Paeds · genetics-dysmorphology-and-metabolism
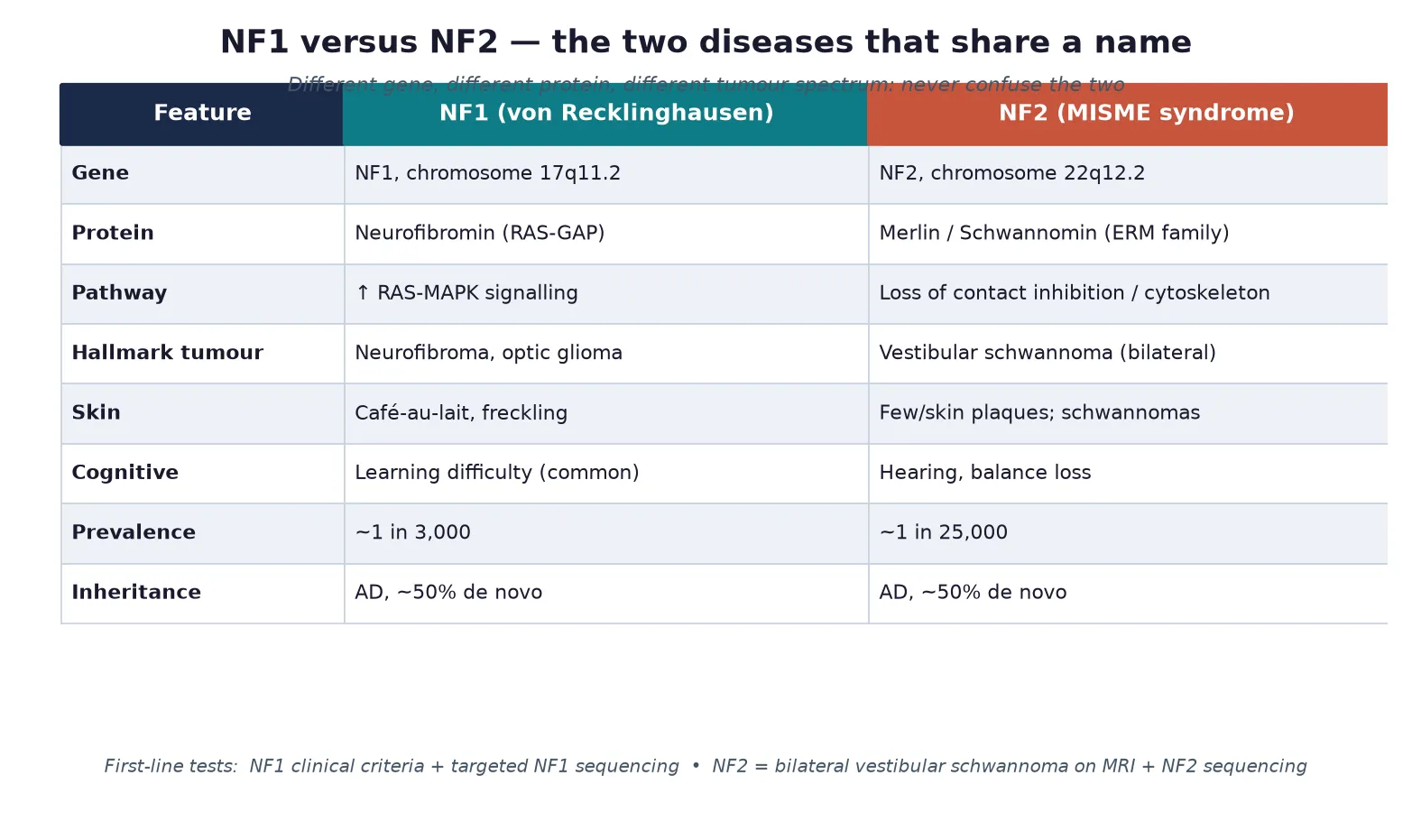
Neurofibromatosis type 1 and type 2
Also known as NF1 · von Recklinghausen disease · NF2 · MISME syndrome · Bilateral acoustic neurofibromatosis · Phakomatoses · RASopathy
A fellowship approach to neurofibromatosis type 1 and type 2: recognise the child with multiple cafe-au-lait macules and freckling as NF1 until proven otherwise, confirm clinically with the NIH criteria and molecularly with NF1 sequencing, build multidisciplinary surveillance around optic glioma, plexiform neurofibroma and learning difficulty, and separate NF2 entirely as a chromosome-22 disorder of bilateral vestibular schwannomas and meningiomas managed in a specialist tumour clinic.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The mark-winning candidate keeps three layers in view at once. The first is the child in front of you: their skin, their vision, their learning, their blood pressure, and the tumours that are and are not there. The second is the molecular mechanism: a lost tumour suppressor that fails to brake cell growth, neurofibromin in NF1 and merlin in NF2, working through completely different pathways. The third is the family: an autosomal-dominant condition with 50 per cent transmission means every first-degree relative deserves assessment and counselling. [1] [3]
Overview & Definition
Neurofibromatosis type 1 is an autosomal-dominant disorder caused by loss-of-function variants in the NF1 gene on chromosome 17q11.2, which encodes neurofibromin, a protein that switches off active RAS and restrains cell proliferation. When neurofibromin is absent, the RAS-MAPK pathway runs unchecked in Schwann cells, melanocytes and glia, producing the cafe-au-lait macules, freckling, neurofibromas and optic pathway gliomas that define the condition. NF1 is the commonest of the phakomatoses and a member of the RASopathies, a family of developmental disorders linked by dysregulated RAS signalling. [1] [10]
Neurofibromatosis type 2 is a clinically and genetically distinct disorder caused by variants in the NF2 gene on chromosome 22q12.2, which encodes merlin (also called schwannomin), a cytoskeletal tumour suppressor that maintains contact-dependent growth inhibition. Loss of merlin drives the growth of bilateral vestibular schwannomas together with meningiomas, spinal ependymomas and peripheral neuropathy. NF2 is roughly ten times rarer than NF1 and is managed as a tumour-predisposition syndrome rather than a dysmorphic one, because the skin findings are minimal and the cognitive profile is usually normal. [2] [3]
The clinical distinction matters because patients and families often assume the two conditions are grades of the same disease. They are not. NF1 brings developmental, dermatological and learning problems that dominate childhood; NF2 brings progressive hearing, balance and cranial-nerve loss that dominates adolescence and young adulthood. Confirming which gene is involved sets the surveillance schedule, the treatment options and the reproductive counselling, and it removes a great deal of needless fear. [2] [4]
Classification
The clinical classification that governs practice is the NIH diagnostic criteria for NF1, set in 1987 and refined by the 2021 international consensus. A child meets criteria for NF1 when two or more of seven features are present, and the 2021 revision adds a pathogenic NF1 variant or an unequivocal plexiform neurofibroma on MRI as standalone criteria, so molecular confirmation can now close the diagnosis even when the skin phenotype is still emerging. NF2 is classified separately by its bilateral vestibular schwannomas or by family history plus unilateral schwannoma and additional tumours. [5] [4]
NF1 and NF2 sit within the broader neurofibromatosis spectrum that now includes the schwannomatoses, themselves classified since 2022 into schwannomatosis, NF2-related schwannomatosis and SMARCB1- or LZK1-related schwannomatosis under the updated consensus nomenclature. The clinically important neighbours are Legius syndrome, caused by SPRED1 variants, which mimics NF1 with cafe-au-lait macules and freckling but lacks neurofibromas, Lisch nodules and optic glioma, and the schwannomatoses, which cause painful peripheral schwannomas without vestibular involvement. Recognising these mimics prevents over-investigation and inappropriate surveillance. [5] [6]
Seven NIH criteria for NF1 \u2014 CLIPCO
Epidemiology & Risk Factors
NF1 is the commonest single-gene disorder of the nervous system, affecting roughly one in 3,000 people across all populations with no major ethnic predilection. NF2 is far rarer at about one in 25,000. Both are autosomal dominant with full penetrance but variable expressivity, so an affected parent has a 50 per cent chance of passing the variant to each child, yet the severity within a family can differ enormously. Around half of NF1 cases and half of NF2 cases arise from de novo variants, which means a negative family history never excludes the diagnosis. [1] [2]
The strongest risk factor is a confirmed affected first-degree relative, which is why a three-generation pedigree is part of the initial assessment. For NF1 the de novo rate is driven by paternal germline mosaicism and advanced paternal age, while for NF2 maternally inherited cases tend to present earlier and more severely. Because penetrance is effectively complete for NF1 by adulthood, an apparently unaffected parent of a sporadic case should be examined for subtle cutaneous signs and offered genetic testing to detect mosaicism, which carries a lower but real recurrence risk. [1] [9]
Population factors shape detection as much as biology. In remote and Indigenous communities, and in migrant and refugee families, limited access to clinical genetics and lower rates of cascade testing mean that affected children and relatives are diagnosed late and tumours present at a larger size, which widens the gap in outcomes that good surveillance is designed to close. [9]
Pathophysiology
The NF1 phenotype begins with loss of neurofibromin, a large GTPase-activating protein that accelerates the conversion of active RAS-GTP back to inactive RAS-GDP. Without neurofibromin, RAS remains constitutively active and the downstream RAF-MEK-ERK (MAPK) cascade is overdriven, driving proliferation in Schwann cells and melanocytes and disturbing glial and neuronal development. The same pathway explains why MEK inhibitors can shrink plexiform neurofibromas, because they cut in downstream of the broken brake. [1] [8]
The cell-type specificity of the lesions reflects where neurofibromin matters most. Melanocytes over-proliferate to form cafe-au-lait macules, Schwann cells form the benign dermal and plexiform neurofibromas, and glial cells around the optic pathway give rise to the low-grade gliomas that are the hallmark nervous-system tumour of childhood NF1. A second hit in a plexiform neurofibroma, often through loss of the remaining NF1 allele and additional driver events, is the route to malignant peripheral nerve sheath tumour, the life-threatening complication that surveillance is built to catch early. [1] [9]
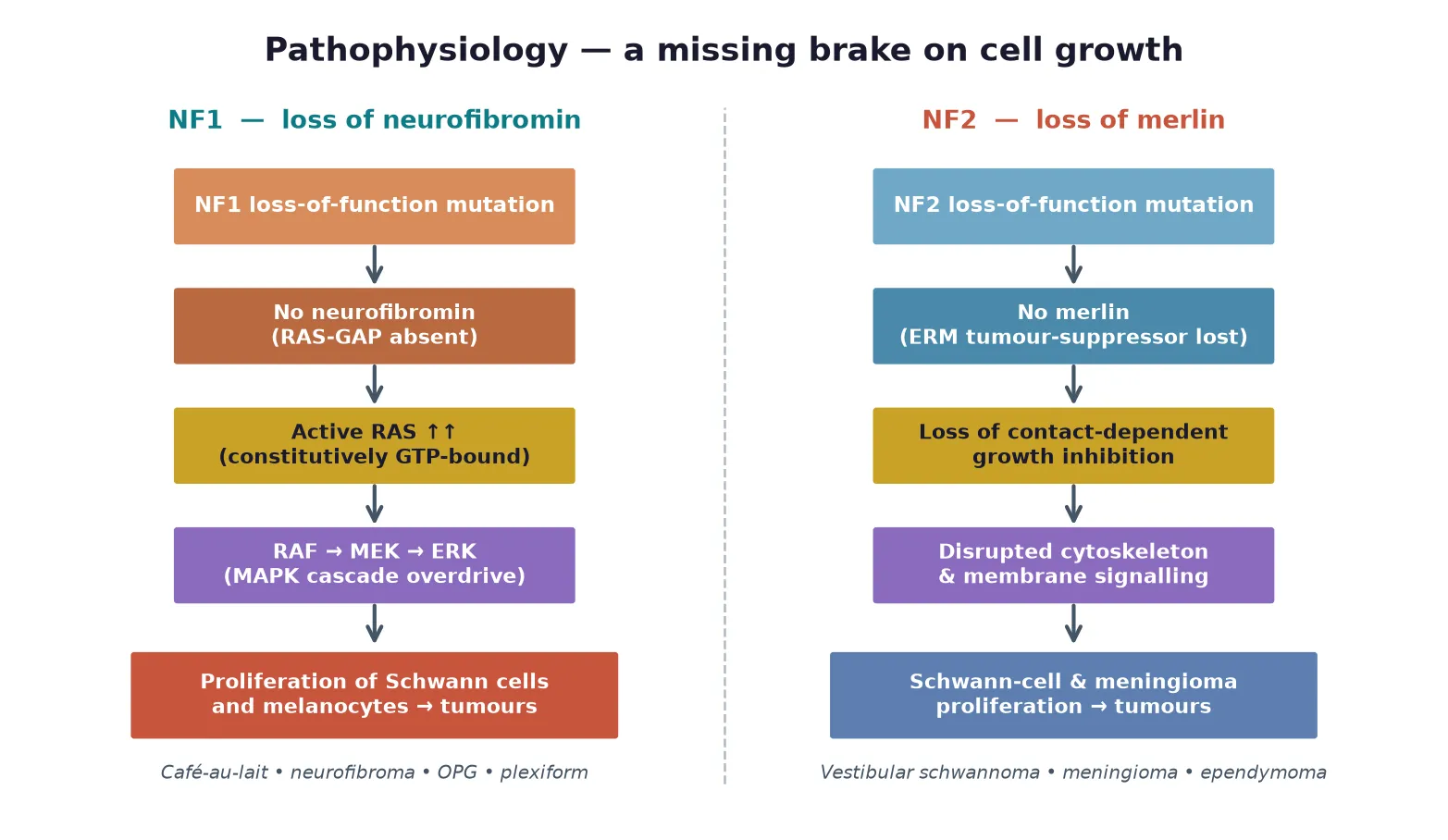
NF2 follows a parallel but distinct logic. Merlin is a member of the ezrin-radixin-moesin family that links the actin cytoskeleton to the cell membrane, and its loss abolishes contact-dependent inhibition of proliferation and disrupts membrane receptor signalling. The result is uncontrolled growth of Schwann cells around the vestibular nerve, of meningothelial cells in the meninges, and of ependymal cells in the spinal cord, producing the bilateral vestibular schwannomas, meningiomas and ependymomas that define the disease. [2] [4]
Clinical Presentation
NF1 presents in an age-dependent sequence that mirrors the emergence of its features. Cafe-au-lait macules are usually the first sign, appearing in infancy, followed by skinfold freckling in the axillary or inguinal regions around age three to five. Dermal neurofibromas and Lisch nodules tend to appear after puberty and accumulate through adulthood, while optic pathway gliomas, when they occur, most often declare themselves before age six. Learning difficulty, attention-deficit/hyperactivity disorder and macrocephaly are the developmental and growth features that bring many children to medical attention in the preschool years. [10] [1]

The complications that change management emerge as the child grows. Optic pathway gliomas cause painless visual decline or precocious puberty and affect roughly 15 per cent of children with NF1, though most remain stable or asymptomatic. Plexiform neurofibromas are congenital, diffuse tumours that can be disfiguring or compress vital structures, and they carry a lifetime risk of malignant transformation of around 8 to 13 per cent. Scoliosis, tibial pseudarthrosis, renal artery stenosis causing hypertension, and short stature complete the multisystem burden that surveillance is designed to detect. [1] [7]
NF2 presents later and more quietly, most often in the late teens or early twenties with progressive hearing loss, tinnitus and balance disturbance from a vestibular schwannoma. Juvenile posterior subcapsular cataracts, cutaneous schwannoma plaques, and a peripheral neuropathy can precede the hearing loss by years. Because the tumours grow slowly, the diagnosis is frequently delayed, and the presenting symptom in a child may be a cataract or a facial-nerve schwannoma rather than deafness, which is why any child with a first-degree relative affected by NF2 needs baseline MRI and audiology regardless of symptoms. [2] [4]
Differential Diagnosis
The differential of multiple cafe-au-lait macules is the first diagnostic fork, and NF1 is the most likely but not the only cause. Legius syndrome is the closest mimic, producing cafe-au-lait macules and freckling without neurofibromas, Lisch nodules or optic glioma because it is caused by SPRED1 variants in the same RAS pathway; the two are separated only by molecular testing and the long-term tumour risk. Other causes of multiple pigmented macules include familial cafe-au-lait macules, Noonan syndrome and other RASopathies, McCune-Albright syndrome (with polyostotic fibrous dysplasia and endocrinopathy), and Bannayan-Riley-Ruvalcaba syndrome. [5] [10]
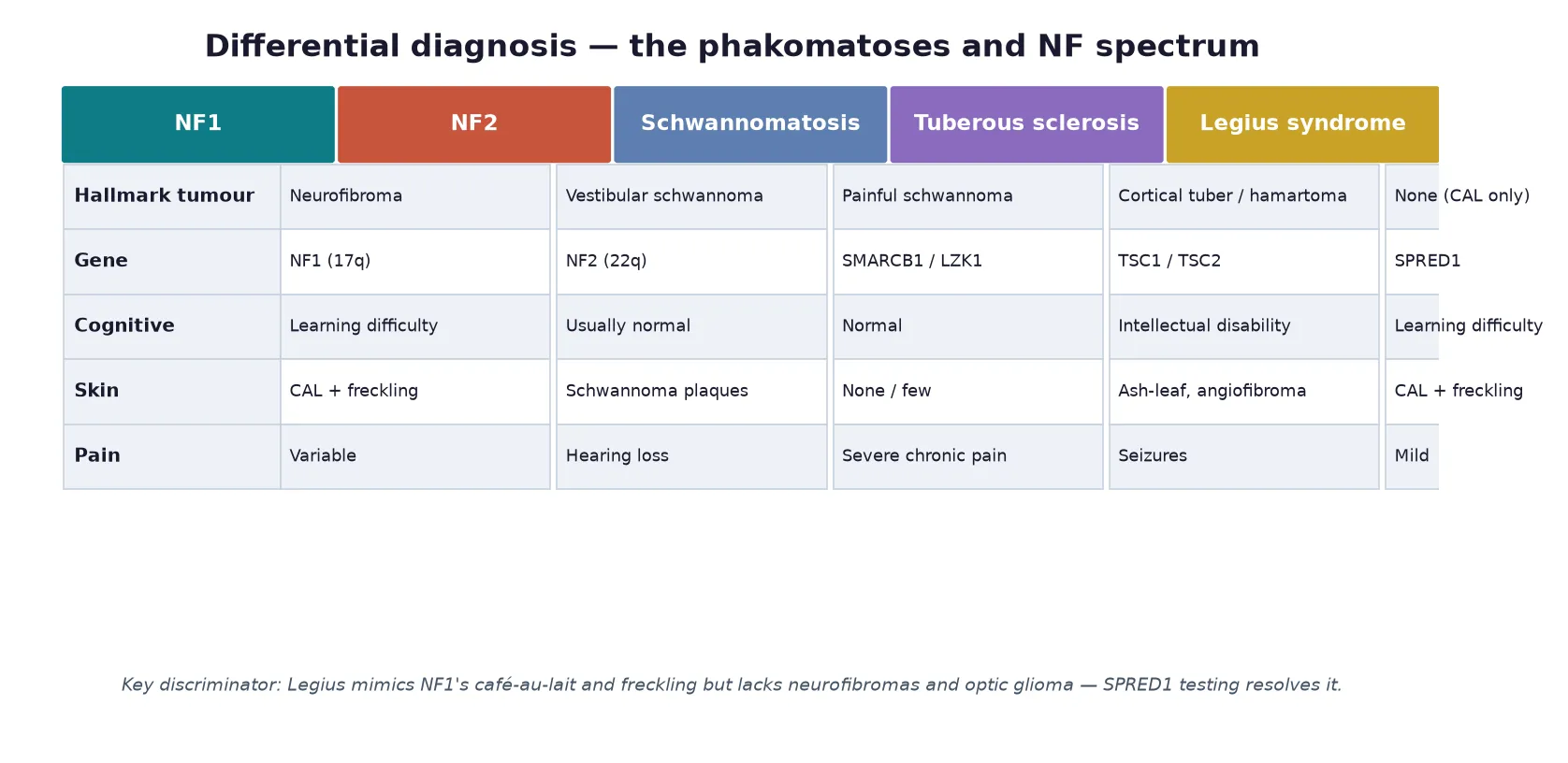
The differential of a nerve-sheath tumour in a child separates benign from malignant and hereditary from sporadic. A solitary neurofibroma or schwannoma in an otherwise well child is usually sporadic, but bilateral or multifocal lesions, early onset, or a family history point to a tumour-predisposition syndrome. Tuberous sclerosis enters the differential of a child with skin findings and brain tumours through its ash-leaf macules, angiofibromas, cortical tubers and epilepsy, and it is separated by its dominant inheritance of TSC1 or TSC2. The schwannomatoses are distinguished from NF2 by painful peripheral schwannomas in the absence of vestibular involvement. [3] [6]
For NF2, the differential of progressive sensorineural hearing loss in a young person includes sporadic vestibular schwannoma, cytomegalovirus or congenital infection, and otosclerosis, but bilateral disease or accompanying meningioma and cataract make NF2 overwhelmingly likely. Molecular testing of NF2 resolves equivocal cases and is essential before attributing a unilateral schwannoma with a family history to chance. [2] [4]
Clinical & Bedside Assessment
The bedside assessment of a child with suspected NF1 is a structured skin, eye, growth and neurological examination that converts findings into a diagnostic and surveillance plan. Count and measure the cafe-au-lait macules against the age-adjusted thresholds (5 mm before puberty, 15 mm after), examine the axillae and groins for freckling, inspect the iris with a slit lamp for Lisch nodules, and palpate the skin and superficial nerves for dermal and plexiform neurofibromas, noting any lesion that is painful, firm, rapidly growing or fixed, because these features raise concern for malignant change. [10] [1]
Measure head circumference, height and weight and plot them, because macrocephaly, short stature and growth attenuation are part of the phenotype, and examine the spine for scoliosis and the long bones for bowing or pseudarthrosis. Check blood pressure at every visit, as hypertension from renal artery stenosis or phaeochromocytoma is a treatable and easily missed complication. Take a three-generation pedigree that explicitly asks about cafe-au-lait spots, neurofibromas, hearing loss, brain tumours and learning difficulty in relatives, and examine both parents if possible, because subtle manifestations in a parent confirm the diagnosis and reveal an affected family. [1] [9]
Developmental and behavioural assessment is not optional. Screen for learning difficulty, attention-deficit/hyperactivity disorder and autism-spectrum features, and assess visual acuity and colour vision annually until at least age eight, because optic pathway glioma is most likely to emerge and threaten vision in early childhood. For suspected NF2 the examination adds a full cranial-nerve assessment, audiometry, and a search for cutaneous schwannoma plaques and cataracts. [2] [7]
Investigations
The diagnosis of NF1 is clinical in most children, made by the NIH criteria, and molecular testing with NF1 sequencing confirms the diagnosis when the phenotype is incomplete, when a mosaic or mild presentation is suspected, or when the result will guide reproductive counselling. The 2021 consensus allows a pathogenic NF1 variant alone to confirm the diagnosis, and chromosomal microarray remains useful to exclude copy-number mimics. Routine brain MRI is not recommended for asymptomatic children, because optic pathway gliomas are managed by ophthalmological surveillance and incidental findings cause harm. [5] [1]
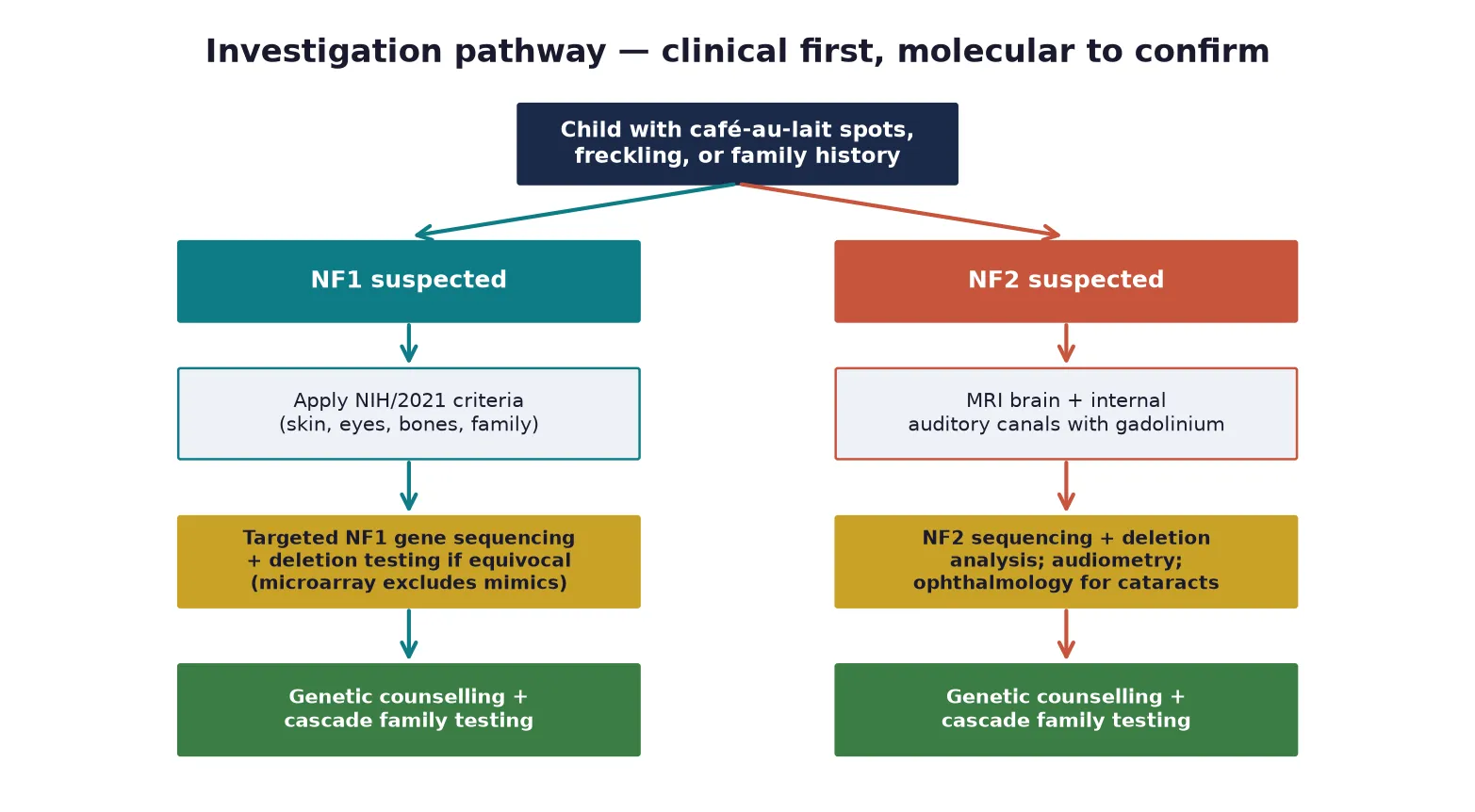
Investigation of complications is symptom-driven. An abnormal vision assessment, proptosis, or precocious puberty prompts an MRI of the optic pathway to define a glioma. A plexiform neurofibroma that is painful, growing or changing in character is imaged with MRI and, when malignant peripheral nerve sheath tumour is suspected, sampled with biopsy and evaluated with positron-emission tomography for metabolic activity and metastatic staging. Hypertension is investigated with renal ultrasound and Doppler of the renal arteries and, when clinical features suggest it, urinary catecholamines for phaeochromocytoma. [1] [9]
For NF2 the investigation is radiological and audiological from the outset. A gadolinium-enhanced MRI of the brain and internal auditory canals defines the vestibular schwannomas and any meningioma, and a whole-neuraxis MRI screens for spinal ependymoma. Pure-tone audiometry and speech discrimination testing establish the baseline hearing, and ophthalmology assessment looks for cataracts and retinal hamartomas. NF2 sequencing and deletion analysis confirm the diagnosis and enable cascade testing. [2] [4]
Management — Resuscitation
Acute resuscitation in NF1 is uncommon, but three scenarios demand urgent action. The first is malignant peripheral nerve sheath tumour, which presents as a rapidly enlarging, painful, hard mass within a pre-existing plexiform neurofibroma and requires urgent imaging, biopsy and oncology referral. The second is severe or rapidly progressive optic pathway glioma, where abrupt visual loss or a large chiasmal lesion with endocrine disturbance needs urgent imaging, ophthalmology and a neuro-oncology opinion. The third is hypertensive emergency from renal artery stenosis, managed with standard paediatric antihypertensive protocols and a nephrology referral. [1] [9]
For NF2 the acute emergency is brainstem compression from a growing vestibular schwannoma or posterior fossa meningioma, presenting with raised intracranial pressure, hydrocephalus or lower cranial-nerve dysfunction. These children need urgent neurosurgical assessment, imaging and often decompression or shunting. Acute hearing loss in an established schwannoma may respond to corticosteroids and warrants expedited review. [2] [3]
Management — Definitive & Stepwise
Definitive management of NF1 is a coordinated surveillance programme that a fellowship candidate can recite as a five-step framework: confirm the diagnosis, assemble a multidisciplinary baseline assessment, run annual surveillance, treat complications as they arise, and cascade-test the family with genetic counselling. No step cures NF1, but each prevents disability and detects life-threatening change early. The backbone is an annual review of growth, blood pressure, skin, spine, puberty, development and behaviour, coordinated through a specialist NF or clinical genetics service. [1] [9]
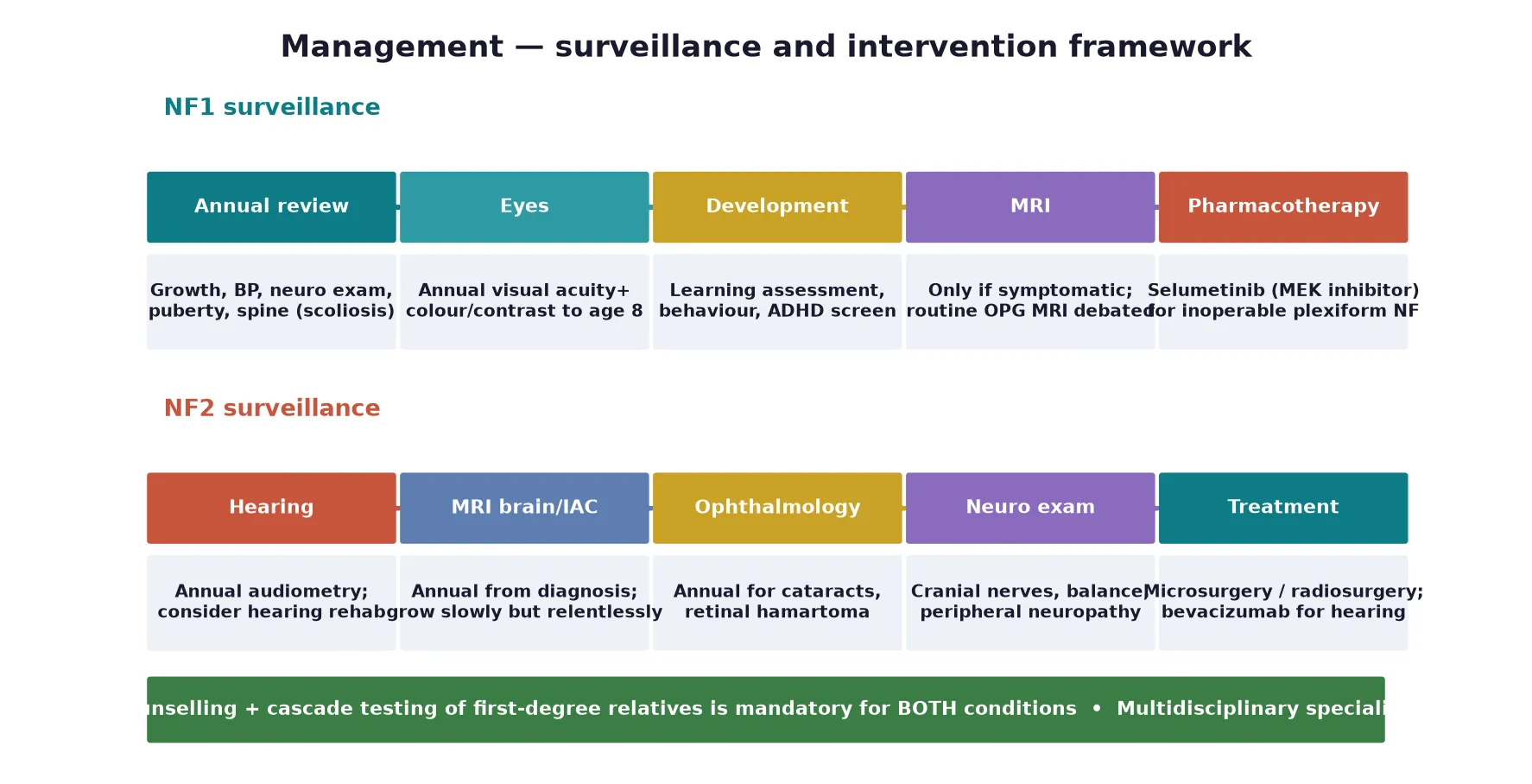
The transformative development of the past decade is targeted medical therapy for plexiform neurofibroma. The MEK inhibitor selumetinib produced objective tumour shrinkage in the majority of children with inoperable, symptomatic plexiform neurofibromas in the SPRINT trial at a dose of 25 mg per square metre twice daily, and it is now a first-line systemic option alongside surgery for disfiguring or compressive lesions. Dermal neurofibromas are managed expectantly or with surgical excision when symptomatic, and optic pathway gliomas that progress on surveillance are treated with carboplatin-based chemotherapy, reserving radiotherapy because of secondary malignancy risk. [8] [7]
NF2 management is surgical and rehabilitative, because the tumours grow slowly but relentlessly and medical options are limited. Vestibular schwannomas are managed by watchful waiting with serial MRI for small asymptomatic lesions, microsurgical resection or stereotactic radiosurgery for growing or symptomatic tumours, and hearing rehabilitation with hearing aids or cochlear or auditory-brainstem implants. The vascular endothelial growth factor inhibitor bevacizumab can improve hearing and reduce tumour volume in some patients and is a consideration for progressive disease. Meningiomas and symptomatic ependymomas are treated surgically when they compress neural structures. [2] [4]
| Domain | NF1 | NF2 |
|---|---|---|
| Lead team | Paediatric/genetics NF clinic | Specialist NF2 / neuro-oncology clinic |
| Surveillance imaging | MRI only if symptomatic | Annual MRI brain and IAC from diagnosis |
| Vision/hearing | Annual visual acuity to age 8 | Annual audiometry |
| Systemic therapy | Selumetinib (MEK inhibitor) | Bevacizumab (selected cases) |
| Family testing | Cascade + counselling | Cascade + counselling |
Specific Subtypes & Scenarios
The mosaic NF1 child is a scenario where under-diagnosis is the chief pitfall. Children with somatic mosaicism carry the NF1 variant in only a proportion of cells, producing pigmentary changes and neurofibromas that follow a segmental or blaschkoid distribution and are milder than germline disease. Because the gonads may or may not be involved, the recurrence risk for offspring is lower than the 50 per cent of germline NF1 but is not zero, and molecular confirmation on affected tissue guides counselling. The reflex to dismiss segmental pigmentary change as a birthmark misses these patients and their families. [1] [5]
The child with an optic pathway glioma is managed jointly by ophthalmology and neuro-oncology. Most NF1-associated optic gliomas are pilocytic astrocytomas that remain stable or regress, so the default is surveillance with serial vision testing rather than intervention, and treatment with carboplatin-based chemotherapy is reserved for progressive visual loss. Radiotherapy is avoided wherever possible because children with NF1 are exquisitely sensitive to radiation-induced vasculopathy and secondary malignancy. [7] [9]
The child at risk of NF2 demands proactive surveillance rather than a wait-and-see approach. A first-degree relative of an affected person should have a baseline gadolinium MRI of the brain and internal auditory canals and audiometry in childhood, because tumours can be present long before symptoms and early detection preserves hearing. Juvenile cataracts in a child with a family history of NF2 are a recognised herald and should trigger full evaluation. [2] [4]
Complications & Pitfalls
The complications of NF1 divide into the clinical comorbidities that accumulate over a lifetime and the cognitive traps that cost marks. The clinical comorbidities are optic pathway glioma, plexiform neurofibroma and its malignant transformation, scoliosis and tibial pseudarthrosis, renal artery stenosis and phaeochromocytoma, learning difficulty and attention-deficit/hyperactivity disorder, and short stature. Each is treatable in its own right, and proactive surveillance prevents the secondary disability that neglect produces. [1] [10]
The most feared complication is malignant peripheral nerve sheath tumour, which arises within a pre-existing plexiform neurofibroma in 8 to 13 per cent of patients over a lifetime. Persistent pain, rapid growth, a change from soft to hard texture, and new neurological deficit are the warning signs, and prompt MRI, positron-emission tomography and biopsy are essential because early complete excision and oncological treatment determine survival. Missing this transformation is the single most dangerous error in NF1 care. [1] [9]
The cognitive traps are five. Assuming NF1 and NF2 are related, waiting for neurofibromas before making the diagnosis in a young child, attributing hypertension to essential causes without excluding renal artery stenosis, ordering routine brain MRI that generates incidentalomas and anxiety, and forgetting to cascade-test the family. Each costs marks in an examination and harms patients in practice. [2] [3]
Prognosis & Disposition
Life expectancy in NF1 is reduced by roughly 8 to 15 years on average, driven largely by malignant peripheral nerve sheath tumour and vascular disease, but most children with good surveillance and management live into adulthood with a good quality of life. Cognitive outcome is shaped by the severity of learning difficulty and attentional problems, which respond to educational and behavioural support more than to any medical therapy, and early intervention in the preschool years measurably improves school function and self-esteem. [1] [10]
NF2 carries a heavier burden because the bilateral vestibular schwannomas and associated tumours are inexorably progressive. Deafness is the dominant long-term outcome, and the goals of care shift over time toward hearing preservation and rehabilitation, facial-nerve function, and the cumulative effects of multiple tumours and surgeries. Modern microsurgery, radiosurgery and targeted agents have improved outcomes, but NF2 remains a condition that demands lifelong specialist care and proactive transition to adult services. [2] [3]
For both conditions, disposition is to a specialist multidisciplinary clinic that can coordinate genetics, ophthalmology, neurology, neurosurgery, oncology, developmental paediatrics and psychology. Transition to adult care is a structured process that should begin in early adolescence, because the fragmented handover of complex chronic disease is a well-recognised cause of harm. [9]
Special Populations
The same NF1 diagnosis behaves differently across populations because access and recognition are unevenly distributed. In remote and Indigenous communities, later presentation, limited genetic-service access, and lower rates of cascade testing mean that affected children and their relatives are diagnosed late, tumours are larger at detection, and culturally safe genetic counselling is essential to rebuild trust and engagement. In migrant, refugee and asylum-seeking families, language barriers, incomplete family histories and interrupted care make the multigenerational assessment that NF1 demands especially hard. [9]
In Australia and New Zealand, specialist neurofibromatosis clinics are concentrated in major centres, and children in rural and remote areas rely on visiting outreach or telehealth for surveillance. For Aboriginal and Torres Strait Islander and Maori whanau, engaging an Indigenous health worker and offering culturally appropriate genetic counselling improves uptake of cascade testing and long-term engagement. Families should be connected to the Neurofibromatosis Associations of Australia and New Zealand for peer and practical support.
Children with disability and neurodiversity make up a large share of the NF1 population, because learning difficulty and attention-deficit/hyperactivity disorder are core features, and their management needs an education plan, behavioural support and a strengths-based, neurodiversity-affirming frame. Children in out-of-home care or with socioeconomic disadvantage face fragmented records and missed surveillance, and they need a named coordinator to maintain continuity. [1]
Evidence, Guidelines & Regional Differences
The evidence base rests on three pillars: consensus diagnostic criteria, prospective natural-history and epidemiology studies, and the emerging targeted-therapy trials. The 2021 revised diagnostic criteria for NF1 and Legius syndrome, and the 2022 updated criteria and nomenclature for NF2 and the schwannomatoses, are the current international reference standards and have simplified diagnosis by allowing molecular confirmation as a standalone criterion. The Gutmann 2017 disease primer and the Asthagiri 2009 Lancet review set the comprehensive clinical and scientific framework for each condition. [5] [4]
The therapeutic evidence is dominated by the SPRINT trial of selumetinib, which showed that MEK inhibition can shrink inoperable plexiform neurofibromas and has transformed the systemic treatment of NF1. For NF2, evidence for bevacizumab and for hearing-preservation surgery guides practice, though randomised trials remain scarce and management continues to draw heavily on cohort data and expert consensus. The European Reference Network GENTURIS guidelines synthesise the surveillance framework adopted across much of Europe. [8] [6]
Regional differences are mainly in service delivery rather than biology. Australia and New Zealand follow RACP and RCPCH-aligned pathways through specialist NF clinics, North American practice is shaped by the National Cancer Institute NF Clinical Trials Consortium, and many low- and middle-income settings adapt the same surveillance principles with more limited access to molecular testing and MEK inhibitors. The principle is universal: coordinated, lifelong, family-aware care. [3] [9]
Exam Pearls
A fellowship candidate answering on neurofibromatosis should land these anchors and avoid these traps, then close with the family. The strongest answer names the gene and pathway, applies the diagnostic criteria precisely, sets out a surveillance plan tailored to the child's age, identifies the red flags for malignant transformation, and frames the whole discussion around autosomal-dominant inheritance and cascade testing. [1] [2]
References
- [1]Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers, 2017.PMID 28230061
- [2]Asthagiri AR, Parry DM, Butman JA, Kim HJ, Tsilou ET, Zhuang Z, Lonser RR. Neurofibromatosis type 2. Lancet, 2009.PMID 19476995
- [3]Plotkin SR, Wick A. Neurofibromatosis and Schwannomatosis. Semin Neurol, 2018.PMID 29548054
- [4]Plotkin SR, Messiaen L, Legius E, Pancza P, Avery RA, Hulf S, et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: an international consensus recommendation. Genet Med, 2022.PMID 35674741
- [5]Legius E, Messiaen L, Wolkenstein P, Pancza P, Avery RA, Berman Y, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med, 2021.PMID 34012067
- [6]Evans DG, Mostaccioli S, Pang D, Papi L, Huson SM, Smith MJ, et al. ERN GENTURIS clinical practice guidelines for the diagnosis, treatment, management and surveillance of people with schwannomatosis. Eur J Hum Genet, 2022.PMID 35361920
- [7]Fisher MJ, Loguidice M, Gutmann DH, Listernick RH, Fisher MJ, Pierz K, et al. Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy: a multicenter retrospective analysis. Neuro-oncology, 2012.PMID 22474213
- [8]Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N Engl J Med, 2020.PMID 32187457
- [9]Ferner RE, Bakker A, Elgersma Y, Fisher MJ, Gutmann DH, Korf BR, et al. From process to progress-2017 International Conference on Neurofibromatosis 1, Neurofibromatosis 2 and Schwannomatosis. Am J Med Genet A, 2019.PMID 30908866
- [10]Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Hamilton RL. Neurofibromatosis type 1 revisited. Pediatrics, 2009.PMID 19117870