metabolic disorders. The Klopstock overview in Deutsches Arzteblatt provides a European clinical framework, and the Chin clinical-approach paper in Neurotherapeutics sets out the contemporary, tiered diagnostic pathway that a fellowship answer is built on. [1] [2] [3]
Consensus statements anchor the practical management. The Mitochondrial Medicine Society consensus on seizure management provides the anticonvulsant framework — levetiracetam and lamotrigine first, valproate avoided until POLG excluded — that is the single most safety-critical guidance in the field. The syndrome-specific reviews — El-Hattab on MELAS, Rahman on Leigh syndrome and on POLG, Hirano on progressive external ophthalmoplegia, Hage on Leber hereditary optic neuropathy, Wang on mtDNA depletion and cardiac disease — provide the depth that distinguishes a fellowship answer from a textbook summary. [4] [6] [9] [12]
Emerging therapies are reshaping the evidence base. Idebenone is the first licensed treatment for Leber hereditary optic neuropathy, and it established the principle that a mitochondrial disorder can be treated. Elamipretide, a cardiolipin-binding peptide investigated for primary mitochondrial myopathy, and nucleoside bypass therapy for TK2-related mtDNA depletion syndrome represent the next generation, though most remain investigational. Mitochondrial replacement therapy (three-parent IVF) offers reproductive prevention for women carrying severe mtDNA mutations, and it is available in a small number of jurisdictions. [11] [2]
In Australia and New Zealand, the diagnosis and management of mitochondrial disease is coordinated through the state-based metabolic and neurogenetics services, with dual-genome testing increasingly available through publicly funded genomic programmes. Every child receives a written, portable illness plan detailing glucose requirements during fasting, mitochondrial-safe anticonvulsants, and the absolute contraindication to valproate in POLG disease. Telehealth and outreach extend the metabolic team into rural and remote communities, and Indigenous-health considerations prompt a lower threshold for aggressive management of the infections that precipitate decompensation. Genetic counselling for maternal inheritance and reproductive options, including prenatal and preimplantation diagnosis for nuclear defects, is integrated into the metabolic service. [2] [3]
Exam Pearls
A fellowship candidate answering on mitochondrial disease should land five anchor points and avoid four classic traps. The anchors are the framework examiners listen for; the traps are where candidates lose easy marks. [2] [3]
Anchor one: the dual-genome logic. Mitochondrial disease can be caused by mtDNA (maternal, heteroplasmic, threshold-governed) or nDNA (Mendelian, mostly recessive, encoding most respiratory-chain proteins). The genome determines the inheritance, the recurrence risk and the counselling — and most paediatric disease is nuclear. Test both genomes. [1] [2]
Anchor two: the threshold effect explains variability. Disease appears only when mutant mtDNA load exceeds a tissue-specific level, and mitotic segregation shifts that load over time. This is why two family members with the same mutation can be asymptomatic or severely affected, and why a low blood heteroplasmy does not exclude disease. [4] [9]
Anchor three: never give valproate in POLG disease. Alpers-Huttenlocher syndrome presents with refractory seizures, regression and liver failure, and valproate can precipitate fatal hepatotoxicity. Assume POLG in any young child with unexplained encephalopathic epilepsy, exclude it, and use levetiracetam first. This is the single most safety-critical fact in the topic. [7] [12]
Anchor four: a normal lactate does not exclude the diagnosis. Blood lactate sensitivity is only around fifty per cent, and it is frequently normal in chronic and tissue-restricted phenotypes. A compatible phenotype with a pathogenic variant is diagnostic; a normal lactate should never close the door. Proceed to neuroimaging and dual-genome testing when suspicion is high. [2] [3]
Anchor five: management is supportive, toxin-avoidant, and decompensation-focused. No disease-modifying therapy exists for most disorders. Avoid valproate and metformin, give the mito cocktail, watch for cardiac conduction disease in Kearns-Sayre, give early L-arginine in MELAS, and treat intercurrent illness aggressively with glucose and fluids. The drugs you do not give matter as much as those you do. [5] [12]
The four traps to avoid are giving valproate before excluding POLG disease, abandoning the diagnosis on a single normal lactate, treating MELAS stroke-like episodes as ischaemic stroke without early L-arginine, and discharging a Kearns-Sayre patient after a single normal ECG. Avoid these and the rest of the answer falls into place. [7] [9]
References
- [1]Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, Feeney C, Horvath R, Yu-Wai-Man P, Chinnery P, Taylor RW, Turnbull DM, McFarland R. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol, 2015.PMID 25652200
- [2]Klopstock T, Mayer C, Koshenov Z, Flögel K, Vill K. Mitochondrial Disorders. Dtsch Arztebl Int, 2021.PMID 34158150
- [3]Chin HL, Nordin S, Ng YS, Taylor RW, McFarland R, Gorman GS, Blakely EL. A clinical approach to diagnosis and management of mitochondrial myopathies. Neurotherapeutics, 2024.PMID 38241155
- [4]El-Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab, 2015.PMID 26095523
- [5]Tetsuka S, Ogawa T, Hashimoto R, Sugishita K, Kon T, Ueno T, Fujita M, Kato H, Iida J, Tomiyasu M, Sakamoto K, Nakamoto H, Suzuki T, Narita N, Yamashita T, Yanai K, Tanji K, Bonaki H, Koyama Y, Toyoshima I, Suzuki H, Kato T, Sueka T, Sato K. Clinical features, pathogenesis, and management of stroke-like episodes due to MELAS. Metab Brain Dis, 2021.PMID 34118021
- [6]Rahman S, Thorburn D. Leigh syndrome. Handb Clin Neurol, 2023.PMID 36813320
- [7]Rahman S, Copeland WC. POLG-related disorders and their neurological manifestations. Nat Rev Neurol, 2019.PMID 30451971
- [8]Lim A, Coebergh JA, Tuppen HA, Blakely EL, McFarland R, Taylor RW, Hadjivassiliou M, Rahman S. The mitochondrial epilepsies. Eur J Paediatr Neurol, 2020.PMID 31973983
- [9]Hirano M, Emmanuele V, Quinzii CM. Progressive external ophthalmoplegia. Handb Clin Neurol, 2023.PMID 36813323
- [10]Karaa A, Goldstein A. The spectrum of clinical presentation, diagnosis, and management of mitochondrial forms of diabetes. Pediatr Diabetes, 2015.PMID 25330715
- [11]Hage R, Vignal-Clermont C. Leber Hereditary Optic Neuropathy: A Review of Treatment and Management. Front Neurol, 2021.PMID 34122299
- [12]Mancuso M, Angelini C, Bertini E, Bruno C, Catteruccia M, Diodato D, Donati MA, Ennio P, Filosto M, Frosini S, Garcia de Deus D, Lamantea E, Manco M, Marchet S, Maria Rizzo MR, Mongini T, Moroni I, Musumeci O, Naef V, Pegoraro E, Piaia NC, Piga E, Rigoldi M, Salani S, Santorelli FM, Scarpelli M, Spinnazzola A, Trama C, Tugnoli V, Verny C, Vercelli L, Vilarinho L, Zeviani M, Catania S, Comi GP, Carelli V, La Morgia C, Tesi N, Laan L, Horvath R, Piscosquito G, Salsano E, Sacco S, D'Angelo R, Zoccolella S, Garone C. Management of seizures in patients with primary mitochondrial diseases: Consensus statement from the mitochondrial medicine society. Eur J Neurol, 2024.PMID 38576261
- [13]Wang J, Li Y, Tian Y, Hua Z, Jiang X. Mitochondrial DNA Depletion Syndrome and Its Associated Cardiac Disease. Front Cardiovasc Med, 2021.PMID 35237671