Paeds · genetics-dysmorphology-and-metabolism
Mucopolysaccharidoses and oligosaccharidoses
Also known as Mucopolysaccharidoses · MPS · Oligosaccharidoses · Glycoproteinoses · Mucolipidoses · Glycosaminoglycan storage disorders
A fellowship approach to the mucopolysaccharidoses and oligosaccharidoses: recognise the coarse-facies child with dysostosis multiplex and hepatosplenomegaly, group the disorders by stored substrate (glycosaminoglycans versus glycoprotein-derived oligosaccharides), confirm with urine metabolic screening then enzyme activity and molecular testing, and match the disease-modifying therapy — enzyme replacement, haemopoietic stem cell transplant, or gene therapy — to the subtype and whether the central nervous system is involved.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A three-year-old who seemed entirely well as a baby is brought in because the face has thickened, the words have slowed, and the herniae never closed. The general practitioner mentions large tonsils, snoring, and a heart murmur. Across the corridor, a six-year-old with striking short-trunk dwarfism and perfectly preserved intellect is referred after a near-miss at intubation for dental work. In both rooms the unifying question is the same: which lysosomal hydrolase is missing, which glycosaminoglycan is accumulating, and is the brain involved. The fellowship task is to reason from the coarse-facies pattern to the substrate, the enzyme, and the treatment — before the airway, the cervical cord, or the cognition is lost. [1] [2]
C · O · A · R · S · E
Overview & Definition
The mucopolysaccharidoses are a family of inherited metabolic diseases in which a deficient lysosomal enzyme blocks the stepwise degradation of glycosaminoglycans — the long, sulphated sugar chains once called mucopolysaccharides that make up much of connective tissue, cartilage, and the extracellular matrix. When a single cleavage step fails, the partially degraded chain accumulates inside the lysosome and the cell distends, malfunctions, and dies. The stored material, not the missing enzyme per se, drives the visible phenotype, which is why a Hurler macrophage engorged with dermatan and heparan sulfate looks and behaves differently from a neuron in Sanfilippo swollen with heparan sulfate alone. [1] [9]
The oligosaccharidoses — sometimes called glycoproteinoses — sit in the same clinical space but store the sugar chains stripped from glycoproteins rather than free glycosaminoglycans. Alpha-mannosidosis, fucosidosis, the sialidoses, aspartylglucosaminuria, and the mucolipidoses (including I-cell disease) share the coarse facies, dysostosis multiplex, and variable neurodegeneration of the mucopolysaccharidoses, and they are screened for on the same urine metabolic panel. Grouping them together is not a taxonomic pedantry: it reflects the reality that a single urine test and a single enzyme-assay strategy covers both families, and that the coarse-facies differential at the bedside spans both. [2] [6]
The inheritance matters because it changes counselling and screening. All the mucopolysaccharidoses except Hunter (MPS II) are autosomal recessive, so a previously affected child reshapes the reproductive risk for every subsequent pregnancy and makes carrier testing and prenatal diagnosis central to management. Hunter syndrome is X-linked recessive, so affected boys present with disease and carrier mothers may show attenuated features, and the screening of at-risk maternal males becomes part of the family workup. Naming the inheritance at the first consultation frames every later conversation about siblings, future children, and the wider family. [1] [4]
Classification
Clinicians classify the mucopolysaccharidoses by the Roman-numeral subtypes I through VII, each defined by a deficient enzyme and the glycosaminoglycan it fails to cleave, because the substrate predicts the organ involvement and points to the first-line assay. The figure lays out the subtypes alongside their enzymes and the discriminating clinical clue; the parallel column for the oligosaccharidoses and mucolipidoses captures the disorders that share the coarse-facies presentation but store glycoprotein-derived sugars. [1] [2]

A second, more practical axis separates the disorders by whether the central nervous system is involved, because that distinction governs treatment choice. Non-neuronopathic disease — Morquio (MPS IV) and Maroteaux–Lamy (MPS VI), and the attenuated Hurler–Scheie form of MPS I — can be reached by intravenous enzyme replacement therapy, which does not cross the blood–brain barrier but rescues the visceral and skeletal burden. Neuronopathic disease — Hurler (severe MPS I), Sanfilippo (MPS III), and the severe form of Hunter (MPS II) — needs a therapy that reaches the brain, and that usually means haemopoietic stem cell transplant given before the injury is fixed. Holding this binary in mind prevents the common error of offering intravenous enzyme to a child whose dominant problem is cognitive decline. [2] [12]
Neuronopathic versus non-neuronopathic MPS — the treatment-defining split
- Non-neuronopathic (ERT reaches the disease): Morquio (MPS IV), Maroteaux–Lamy (MPS VI), attenuated Hurler–Scheie (MPS I). Intellect is preserved; the skeleton, airway, and valves bear the burden.
- Neuronopathic (ERT cannot reach brain; HSCT or trial therapy): Hurler (severe MPS I), Sanfilippo (MPS III), severe Hunter (MPS II). Cognition, behaviour, and sleep decline; transplant is most effective before symptoms in Hurler.
- Mixed / glycoproteinoses: alpha-mannosidosis, fucosidosis, and the sialidoses combine visceral storage with variable neurodegeneration; treatment is supportive, with transplant or ERT in selected early cases. [1] [2]
Epidemiology & Risk Factors
Individually each mucopolysaccharidosis is rare, with birth prevalence figures between one in forty thousand and one in two hundred thousand, but collectively the mucopolysaccharidoses occur in roughly one in twenty-five thousand live births, and the oligosaccharidoses add a further layer of individually rare disease. Several subtypes are markedly more common in particular populations: Hurler syndrome is over-represented in Northern European and Anglo-Celtic founders, aspartylglucosaminuria is a Finnish founder disease, and the sialidoses and mucolipidoses cluster in consanguineous communities. Consanguinity and a previously affected sibling are the two strongest risk factors and should lower the threshold for screening in any child with compatible features. [1] [2]
The epidemiology is shifting because of newborn screening. Mucopolysaccharidosis type I has been added to bloodspot panels in a growing number of regions, and Hunter and the oligosaccharidoses are being progressively evaluated as treatment and assay technology mature. This presymptomatic detection is a double-edged benefit: it opens the transplant window for Hurler and the enzyme window for attenuated disease, but it also surfaces infants with pseudodeficiency alleles or late-onset variants whose clinical course is uncertain and who may never become unwell. [10]
Pathophysiology
The lysosome maintains an acidic interior near pH 4.5 that lets its hydrolases cleave complex macromolecules into reusable building blocks. A pathogenic variant in the gene encoding one of these enzymes either abolishes its activity or, more often, causes misfolding so the enzyme is retained and degraded in the endoplasmic reticulum rather than trafficked to the lysosome. Either way the glycosaminoglycan cannot be processed, and it accumulates. The figure traces the cascade from gene change to multi-organ disease and overlays the therapeutic levers. [1] [9]
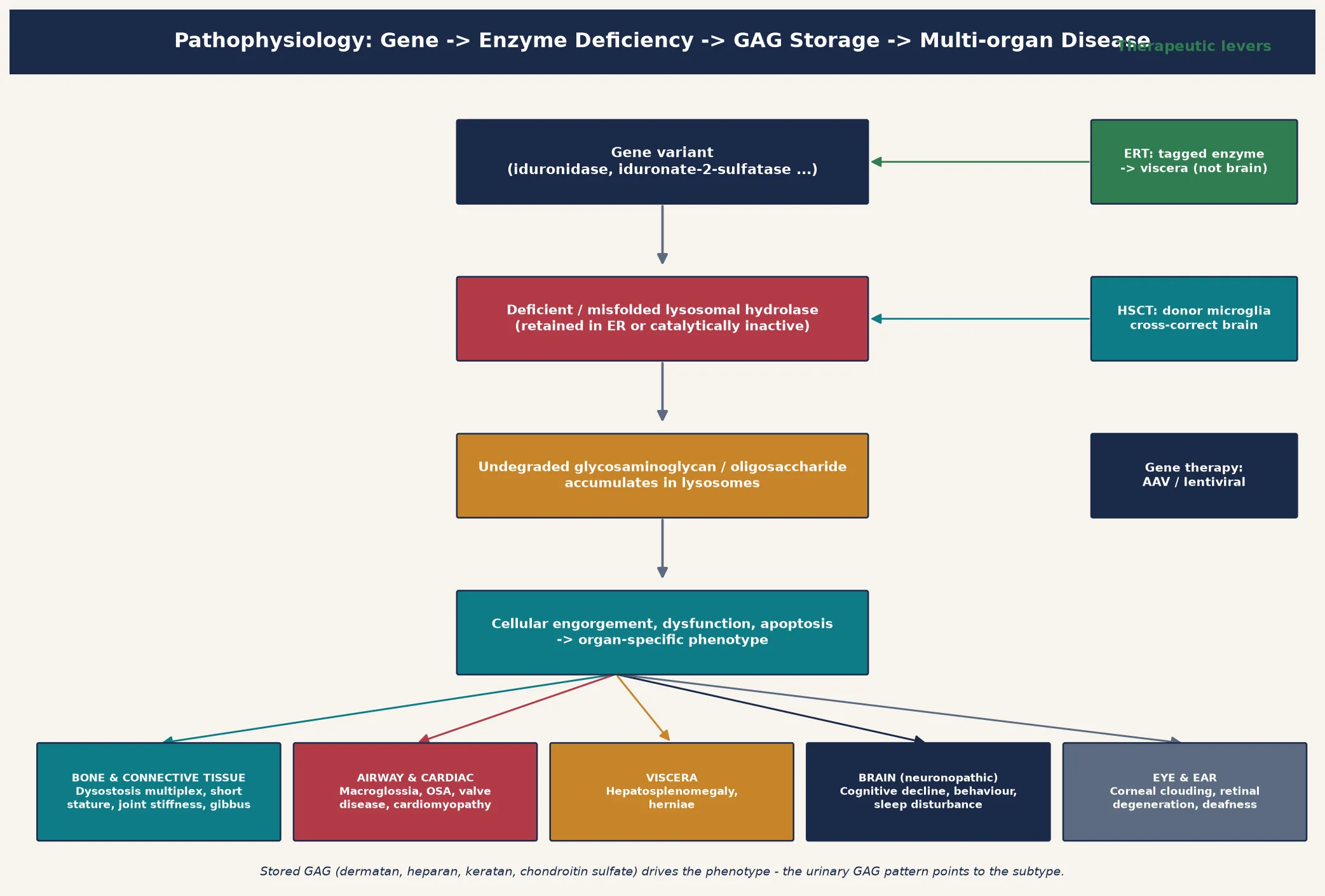
The organ phenotype follows where the glycosaminoglycan lands. Dermatan and heparan sulfate are the dominant storage material in MPS I, II, and VI, where they load connective tissue, bone, airway, cardiac valves, and viscera to produce the coarse facies, dysostosis multiplex, organomegaly, and valvular disease. Heparan sulfate alone predominates in Sanfilippo (MPS III), where the brain bears the brunt and the severe behavioural and sleep disturbance far outweighs the somatic change. Keratan sulfate accumulates in Morquio (MPS IV), loading cartilage and cornea to produce the striking skeletal dysplasia with spared intellect. Understanding that the stored material itself is the injurious agent explains why these diseases progress even when the enzyme is only partially deficient, and why the urinary glycosaminoglycan pattern points to the subtype. [1] [2]
Treatment is built on this same biology. Recombinant enzyme replacement therapy delivers a mannose-6-phosphate-tagged enzyme intravenously; the tag lets cells bind and internalise it, delivering functional enzyme to lysosomes in viscera — but the large molecule does not cross the blood–brain barrier, which is why ERT transforms Morquio, Maroteaux–Lamy, and the somatic burden of Hurler–Scheie yet cannot halt Sanfilippo. Haemopoietic stem cell transplant works differently: engrafted donor microglia and macrophages secrete functional enzyme that neighbouring cells take up by cross-correction, which is why transplant can preserve cognition in Hurler when it is given before the brain is injured. Gene therapy — autologous haemopoietic stem cells corrected with a lentiviral or adeno-associated-virus vector — extends cross-correction and has shown durable enzyme expression in early Hurler trials. [11] [12]
Clinical Presentation
The presentation depends on the subtype and the age at onset, but the unifying theme is the child who seemed normal at birth and then progressively coarsens. The face thickens — coarse hair, low nasal bridge, flaring nostrils, thick lips and alae, macroglossia. The head enlarges, the brow ridges prominent. Umbilical and inguinal herniae persist or recur. Joint stiffness, claw-hand deformity, and short stature develop insidiously, and the radiograph reveals dysostosis multiplex — thickened and oar-shaped ribs, bullet-shaped phalanges, an abnormally small and J-shaped sella turcica, and a thoracolumbar gibbus. Hepatosplenomegaly is the single most useful abdominal sign. This prototype is Hurler syndrome (severe MPS I), and most other subtypes are recognised as variations on it. [1] [2]
The discriminating features between subtypes are where the marks live. Hunter syndrome (MPS II) resembles a Hurler phenotype but is X-linked and the cornea is clear — there is no corneal clouding — and affected boys may have pebbly skin lesions (peau d'orange). Sanfilippo syndrome (MPS III) is distinguished by disproportionately severe behavioural disturbance — hyperactivity, aggression, sleep reversal, and developmental regression — with milder somatic change, so the parents complain of behaviour and sleep long before the face looks coarse. Morquio syndrome (MPS IV) produces the most striking skeletal dysplasia — short trunk, short neck, genu valgum, flat feet, and bell-shaped chest — with intellect entirely preserved, and cervical spine instability that threatens cord compression. Maroteaux–Lamy (MPS VI) resembles Hurler somatically but spares the intellect. [1] [5]
The oligosaccharidoses and mucolipidoses declare themselves in the same coarse-facies space. Alpha-mannosidosis combines coarse features, dysostosis multiplex, hearing loss, immunodeficiency, and variable intellectual disability, sometimes with a slow progressive course. Fucosidosis adds angiokeratomata, spasticity, and a sweat-gland abnormality. The sialidoses present with a cherry-red spot and action myoclonus in the normosomatic type I form, or with coarse dysostotic features and hydrops in the severe type II infantile form. I-cell disease (mucolipidosis II) is the most devastating of the group, presenting in early infancy with severe coarse features, striking gingival hyperplasia, restricted joint movement, and a lethal cardiopulmonary course, while the plasma enzymes are paradoxically elevated because the missing trafficking tag sends hydrolases out of the cell instead of into the lysosome. [6] [7]
Differential Diagnosis
When a mucopolysaccharidosis is suspected, the differential depends on which feature brought the child to attention. A coarse-facies child with dysostosis multiplex must be separated from the oligosaccharidoses (mannosidosis, fucosidosis, sialidosis), the mucolipidoses (I-cell disease, mucolipidosis III), aspartylglucosaminuria, and multiple sulfatase deficiency, all of which share the somatic phenotype. Urine glycosaminoglycan and oligosaccharide analysis is the single most useful first-line test, because the mucopolysaccharidoses elevate the glycosaminoglycan fraction while the oligosaccharidoses and mucolipidoses instead show a characteristic pattern of excreted oligosaccharides on thin-layer chromatography. [2] [8]
Non-storage causes of coarse features enter the differential when the phenotype is mild or atypical. Congenital hypothyroidism presenting late can coarsen the face and slow development, and is excluded by thyroid function testing — a cheap, fast, and essential check. The congenital disorders of glycosylation can mimic the multisystem and coarse-facies presentation and are screened by transferrin isoelectric focusing. GM1 gangliosidosis shares dysostosis multiplex and coarse features but adds a cherry-red spot and rapid neurodegeneration. When the urine glycosaminoglycan screen is negative but the phenotype is convincing, the answer is usually an oligosaccharidosis or mucolipidosis, not a missed MPS. [2] [7]
The behavioural presentation of Sanfilippo raises a different list. A child with hyperactivity, aggression, sleep reversal, and developmental regression may be evaluated for autism spectrum disorder, attention-deficit hyperactivity disorder, or a primary sleep disorder before the storage cause is considered. The clue that shifts the workup is the coexistence of any somatic feature — mild coarsening, hepatosplenomegaly, hirsutism, or mild dysostosis — with the behavioural change, which should prompt urine glycosaminoglycan testing rather than a behavioural assessment alone. [2] [1]
Clinical & Bedside Assessment
The bedside assessment is built around three questions. First, is the brain involved, and how fast is it declining — because this sets the urgency and the treatment modality. Second, which viscera are affected, and how badly — because this sets the surveillance and the anaesthetic risk. Third, is there a family history or a known affected sibling, and what is the sex of the child — because Hunter is X-linked and the reproductive counselling follows. A focused history and examination answer all three in a single consultation and direct the laboratory workup. [1] [3]
The developmental history maps the pace and pattern of any cognitive change. Documenting the trajectory — what the child could do six and twelve months ago, and what has been lost — is more informative than any single finding. In Sanfilippo, behaviour and sleep disturbance typically precede overt developmental regression by months to years; in Hurler, cognitive slowing and language delay accompany the somatic coarsening; in Morquio and Maroteaux–Lamy, intellect is preserved and the history is dominated by skeletal, airway, and cardiac symptoms. A sleep history — snoring, witnessed apnoea, daytime somnolence — is mandatory because obstructive sleep apnoea from glycosaminoglycan deposition in the upper airway is near-universal and treatable. [1] [2]
The systemic examination records hepatosplenomegaly (measure the liver and spleen edges in centimetres below the costal margin), growth parameters including head circumference, facial coarsening, corneal clouding (at the slit lamp or by direct inspection), joint range of movement, herniae, and any cardiac murmur or gallop. The spine is examined for a thoracolumbar gibbus, the hands for claw deformity, and the chest for deformity and restriction. A full cardiac and respiratory examination is essential: cardiomyopathy and valvular regurgitation are treatable complications, and the combination of macroglossia, short neck, and cervical spine instability makes anaesthesia genuinely dangerous. [3] [5]
Investigations
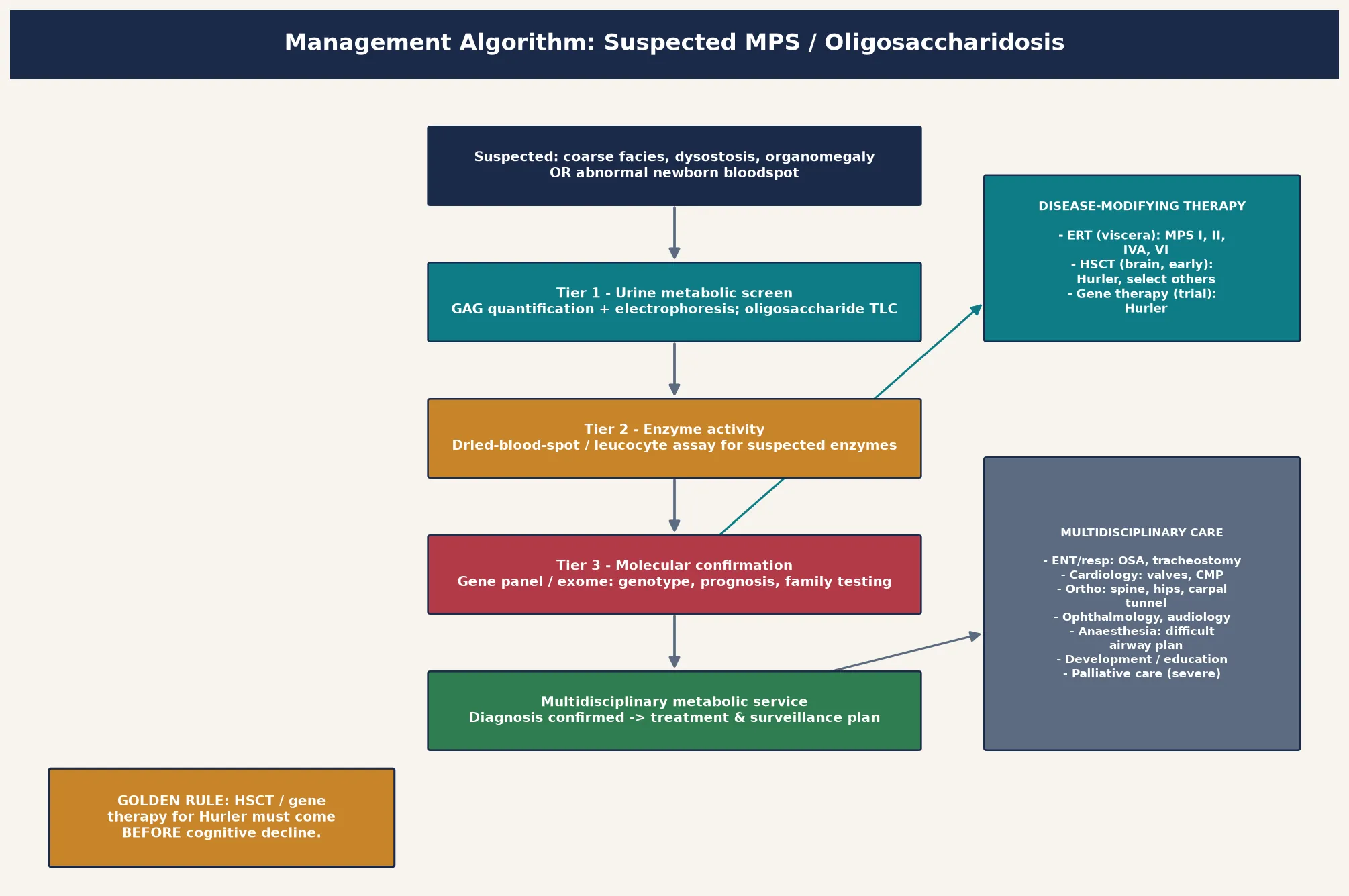
The investigation strategy is layered and escalates from cheap screening tests to definitive enzyme and molecular confirmation. The first tier is urine metabolic screening — glycosaminoglycan quantification with electrophoretic or chromatographic separation of dermatan, heparan, keratan, and chondroitin sulfate, alongside oligosaccharide thin-layer chromatography. The glycosaminoglycan pattern points to the subtype: dermatan and heparan sulfate elevation suggests MPS I, II, or VI; heparan sulfate alone suggests Sanfilippo (MPS III); keratan sulfate suggests Morquio (MPS IV). The second tier is enzyme activity measurement, now feasible on a dried blood spot or a small leucocyte sample for all the common subtypes, and this usually identifies the deficient enzyme. [1] [2]
The third and confirmatory tier is molecular genetic testing — sequencing the relevant gene or a targeted lysosomal-disease gene panel — which confirms the diagnosis, defines the variant, and enables family testing, prenatal diagnosis, and genotype-based prognostication. Molecular confirmation matters because enzyme activity alone cannot distinguish severe from attenuated Hurler phenotypes reliably, and because genotype informs whether a child is a candidate for enzyme replacement, transplant, or a gene-therapy trial. Newborn bloodspot screening for MPS I has reshaped the entry point for Hurler, and a low enzyme activity on the bloodspot is a screening result, not a diagnosis, that must be confirmed urgently with the layered pathway because pseudodeficiency alleles and carriers all produce abnormal screens. [10] [9]
Supportive investigations quantify organ involvement and set the surveillance baseline. Echocardiography defines cardiomyopathy and valvular disease and is mandatory at diagnosis and periodically thereafter. Sleep studies quantify obstructive sleep apnoea. Cervical spine imaging (flexion–extension and magnetic resonance imaging) assesses atlanto-axial instability and cord compression, especially in Morquio. A skeletal survey documents dysostosis multiplex, audiology screens the hearing loss that is common in mannosidosis and several MPS subtypes, and ophthalmology assesses the cornea and retina. Biochemistry tracks organ function — liver function, renal function, and the full blood count. The goal is to stage the disease precisely so that therapy and surveillance are matched to the individual child. [3] [5]
Management — Resuscitation
Most children with a mucopolysaccharidosis do not present in collapse, but several acute scenarios demand prompt recognition and skill. The difficult airway is the prototypical MPS emergency: glycosaminoglycan deposition produces macroglossia, a short neck, supraglottic narrowing, and temporomandibular joint restriction, and the combination with cervical spine instability makes intubation genuinely hazardous. Any MPS child needing sedation or intubation — for dental work, imaging, tonsillectomy, or an acute illness — is managed by a team experienced in metabolic airways, with cervical spine imaging, a difficult-airway strategy, and senior anaesthetic cover in place, because airway loss is a leading cause of death in this population. [1] [3]
Sleep-disordered breathing and cervical cord compression carry their own urgency. Severe obstructive sleep apnoea may present with nocturnal desaturation, cor pulmonale, or daytime somnolence, and is managed with adenotonsillectomy, continuous positive airway pressure, and in severe cases tracheostomy. Cervical spinal cord compression — from ligamentous thickening, atlanto-axial instability, or odontoid hypoplasia — may present with progressive weakness, sensory change, or sudden deterioration after minor trauma, and demands urgent neurosurgical and spinal opinion, sometimes decompression and fusion. In every case the resuscitation phase stabilises the child and secures the diagnosis and the disease-modifying pathway in parallel. [2] [5]
Management — Definitive & Stepwise
The definitive management matches the therapy to the subtype and the central-nervous-system burden. The figure lays out the decision pathway from suspicion to confirmed genotype to the treatment modality, and the guiding principle is that enzyme replacement therapy reaches the viscera but not the brain while haemopoietic stem cell transplant reaches both — but only if given early. [2] [12]
For non-neuronopathic disease, enzyme replacement therapy is the cornerstone. Intravenous recombinant enzyme every one to two weeks reduces organomegaly, improves respiratory and joint symptoms, and stabilises the somatic burden in MPS I (attenuated Hurler–Scheie), MPS II (Hunter), MPS IVA (Morquio), and MPS VI (Maroteaux–Lamy), with ERT established for laronidase, idursulfase, elosulfase alfa, and galsulfase respectively. The enzyme does not cross the blood–brain barrier, so it does not halt cognitive decline in neuronopathic disease, and infusion reactions, antibody formation, and the lifelong infusion burden are real limitations. ERT is most effective for the visceral, airway, and skeletal complications, and it does not reliably rescue established cardiac valve disease or bony dysostosis, so it complements rather than replaces multidisciplinary care. [4] [12]
For neuronopathic disease where a disease-modifying option exists, haemopoietic stem cell transplant is the established therapy. Transplant works because engrafted donor microglia and macrophages deliver functional enzyme to the brain and viscera by cross-correction, and it is most effective in Hurler (severe MPS I), where transplant given before two years of age and before cognitive decline preserves intellect and improves survival, airway, and cardiac outcomes. Its success is exquisitely time-sensitive, which is the rationale for newborn screening for MPS I. Transplant carries significant morbidity and mortality and is made at a specialist centre with metabolic and transplant expertise, ideally before the child is two years old and before neurological symptoms. Gene therapy — autologous haemopoietic stem cells corrected ex vivo and reinfused — extends the cross-correction principle and has shown durable, supraphysiological enzyme expression in early Hurler trials, with the prospect of a single treatment that reaches the brain. [11] [2]
Enzyme replacement therapy — representative regimens (specialist-initiated, weight-based, confirm current dosing)
Symptom-directed multidisciplinary care runs in parallel with disease-modifying therapy and is often what determines quality of life. Cardiology manages cardiomyopathy, arrhythmia, and valvular disease; respiratory and ENT manage airway obstruction, sleep-disordered breathing, and recurrent infections, including adenotonsillectomy, continuous positive airway pressure, and tracheostomy in severe cases; orthopaedics manages hip dysplasia, gibbus, carpal tunnel, and cervical instability; ophthalmology follows the cornea; audiology the hearing loss; developmental and educational services support learning and communication; and palliative care is involved early in the most severe neuronopathic forms. The medical home coordinates surveillance and integrates the family's psychosocial, educational, and genetic-counselling needs. [1] [5]
Specific Subtypes & Scenarios
Hurler syndrome (severe MPS I) is the prototype of the neuronopathic mucopolysaccharidosis and the subtype most likely to appear as a long-case vignette. A child who seems normal at birth develops coarse features, macroglossia, corneal clouding, gibbus, claw hands, hepatosplenomegaly, herniae, and progressive cognitive and airway involvement within the first one to two years. Untreated it is fatal in the first decade from cardiopulmonary disease. Haemopoietic stem cell transplant given before two years and before cognitive decline preserves intellect and modifies survival, and intravenous laronidase addresses the visceral burden of the attenuated Hurler–Scheie phenotype. The Hurler-versus-Hurler–Scheie distinction, and the timing of transplant, are the central teaching points. [1] [11]
Hunter syndrome (MPS II) is the X-linked exception and the most likely MPS to appear as an inheritance question. Affected boys resemble a Hurler phenotype but without corneal clouding, and they may show pebbly skin lesions. Two phenotypic extremes exist: an attenuated form with near-normal intellect and survival into adulthood, and a severe neuronopathic form with cognitive decline, behavioural disturbance, and a more rapid course. Intravenous idursulfase addresses the visceral burden, and early trials of intrathecal and gene-therapy approaches aim to reach the brain. Because it is X-linked recessive, carrier mothers may show mild features, and screening of at-risk maternal males is part of the family workup. [4] [2]
Sanfilippo syndrome (MPS III) is the great behavioural mimic. Four enzyme defects (subtypes A–D) all lead to heparan-sulfate accumulation, and the clinical picture is dominated by severe behavioural disturbance — hyperactivity, aggression, sleep reversal — followed by progressive developmental regression, seizures, and loss of mobility, with only mild somatic change. The parents complain of behaviour and sleep long before the face looks coarse, and the diagnosis is often delayed for years. There is currently no approved disease-modifying therapy, though ERT, substrate reduction, and gene-therapy trials are in development, so management is supportive and palliative, with a strong emphasis on sleep, behaviour, and family support. [1] [2]
Morquio syndrome (MPS IV) is the skeletal-dysplasia extreme with preserved intellect. Keratan-sulfate accumulation produces striking short-trunk dwarfism, genu valgum, flat feet, bell-shaped chest, and corneal clouding, but cognition is entirely spared. The dominant risks are cervical spinal cord compression from atlanto-axial instability and odontoid hypoplasia — a cause of sudden deterioration or death — and obstructive airway and cardiac-valve disease. Intravenous elosulfase alfa addresses the skeletal and respiratory burden, and the multidisciplinary plan centres on spinal surveillance, airway, and orthopaedic management. [5]
The oligosaccharidoses round out the family. Alpha-mannosidosis combines coarse features, dysostosis multiplex, sensorineural hearing loss, immunodeficiency, and variable intellectual disability, and may respond to ERT (velmanase alfa) in early disease. The sialidoses present with a cherry-red spot and action myoclonus in the normosomatic type I form, or with severe coarse dysostotic features and hydrops in the infantile type II form; the normosomatic type is an important and often-missed cause of late-onset ataxia and myoclonus. I-cell disease (mucolipidosis II) is the most severe of the group, presenting in early infancy with coarse features, striking gingival hyperplasia, restricted joints, and a lethal course, while plasma lysosomal enzymes are paradoxically elevated because the missing mannose-6-phosphate trafficking tag sends hydrolases out of the cell instead of into the lysosome. [6] [7] [8]
Complications & Pitfalls
The complications of the mucopolysaccharidoses are the complications of untreated or progressive glycosaminoglycan storage, and they cluster by organ. The airway narrows from glycosaminoglycan deposition, and the combination of macroglossia, short neck, temporomandibular joint restriction, and cervical spine instability makes anaesthesia genuinely dangerous, with difficult intubation and airway loss a leading cause of death. The heart hypertrophies and its valves thicken and regurgitate, and cardiomyopathy and valvular disease dominate the late course of Hurler, Hunter, and Maroteaux–Lamy. The bones infarct and deform, producing the painful carpal tunnel, hip dysplasia, and gibbus that dominate quality of life, and the cervical cord compresses in Morquio. The brain, once injured by storage, does not recover — which is why timing is the single most important determinant of outcome in the neuronopathic forms. [1] [5]
The most consequential pitfall is delay. Treating Sanfilippo as a primary behavioural or sleep disorder for years, or Hurler as a feeding or developmental problem, forfeits the transplant window and converts a potentially modifiable disease into a fatal one. A second pitfall is misreading a screening result: a low enzyme activity on a newborn bloodspot reflects enzyme level, not clinical disease, and pseudodeficiency alleles, late-onset variants, and carriers all produce abnormal screens in children who may never become unwell. Confirmatory enzyme assay in leucocytes and molecular testing are essential before any irreversible decision such as transplant, and before reassuring or alarming a family. [10] [9]
A third pitfall is underestimating the anaesthetic and perioperative risk. Children with mucopolysaccharidoses have difficult airways, cervical spinal cord compression, and restrictive lung disease, and even routine procedures such as dental work or tonsillectomy carry a significant risk of airway loss or spinal cord injury. Any procedure should be planned with an anaesthetic team experienced in metabolic airways, with cervical spine imaging and a difficult-airway strategy in place. Finally, assuming that enzyme replacement therapy will halt neurological decline — when it cannot cross the blood–brain barrier — sets up both clinician and family for avoidable disappointment, and assuming that transplant rescues the skeleton and valves fully — when it primarily rescues the brain and viscera — overstates its benefit. [2] [12]
Prognosis & Disposition
Prognosis is determined by the subtype, the age at onset, the genotype, and crucially the timing of treatment. Hurler syndrome, untreated, is fatal in the first decade from cardiopulmonary disease; transplanted early, it is transformed into a survivable chronic disease with preserved intellect, albeit with residual skeletal, airway, and valvular complications that need lifelong care. Hunter and Maroteaux–Lamy, treated with ERT, approach a near-normal life expectancy for the somatic disease, though the severe neuronopathic Hunter form retains a guarded cognitive prognosis. Morquio, treated with elosulfase alfa and careful spinal and airway management, retains normal intellect but remains at lifelong risk of cervical cord and airway events. [1] [12]
The neuronopathic disorders without an effective CNS-directed therapy remain the most challenging. Sanfilippo (MPS III) progresses over a decade through behavioural disturbance, loss of skills, loss of mobility, and finally a vegetative state, and management is supportive and palliative, with gene-therapy and substrate-reduction trials the main hope. I-cell disease is lethal in the first years of life. For all the disorders, honest, genotype-grounded prognostic conversations with the family, set against the realistic limits of the available therapy, are the foundation of good care. [2] [7]
Disposition is to a specialist metabolic service with a structured transition to adult care, because these are lifelong diseases that cross every organ system and every life stage. The medical home coordinates surveillance — growth, development, audiology, ophthalmology, cardiology, respiratory, sleep, and bone and spinal health — and integrates the family's psychosocial, educational, and genetic-counselling needs. For the most severe neuronopathic forms, early involvement of paediatric palliative care alongside disease-directed therapy supports the child and the family through a progressive illness, and does not preclude continuing disease-modifying treatment where it is available. [3] [5]
Special Populations
Newborn screening for MPS I has created a distinct population — the asymptomatic neonate with an abnormal screen — whose management differs from the symptomatic child. The metabolic service must distinguish true severe Hurler disease, which demands urgent confirmatory testing and a rapid transplant decision, from attenuated Hurler–Scheie variants and pseudodeficiency alleles that may never cause disease. Families need honest, calibrated counselling: not every abnormal screen means a sick child, but every abnormal screen demands a thorough, time-sensitive workup, because the cost of missing a transplantable Hurler is irreversible cognitive injury. [10] [11]
The conditions screened and the therapies funded vary between jurisdictions and change over time, so the examining clinician should state the local panel and funding rather than assume a universal list. In Australia and New Zealand, mucopolysaccharidosis type I is being progressively added to bloodspot panels as evidence and treatment evolve, and the access to and funding of expensive enzyme therapies and transplant services differs between regions. The principles are constant: a low enzyme activity is a screen, not a diagnosis; confirm with leucocyte enzyme assay and molecular testing within days; and refer urgently to the regional metabolic service for any screen positive for a transplantable disorder. [10] [3]
Consanguineous communities and founder populations carry a higher burden of the autosomal recessive mucopolysaccharidoses and oligosaccharidoses, and a previously affected sibling reshapes the reproductive risk for every subsequent pregnancy. In these families carrier testing, prenatal diagnosis by chorionic villus sampling or amniocentesis, and preimplantation genetic testing are central to management, and the metabolic and genetics services should be involved before the next pregnancy rather than after it. Indigenous, migrant, refugee, and remote populations face additional barriers — distance from specialist centres, language and cultural considerations, and inequitable access to expensive enzyme therapies — and the medical home must actively coordinate equitable, culturally safe care, often through telehealth and shared care with regional services. [1] [2]
Evidence, Guidelines & Regional Differences
The evidence base for treatment has grown substantially but remains uneven across the subtypes. Enzyme replacement therapy for MPS I, II, IVA, and VI is supported by randomised trials and registry data showing reduced organomegaly, improved respiratory function, and stabilised urinary glycosaminoglycans, though residual skeletal, airway, and valvular disease persists and the brain is not reached. Haemopoietic stem cell transplant for Hurler rests on decades of cohort and registry evidence showing that transplant given before two years and before cognitive decline preserves intellect — the rationale that underpins newborn screening for MPS I. Gene therapy, in which autologous haemopoietic stem cells are corrected ex vivo and reinfused, has shown durable and supraphysiological enzyme expression in early Hurler trials, with the prospect of a single treatment that reaches the brain. [11] [12]
Guidelines are subtype-specific and specialist-led. The management guidelines for mucopolysaccharidosis VI set the framework for surveillance and galsulfase therapy; the systematic consensus recommendations for MPS IVA address spinal, airway, and elosulfase management; and the phase II/III idursulfase study underpins Hunter ERT. [3] [5] Regional differences persist in what is screened, what therapies are funded, and how transplant is accessed, so the clinician should ground recommendations in local policy while applying the universal principles: screen early, confirm precisely, treat before the brain is injured, and support the family across the lifespan. [4] [10]
The frontier is brain-directed therapy. Intrathecal enzyme delivery, substrate reduction, and gene therapy are in active development for Sanfilippo and severe Hunter, where intravenous ERT cannot reach the central nervous system, and these advances make early, accurate diagnosis more valuable than ever. A subtype that was untreatable a decade ago may be modifiable today, provided it is found before irreversible injury — so the fellowship answer closes with an active posture: keep the mucopolysaccharidoses and oligosaccharidoses on the differential of any unexplained coarsening, organomegaly, or behavioural regression, confirm fast, and refer early. [2] [9]
Exam Pearls
In a viva, lead with the clinical pattern — the coarsening child with dysostosis multiplex — name the stored glycosaminoglycan and deficient enzyme, and then state the confirmatory test and the treatment modality. That four-step structure answers almost any mucopolysaccharidosis question. For a short case, the cornea and the intellect are the findings that earn marks: clouding versus clear splits Hurler from Hunter, and preserved intellect with striking skeletal dysplasia splits Morquio from Hurler. For a long case, the management plan that matches the therapy to the central-nervous-system burden — ERT for the viscera, transplant for the brain — and sets out airway, spinal, cardiac, and genetic counselling is what distinguishes a pass from a commendation. [1] [5]
The single most testable principle is that enzyme replacement therapy does not cross the blood–brain barrier, so it cannot halt neuronopathic decline in Sanfilippo and severe Hunter, whereas haemopoietic stem cell transplant and gene therapy can — but only before the brain is injured. Couple that with the recognition that newborn screening for MPS I exists precisely to find Hurler children pre-symptomatically and refer them for transplant, and you have the framework that ties the whole topic together. Every mucopolysaccharidosis question ultimately reduces to: which enzyme, which glycosaminoglycan, is the brain involved, and what is the window for the therapy that reaches it. [2] [11]
References
- [1]Muenzer J. Overview of the mucopolysaccharidoses. Rheumatology (Oxford), 2011.PMID 22210669
- [2]Wraith JE. Mucopolysaccharidoses and mucolipidoses. Handb Clin Neurol, 2013.PMID 23622395
- [3]Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics, 2007.PMID 17671068
- [4]Muenzer J, Gucsavas-Calkoglu M, McCandless SE, Schuetz TJ, Kimura A. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med, 2006.PMID 16912578
- [5]Akyol MU, Alden TD, Amartino H, et al. Recommendations for the management of MPS IVA: systematic evidence- and consensus-based guidance. Orphanet J Rare Dis, 2019.PMID 31196221
- [6]Malm D, Nilssen O. Alpha-mannosidosis. Orphanet J Rare Dis, 2008.PMID 18651971
- [7]David-Vizcarra G, Briody J, Crombie G, et al. The natural history and osteodystrophy of mucolipidosis types II and III. J Paediatr Child Health, 2010.PMID 20367762
- [8]Caciotti A, Di Rocco M, Filocamo M, et al. Type I sialidosis, a normosomatic lysosomal disease, in the differential diagnosis of late-onset ataxia and myoclonus: an overview. Mol Genet Metab, 2020.PMID 31711734
- [9]Platt FM, d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primers, 2018.PMID 30275469
- [10]Matern D, Gavrilov D, Oglesbee D, Raymond K, Rinaldo P, Tortorelli S. Newborn screening for lysosomal storage disorders. Semin Perinatol, 2015.PMID 25891428
- [11]Gentner B, Spinozzi G, Cordes S, et al. Hematopoietic stem- and progenitor-cell gene therapy for Hurler syndrome. N Engl J Med, 2021.PMID 34788506
- [12]Parini R, Rigoldi M, Tedesco L, Boffi L. Intravenous enzyme replacement therapy in mucopolysaccharidoses: clinical effectiveness and limitations. Int J Mol Sci, 2020.PMID 32340185