Paeds · genetics-dysmorphology-and-metabolism
Noonan syndrome and RASopathies
Also known as Noonan syndrome · RASopathies · RAS/MAPK pathway syndromes · NSML (LEOPARD syndrome) · Costello syndrome · Cardio-facio-cutaneous syndrome · Male Turner syndrome
A fellowship approach to Noonan syndrome and the RASopathies: recognise the shared facio-cardio-cutaneous phenotype produced by gain-of-function germline variants in the RAS/MAPK pathway, distinguish Noonan syndrome from Noonan syndrome with multiple lentigines, Costello syndrome, cardio-facio-cutaneous syndrome, and neurofibromatosis type 1 by gene and cardiac profile, and apply a genotype-aware surveillance schedule anchored by an early echocardiogram.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A newborn is noted to have a Turner-like phenotype — webbed neck, low posterior hairline, widely spaced nipples — but the karyotype is 46,XX or 46,XY. The same infant may have a heart murmur that points to pulmonary valve stenosis rather than coarctation, or no murmur at all yet a thickened ventricular wall on ultrasound. The fellowship task is not merely to name the syndrome — the facies and the cardiac lesion begin to do that — but to place it within the RASopathy family, to confirm the gene, and to construct a surveillance plan that prevents the two preventable harms: undiagnosed cardiomyopathy and missed malignancy. [1] [7]
R.A.S.O.P.A.T.H.Y. — the syndromes on one pathway
Overview & Definition
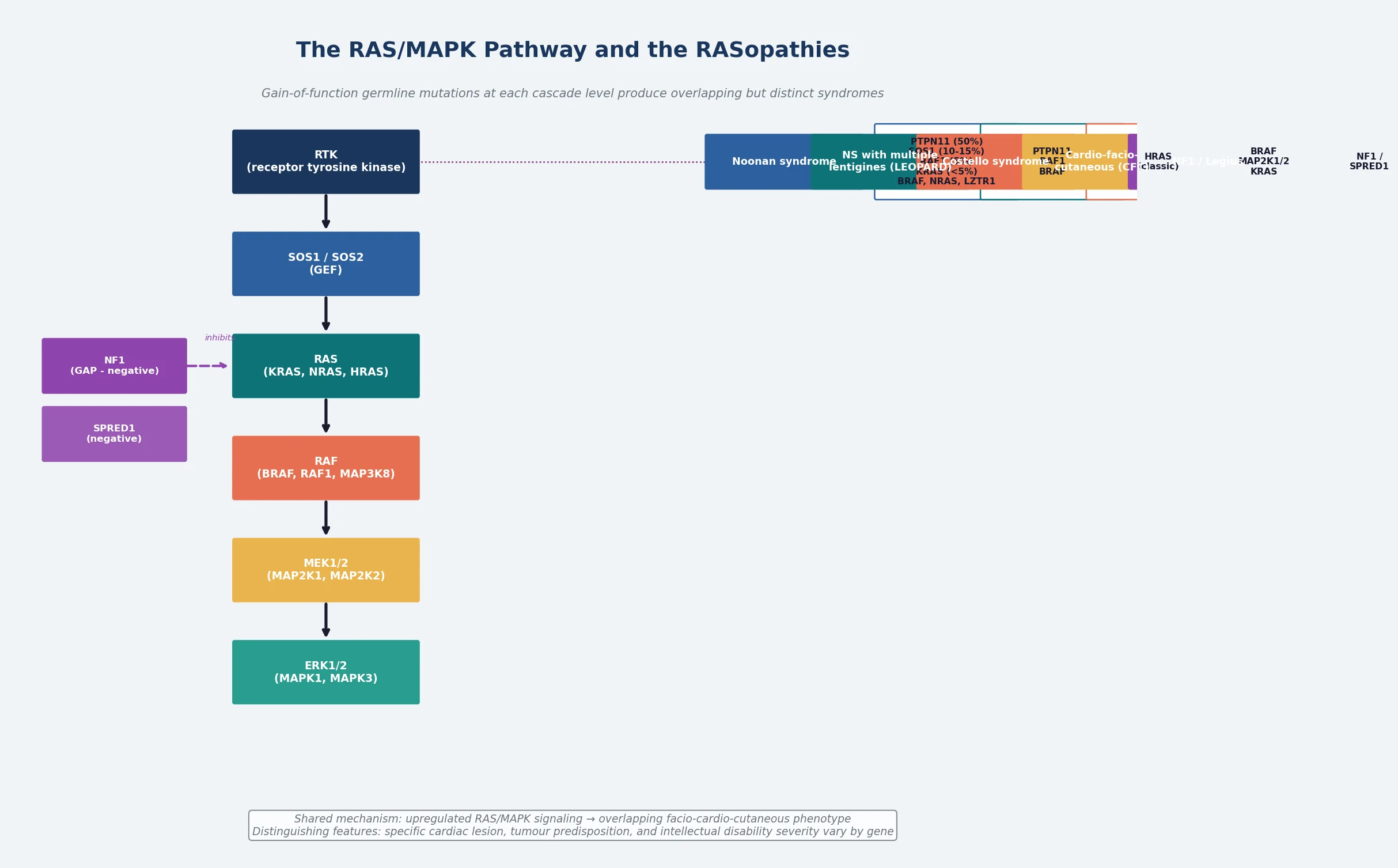
Noonan syndrome is the prototypical member of the RASopathies — a family of developmental disorders caused by germline gain-of-function variants in the RAS/MAPK mitogen-activated protein kinase signal transduction pathway. It is one of the most common syndromic causes of congenital heart disease encountered in general paediatric practice, with an estimated incidence of one in one thousand to one in two thousand five hundred live births. The face, the short stature, and the cardiac lesion make the diagnosis suspected at the bedside, but the confirmation is molecular and the gene matters — the gene predicts the cardiac profile, the tumour risk, and the genetic counselling. [1] [3]
The RASopathies share a unifying pathophysiology: upregulated RAS/MAPK signalling during embryonic development disrupts the carefully choreographed processes of cell proliferation, differentiation, migration, and survival. Because the pathway is active in nearly every tissue during development, the phenotype is multisystem, and because different genes operate at different levels of the same cascade, the syndromes overlap but retain discriminating features. Noonan syndrome itself is caused most often by variants in PTPN11 — encoding the SHP-2 phosphatase — but a substantial minority carry variants in SOS1, RAF1, KRAS, BRAF, NRAS, LZTR1, or other pathway genes. [2] [4]
The lifespan trajectory is shaped less by intellectual disability — which is usually mild or absent — and more by the cardiovascular and haematological risks that run through childhood and into adult life. Structured surveillance has improved outcomes, and the clinical task has shifted from making the diagnosis in the newborn to sustaining a surveillance net across the lifespan. A fellowship answer that lists features without linking them to the RAS/MAPK pathway reads as a catalogue; the pathway link is the teaching that holds the topic together. [1] [7]
Classification
The RASopathies are classified by the gene mutated within the RAS/MAPK cascade, and each gene produces a syndrome with an overlapping but distinct clinical signature. Noonan syndrome is the most common and the mildest; Costello syndrome and cardio-facio-cutaneous syndrome sit at the more severe end of the spectrum; Noonan syndrome with multiple lentigines, formerly LEOPARD syndrome, is distinguished by its lentigines and electrocardiographic abnormalities; and neurofibromatosis type 1, caused by NF1 loss-of-function, and Legius syndrome, caused by SPRED1 loss-of-function, share the pathway because their gene products normally down-regulate RAS signalling. [1] [4]
Within Noonan syndrome itself, genotype-phenotype correlations are clinically useful even though they are not absolute. PTPN11 variants, found in approximately fifty per cent of cases, are strongly associated with pulmonary valve stenosis and a bleeding diathesis but are relatively protective against hypertrophic cardiomyopathy. RAF1 variants, found in five to ten per cent, are associated with hypertrophic cardiomyopathy and a higher risk of arrhythmia. SOS1 variants are associated with more prominent ectodermal features and a relatively favourable neurocognitive outcome. KRAS and BRAF variants are rarer and tend to produce a more severe cognitive phenotype. LZTR1 variants are an increasingly recognised cause and may be inherited recessively in some families. [1] [4]
The reason a multigene RASopathy panel is preferred over single-gene PTPN11 testing is that phenotypic overlap makes clinical prediction unreliable. A child with a suspected RASopathy may carry a variant in any of a dozen genes, and the specific gene changes the counselling — RAF1 demands more intensive cardiac surveillance, HRAS suggests Costello syndrome and a substantial tumour risk, and NF1 or SPRED1 changes the diagnostic criteria entirely. A panel that covers the full pathway, interpreted with the ClinGen RASopathy Expert Panel specifications for variant classification, is the standard approach. [5] [3]
Epidemiology & Risk Factors
Noonan syndrome occurs in approximately one in one thousand to one in two thousand five hundred live births, making it one of the most common single-gene syndromic causes of congenital heart disease. Unlike Down syndrome, it is not associated with advancing maternal age, because the causative variants are germline and autosomal dominant, and the majority arise as de novo mutations in families with no prior history. An affected parent transmits the condition to half of their children, and familial Noonan syndrome with vertical transmission through several generations is well described. [1] [3]
The prevalence is likely underestimated because mildly affected individuals — particularly those with subtle facial features and no cardiac lesion — may never come to medical attention, and the diagnosis in a parent is often made only after a more severely affected child is born. Population-level data are limited because Noonan syndrome is not included in most newborn screening programmes and does not have a chromosomal signature detectable on routine karyotype or microarray. The clinical consequence is that a diagnosis made in a child should prompt examination and genetic testing of both parents, because an affected parent changes the recurrence risk from the de novo background to fifty per cent. [1]
The risk factors for individual complications, separate from the molecular diagnosis, are the risk factors the surveillance schedule is built to modify. A child with an undiagnosed hypertrophic cardiomyopathy is at risk of sudden cardiac death; a child with a bleeding diathesis is at risk of perioperative haemorrhage; a child with Costello syndrome is at risk of malignancy. Identifying these risks early through genotype-aware surveillance is the epidemiology that matters at the bedside. [7] [8]
Pathophysiology
The pathophysiology of the RASopathies is a signal gain problem. The RAS/MAPK cascade is a fundamental signal transduction pathway that transmits extracellular growth and differentiation signals from receptor tyrosine kinases at the cell surface to the nucleus, where they regulate gene expression governing cell proliferation, differentiation, survival, and migration. Gain-of-function germline variants in pathway components lead to constitutive or hyperactive signalling during embryogenesis, and the resulting dysregulation of cellular processes produces the multisystem developmental phenotype. [2] [1]
At the molecular level, the pathway flows from receptor tyrosine kinases through the adaptor protein complex to SOS1 — a guanine nucleotide exchange factor that activates RAS by promoting the exchange of GDP for GTP — then through the RAF kinases (BRAF, RAF1) to MEK and ERK, the terminal kinases that enter the nucleus to regulate transcription. PTPN11 encodes SHP-2, a protein tyrosine phosphatase that positively regulates the pathway upstream of RAS, and its gain-of-function variants are the commonest cause of Noonan syndrome. NF1 encodes neurofibromin, a GTPase-activating protein that negatively regulates RAS by accelerating its return to the inactive GDP-bound state; loss-of-function in NF1 causes neurofibromatosis type 1, which is a RASopathy by virtue of the same pathway dysregulation. [2] [4]
The cardiac pathophysiology is distinctive and high-yield. Gain-of-function signalling in cardiac neural crest and secondary heart field cells produces the characteristic congenital heart defects: pulmonary valve stenosis with dysplastic leaflets, hypertrophic cardiomyopathy, atrioventricular septal defects, and septal defects. The gene determines the dominant lesion — PTPN11 drives pulmonary stenosis, RAF1 drives hypertrophic cardiomyopathy — and the cardiac lesion in turn determines the surveillance intensity and the timing of intervention. A fellowship candidate who links the gene to the cardiac lesion demonstrates the depth that the examiners expect. [7] [8]
The haematological pathophysiology reflects the same pathway gain. Somatic PTPN11, KRAS, and NRAS variants are among the commonest driver mutations in juvenile myelomonocytic leukaemia, and children with germline RASopathy variants carry an elevated risk of myeloproliferative disorders and, to a lesser extent, other haematological malignancies. The bleeding diathesis in Noonan syndrome is multifactorial, involving factor XI deficiency, platelet dysfunction, and coagulation factor deficiencies, and it is the reason every child with a RASopathy needs a coagulation assessment before surgery or dental extraction. [1] [9]

Clinical Presentation
The clinical presentation of Noonan syndrome depends on the age at which the child is seen and on the specific gene involved, and a fellowship answer earns depth by handling each life stage separately rather than collapsing the syndrome into a single newborn snapshot. The diagnosis may be suspected antenatally when increased nuchal translucency, hydrops, or a cardiac anomaly is detected on prenatal ultrasound, or it may declare itself only when short stature or a learning difficulty brings a school-age child to attention. [1] [3]
In the newborn and infant, the facial gestalt is the dominant clue: a triangular face with a broad forehead, hypertelorism with downslanting palpebral fissures, ptosis, a short nose with a depressed bridge, a deeply grooved philtrum, and low-set, posteriorly rotated ears with a thickened helix. The neck is short with redundant skin folds or webbing, the posterior hairline is low, the chest is broad with widely spaced nipples, and there may be a sternal deformity — pectus carinatum superiorly and pectus excavatum inferiorly. None of these signs is individually diagnostic, but their pattern is, and the combination of the facies with a cardiac lesion makes the diagnosis highly likely. [1] [3]
Cardiac presentation dominates the early weeks. Pulmonary valve stenosis, the commonest lesion, may produce a systolic murmur at the upper left sternal border, but the valve is often dysplastic rather than simply stenotic, and surgical valvotomy or balloon valvuloplasty may be needed. Hypertrophic cardiomyopathy, the second commonest lesion, may be entirely asymptomatic in the newborn yet carry a risk of dynamic left ventricular outflow tract obstruction, arrhythmia, and sudden death, and it is the reason an echocardiogram is mandatory in every infant regardless of how well the baby looks. Atrial and ventricular septal defects, atrioventricular septal defects, and coarctation of the aorta complete the cardiac spectrum. [7] [8]
Across infancy and childhood, the presentation shifts to growth and development. Birth weight is often normal, but postnatal growth decelerates and stature tracks below the third centile on standard charts, which is why Noonan-specific growth charts are essential. Motor milestones are delayed by a combination of hypotonia and joint hyperextensibility, and a variable degree of learning difficulty or intellectual disability becomes apparent through the school years, though many children attend mainstream school with support. Feeding difficulty, gastro-oesophageal reflux, and constipation are common in infancy, and easy bruising or prolonged bleeding after minor procedures may reveal the coagulation abnormality. [3] [6]
Differential Diagnosis
The differential diagnosis of a suspected RASopathy splits into two questions: what else produces the Turner-like phenotype with a normal karyotype, and which RASopathy is it. Most of the time the pattern and the gene test settle both questions, but the discriminating skill is to know when the clinical picture alone is insufficient and the panel is mandatory. [1] [3]
Turner syndrome is the classical differential. The webbed neck, low posterior hairline, widely spaced nipples, short stature, and cardiac lesion are shared, but Turner syndrome occurs only in females, the karyotype shows complete or partial monosomy X, and the cardiac lesion is left-sided — coarctation of the aorta and bicuspid aortic valve — rather than the right-sided lesions of Noonan syndrome. A karyotype in any child with a Turner-like phenotype and a normal sex chromosome complement redirects the diagnosis to Noonan syndrome or another RASopathy. [1]
Noonan syndrome must be distinguished from the other RASopathies. Costello syndrome shares the facies and the cardiac lesion but adds severe feeding difficulty and failure to thrive in infancy, deep palmar and plantar creases, papillomata around the nose and mouth, and a markedly elevated tumour risk; it is caused by HRAS variants. Cardio-facio-cutaneous syndrome shares the facies and the cardiac lesion but adds more severe developmental delay, characteristic sparse and curly hair, and skin abnormalities including ichthyosis and keratosis pilaris; it is caused by BRAF, MAP2K1, MAP2K2, or KRAS variants. Noonan syndrome with multiple lentigines, formerly LEOPARD syndrome, is distinguished by the development of diffuse lentigines in childhood and electrocardiographic conduction abnormalities; it is caused by PTPN11 or RAF1 variants. [4] [9]
The trap that costs marks is ordering a single-gene test — typically PTPN11 alone — rather than a multigene RASopathy panel. Single-gene testing misses the substantial minority of cases caused by SOS1, RAF1, KRAS, BRAF, LZTR1, or other genes, and it fails to identify the rarer syndromes whose management differs. The safe habit is a panel that covers the full pathway, ordered through a clinical genetics service with the ClinGen RASopathy variant interpretation specifications in mind. [5] [3]
| Feature | Noonan syndrome | Costello syndrome | CFC syndrome | NF1 |
|---|---|---|---|---|
| Dominant gene | PTPN11 (50%) | HRAS (>80%) | BRAF (~75%) | NF1 |
| Cardiac lesion | Pulmonary stenosis, HCM | HCM, arrhythmia | HCM, PS, ASD/VSD | None typical |
| Intellectual disability | Mild or none | Moderate-severe | Moderate-severe | Usually none |
| Tumour risk | Low (JMML) | High (rhabdomyosarcoma) | Low | High (MPNST, glioma) |
| Distinguishing feature | Classic Noonan facies | Papillomata, deep creases | Curly hair, ichthyosis | Cafe-au-lait, neurofibromas |
Clinical & Bedside Assessment
The bedside assessment of a child with a suspected or confirmed RASopathy is structured around the multisystem comorbidity map rather than a single complaint, because the point of the visit is to run the surveillance net as well as to address the presenting problem. Plot every child on Noonan-specific growth charts, because standard charts will mislabel normal growth as faltering. A complete assessment covers cardiac, growth, developmental, haematological, musculoskeletal, ophthalmic, and auditory systems at every relevant visit. [3] [1]
The history explores feeding and growth in infancy, developmental milestones and school progress, exercise tolerance and any syncopal episodes (which raise concern for arrhythmia or outflow tract obstruction), bleeding or bruising tendency, and any new neurological symptoms. A careful three-generation family history identifies affected relatives, because vertical transmission through a mildly affected parent is common, and parental examination may reveal an unrecognised diagnosis. Ask directly about sudden cardiac events in the family, because hypertrophic cardiomyopathy in an affected parent may have gone undiagnosed. [3] [7]
Examination begins with growth and the facial gestalt and moves through the systems. Listen carefully to the heart, recognising that pulmonary stenosis produces a systolic murmur at the upper left sternal border while hypertrophic cardiomyopathy may produce no murmur or a dynamic ejection murmur that changes with manoeuvres. Examine for the characteristic skeletal features — pectus deformity, wide-spaced nipples, cubitus valgus, clinodactyly — and the dermatological features including lentigines in Noonan syndrome with multiple lentigines, cafe-au-lait patches in NF1 or Legius syndrome, and papillomata or deep creases in Costello syndrome. A brief developmental assessment screens for the learning difficulties that are common but variable. [1] [8]
Assess development with a standardised tool appropriate to the child's age, and interpret the result against RASopathy-specific expectations. Motor delay driven by hypotonia and joint hyperextensibility is expected in early childhood; a language or learning difficulty is common but usually mild in Noonan syndrome and more significant in Costello and CFC syndromes. Regression or loss of skills is not expected and demands investigation for a complication. The bleeding assessment is mandatory and should document any history of bruising, epistaxis, prolonged bleeding after procedures, or menorrhagia, and trigger a coagulation screen before any invasive intervention. [3] [6]
Investigations
The investigation plan for a suspected RASopathy is a two-track process: molecular confirmation and system-specific surveillance. Molecular confirmation is by multigene panel testing of the RAS/MAPK pathway, which should be ordered through a clinical genetics service and interpreted using the ClinGen RASopathy Expert Panel variant specifications. The system-specific surveillance follows the genotype, because the gene determines which complications to screen for and how intensively. [5] [3]
Molecular testing should not be delayed by waiting for the clinical picture to mature, because an early genetic diagnosis enables targeted surveillance from the newborn period. A multigene RASopathy panel — typically covering PTPN11, SOS1, SOS2, RAF1, BRAF, KRAS, NRAS, MAP2K1, MAP2K2, HRAS, LZTR1, RIT1, NF1, and SPRED1 — is the standard first-line test. If the panel is negative but the clinical suspicion is high, exome or genome sequencing may identify rarer pathway genes. In families with an affected parent and a known familial variant, targeted single-variant testing may be sufficient. [5] [1]
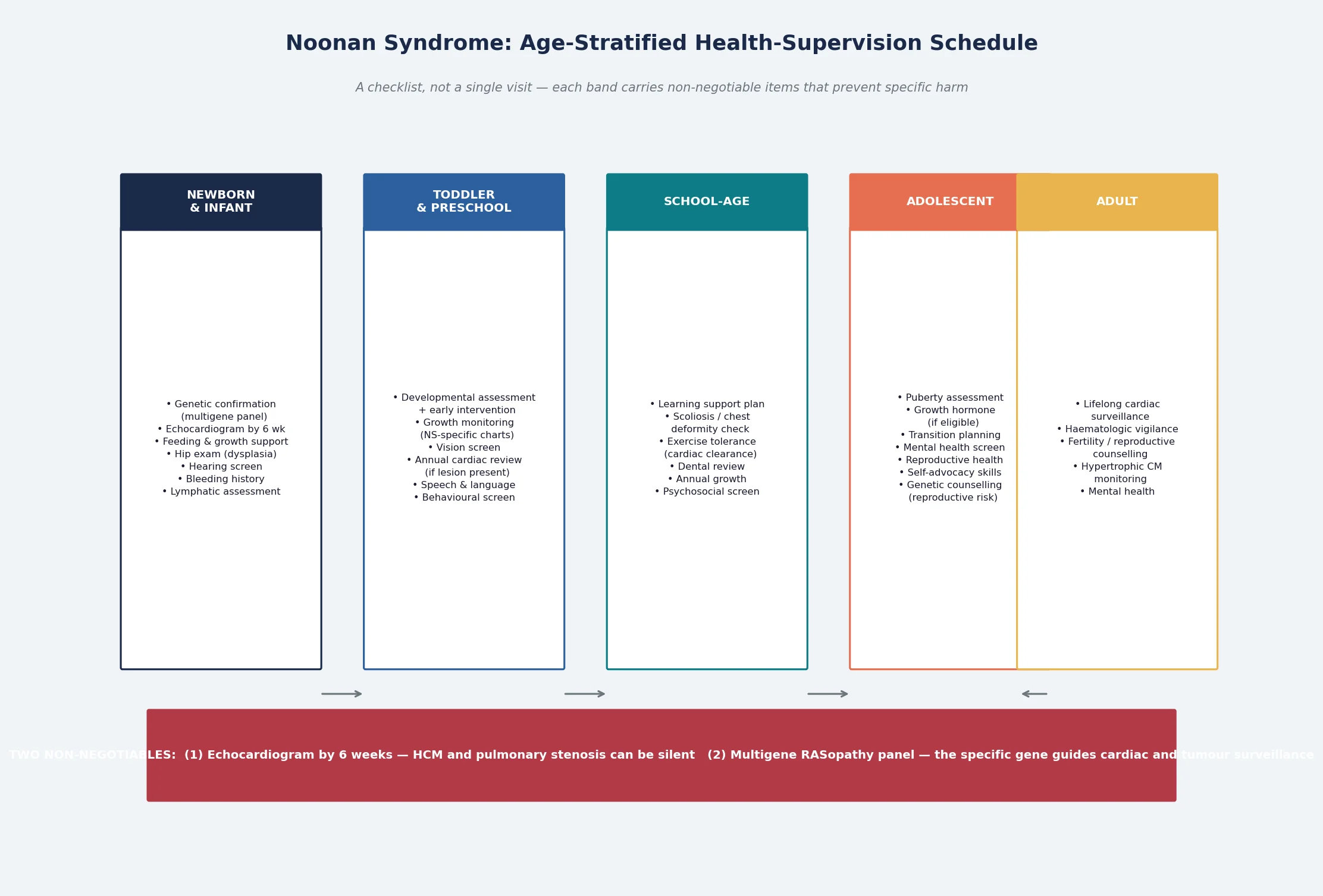
Cardiac investigation is an echocardiogram for every infant, performed by six weeks of age and regardless of symptoms, because hypertrophic cardiomyopathy can be silent. The echocardiogram defines the cardiac lesion — pulmonary stenosis, hypertrophic cardiomyopathy, septal defects — and establishes a baseline for serial follow-up. An electrocardiogram screens for conduction abnormalities and pre-excitation patterns. Children with a cardiac lesion require cardiology follow-up at a frequency determined by the lesion and its severity, and children with hypertrophic cardiomyopathy require ongoing surveillance for arrhythmia and outflow tract obstruction throughout childhood and into adult life. [7] [8]
Haematological investigation begins with a coagulation screen — prothrombin time, activated partial thromboplastin time, and factor XI level — in every child, because the bleeding diathesis is common and may be clinically silent until a procedure exposes it. A full blood count with a blood film screens for myeloproliferative features, and children with Costello syndrome, who carry the highest tumour risk, require periodic abdominal and pelvic ultrasound for rhabdomyosarcoma and neuroblastoma surveillance, and urinalysis for catecholamines. Growth hormone stimulation testing may be indicated in children with documented growth failure, and developmental assessment with a standardised tool quantifies the cognitive profile. Vision and hearing screening complete the baseline work-up. [3] [9]
Management — Resuscitation
Resuscitation in a newborn or infant with a RASopathy means managing the acute cardiac presentations that bring the child to attention in the first days and weeks of life. Two scenarios dominate: symptomatic hypertrophic cardiomyopathy with cardiovascular compromise, and severe pulmonary valve stenosis with right-sided heart failure. Both demand prompt recognition, cardiology involvement, and a defined pathway. [7] [8]
A newborn with hypertrophic cardiomyopathy may present with respiratory distress, poor feeding, and signs of low cardiac output as the dynamic left ventricular outflow tract obstruction worsens. Management is medical in the first instance — beta-blockade to reduce the dynamic obstruction and control the heart rate, fluid management to maintain preload, and avoidance of inotropes that worsen the obstruction. Intervention — surgical myectomy or, in selected cases, pharmacological modulation of the signalling pathway — follows cardiology assessment, and the child requires ongoing monitoring for arrhythmia. [7]
Severe pulmonary valve stenosis may present in the neonate with a loud murmur, right ventricular hypertrophy on echocardiography, and signs of right heart failure. Balloon valvuloplasty is the first-line intervention for membranous stenosis, but Noonan syndrome pulmonary valves are often dysplastic and thickened, which reduces the success rate of balloon valvuloplasty and may necessitate surgical valvotomy. The key teaching point is that the pulmonary valve in Noonan syndrome is anatomically distinct from the typical stenotic valve, and the intervention must be tailored to the valve morphology. [7] [8]
A neonate with Costello syndrome may present with severe feeding difficulty and failure to thrive, refractory hypoglycaemia, cardiac arrhythmia — supraventricular tachycardia is characteristic — and hypertrophic cardiomyopathy. The early mortality is substantial, and the management is supportive and intensive: nasogastric or gastrostomy feeding, arrhythmia control, cardiac surveillance, and early involvement of the multidisciplinary team. The recognition of Costello syndrome in the neonatal period enables the tumour surveillance that is the other pillar of management in this high-risk condition. [9] [4]
Management — Definitive & Stepwise
Definitive management of Noonan syndrome and the RASopathies is a genotype-aware, age-stratified health-supervision schedule, and it is the core of fellowship-level competence. The schedule moves with the child from the newborn period through infancy, childhood, adolescence, and adulthood, and each band carries items that prevent specific, predictable harm. The gene determines the intensity of surveillance: RAF1 and HRAS demand more intensive cardiac and tumour monitoring, while a PTPN11-positive Noonan syndrome with a normal echocardiogram may follow a lighter schedule. [3] [1]
In the newborn period, the priorities are molecular confirmation by multigene panel, an echocardiogram by six weeks, a coagulation screen, feeding support, hip examination for developmental dysplasia, hearing screening, and a baseline developmental assessment. The family is given honest, strengths-based information and connected to a Noonan syndrome or RASopathy support organisation. Both parents should be examined and offered genetic testing, because an affected parent changes the recurrence risk from the de novo background to fifty per cent. [1] [3]
Across infancy and childhood, the schedule layers growth monitoring on Noonan-specific charts, developmental and educational support with early intervention referral, annual or biennial cardiology review determined by the cardiac lesion, vision and hearing screening, dental review, and a symptom review for exercise intolerance, palpitation, or syncope. Growth hormone therapy is licensed for Noonan syndrome in many jurisdictions and produces a modest but significant improvement in adult height; it should be initiated by a paediatric endocrinologist after a careful cardiac assessment, because there is a theoretical concern about worsening hypertrophic cardiomyopathy. [6]
Adolescence brings transition planning, reproductive and menstrual health, mental health screening, and increasing attention to self-advocacy. The transition to adult care should be planned and documented well before the eighteenth birthday, with a structured handover that preserves continuity of cardiac, haematological, and developmental surveillance. In adulthood, the schedule adds lifelong cardiac surveillance for those with cardiomyopathy, reproductive counselling on the fifty per cent transmission risk, and ongoing attention to psychosocial wellbeing. The schedule does not end at twenty-one; it changes shape. [1] [6]
Specific Subtypes & Scenarios
Each major RASopathy carries a distinctive management point, and a fellowship answer earns depth by handling them individually rather than as a single block. The surveillance generalises, but the management is syndrome-specific. [1] [4]
The cardiac scenario is the defining feature of the RASopathies and the reason for the echocardiogram by six weeks. In Noonan syndrome, pulmonary valve stenosis is managed by interventional cardiology — balloon valvuloplasty for membranous stenosis, surgical valvotomy for dysplastic valves — and hypertrophic cardiomyopathy is managed by beta-blockade, surveillance for arrhythmia, and, in selected cases, surgical myectomy. Children with RAF1 variants and hypertrophic cardiomyopathy require the most intensive cardiac surveillance, because their cardiomyopathy carries a higher risk of progression and sudden death. The principle is that the gene predicts the cardiac lesion, the cardiac lesion predicts the risk, and the risk determines the surveillance intensity. [7] [8]
The growth scenario centres on the use of growth hormone. Noonan syndrome is one of the few genetic syndromes for which growth hormone therapy is licensed, and trials demonstrate a modest but significant improvement in adult height — typically adding five to ten centimetres to the final height prediction. The decision to treat should be made by a paediatric endocrinologist, after a careful cardiac assessment and a discussion of the benefits and risks with the family. Growth hormone should be used cautiously in children with hypertrophic cardiomyopathy, and the cardiac status should be monitored during treatment. [6]
The haematological and oncological scenario is dominated by the risk of juvenile myelomonocytic leukaemia and other malignancies. Children with PTPN11 variants carry an elevated risk of juvenile myelomonocytic leukaemia, and any persistent monocytosis, hepatosplenomegaly, or cytopenia warrants urgent haematology assessment. Children with Costello syndrome carry the highest tumour risk in the RASopathy family — rhabdomyosarcoma, neuroblastoma, and bladder carcinoma — and require periodic abdominal and pelvic ultrasound and urinalysis for catecholamines. The principle is that the gene determines the tumour risk, and the tumour risk determines the surveillance schedule. [9] [1]
The bleeding scenario demands a pre-procedure coagulation assessment in every child with a RASopathy. Factor XI deficiency, platelet dysfunction, and combined abnormalities are all common, and the bleeding may be severe enough to require factor replacement or platelet transfusion before surgery. A haematology referral should be made when the coagulation screen is abnormal, and the family should be advised to inform any treating clinician of the bleeding tendency before any procedure. [3] [1]
Why Costello syndrome demands the most aggressive tumour surveillance
Costello syndrome, caused by HRAS gain-of-function variants, carries the highest tumour predisposition of any RASopathy. Approximately fifteen to twenty per cent of affected children develop a malignancy, most commonly rhabdomyosarcoma (particularly pelvic or abdominal), neuroblastoma, and bladder carcinoma. The cumulative risk is high enough to justify routine surveillance — abdominal and pelvic ultrasound every three to six months until age eight to ten, and urinalysis for catecholamine metabolites — because early detection of these tumours improves outcome. The molecular basis is the constitutive activation of the RAS/MAPK pathway that drives uncontrolled cell proliferation, and the senescence response to HRAS gain-of-function paradoxically creates a permissive environment for additional oncogenic hits. [9] [4]
Complications & Pitfalls
The complications of the RASopathies are the comorbidities left undetected, and the pitfalls are the assumptions that lead clinicians to miss them. A fellowship answer handles both, because the harm is rarely the molecular diagnosis itself — it is the preventable complication that the surveillance schedule was designed to catch. [1] [3]
Cardiac complications dominate the infant and childhood period. Undiagnosed hypertrophic cardiomyopathy carries a risk of sudden cardiac death from dynamic outflow tract obstruction or ventricular arrhythmia, and the pitfall is assuming that a well-looking infant with no murmur does not need an echocardiogram. The safeguard is the echocardiogram by six weeks in every infant with a confirmed or suspected RASopathy, followed by serial echocardiography determined by the cardiac lesion and the genotype. Pulmonary valve stenosis that is managed expectantly may progress, and the pitfall is failing to re-evaluate a child with a known lesion at the intervals the cardiology service recommends. [7] [8]
Haematological and oncological complications run through childhood. Juvenile myelomonocytic leukaemia presents with monocytosis, hepatosplenomegaly, and cytopenia, and the pitfall is attributing the blood count abnormality to a benign reactive process. Rhabdomyosarcoma in Costello syndrome presents with a mass, and the pitfall is failing to image a child with an abdominal or pelvic mass because the tumour risk was not recognised. The safeguard is genotype-aware tumour surveillance in the high-risk syndromes and a low threshold for haematology referral in any child with unexplained blood count abnormalities. [9] [1]
Bleeding complications are perioperative rather than spontaneous in most cases, and the pitfall is proceeding with surgery or dental extraction without a coagulation assessment. The safeguard is a pre-procedure coagulation screen and factor XI level in every child with a confirmed RASopathy, with haematology involvement when the screen is abnormal. [1] [3]
Developmental and psychosocial complications include learning difficulties, behavioural differences, and, in adolescence and adulthood, anxiety, depression, and challenges with social participation. The pitfall is attributing all functional difficulties to the underlying syndrome and missing treatable contributors such as sleep-disordered breathing, thyroid dysfunction, or unaddressed sensory impairment. The safeguard is a holistic assessment at every visit and the involvement of psychology and educational support services when indicated. [3] [6]
Prognosis & Disposition
The prognosis of Noonan syndrome has improved across a generation, driven by early genetic diagnosis, structured cardiac surveillance, and improved management of the complications. Life expectancy in Noonan syndrome with appropriate surveillance approaches that of the general population, and most affected individuals live independent or semi-independent adult lives with varying degrees of educational and psychosocial support. The prognosis in Costello syndrome and cardio-facio-cutaneous syndrome is more guarded, with higher early mortality from cardiac complications and malignancy, and a greater need for lifelong support. [1] [9]
The determinants of prognosis are the cardiac lesion, the tumour risk, and the developmental profile, and all three are shaped by the specific gene. A child with Noonan syndrome and a normal echocardiogram, a PTPN11 variant, and no developmental concerns follows a trajectory close to population norms. A child with RAF1-associated hypertrophic cardiomyopathy, or with Costello syndrome and its tumour predisposition, follows a more intensive and more uncertain trajectory. The prognosis is therefore not fixed by the family name of the syndrome; it is shaped by the gene and by the quality of the surveillance. [7] [8]
Quality of life is shaped as much by educational and psychosocial factors as by medical ones. Mainstream schooling with learning support, family support, employment, and meaningful social participation are achievable for most individuals with Noonan syndrome, and the fellowship answer frames prognosis in those terms rather than in deficit language alone. The disposition plan should support the developmental and psychosocial trajectory as actively as the medical surveillance. [3] [6]
Disposition for a general paediatrician is shared, structured care. The paediatrician owns the coordination of subspecialty input, the developmental and educational support, and the transition to adult care. Clinical genetics, cardiology, endocrinology, haematology or oncology, ophthalmology, and audiology each contribute at the relevant point, and a named coordinator prevents the fragmentation that is the enemy of a genotype-aware surveillance schedule. Early referral to a structured RASopathy or genetics clinic, where one exists, supports the family from the point of diagnosis. [1] [3]
Special Populations
The RASopathies interact with the child's social, cultural, and developmental context, and the same molecular diagnosis behaves differently across populations. Access, adherence, and late presentation all shape outcome, and a fellowship answer recognises that the surveillance schedule is only as good as the family's ability to engage with it. [3] [1]
Indigenous children, particularly in Australia and New Zealand, may face a higher background burden of rheumatic heart disease, otitis media, and developmental disadvantage, alongside reduced access to specialist genetic and cardiac services in remote communities. These factors intensify the need for early, structured surveillance and for a low threshold to investigate symptoms. Telehealth and outreach services extend the surveillance net into communities that a clinic-based model would miss, and the collaboration between general paediatric and genetic services is essential to ensure that a molecular diagnosis made in a major centre is translated into a surveillance plan that reaches the community. [8]
Migrant, refugee, and asylum-seeking families may have had no antenatal screening, may arrive with an unrecognised diagnosis, and may face language and trauma barriers to engaging with a complex surveillance schedule. A careful reconstruction of the history, confirmation of the diagnosis and the gene, an interpreter-mediated explanation, and a written schedule in the family's language are the foundations. Genetic counselling must be culturally sensitive, and the family should be connected to support services that understand their circumstances. [1]
Socioeconomically disadvantaged families carry the burden that adherence to a multi-specialist schedule is harder, and the limiting step is often attendance rather than the medicine. Structuring the schedule around a single coordinated visit, providing written and visual materials, and linking the family to a support organisation and to transport and appointment support all improve engagement. The aim is to fit the surveillance to the family's reality rather than the reverse. [3]
Adolescents and adults in transition are a population in their own right. The handover to adult care is a vulnerable point, because the structured paediatric surveillance ends and adult services may not inherit the schedule. A planned, documented transition that transfers the checklist into adult medicine, preserves the cardiac and reproductive counselling, and addresses mental health, fertility, and psychosocial wellbeing is the safeguard. The schedule does not stop at the transition; it changes hands. [1] [6]
Evidence, Guidelines & Regional Differences
The evidence base for the RASopathies rests on landmark molecular discoveries, the Romano clinical practice guidelines published in Pediatrics, and the ClinGen RASopathy Expert Panel consensus specifications for variant interpretation. The Roberts review in the Lancet remains the framing reference for Noonan syndrome, organising the genetics, the clinical features, and the management in a way that a fellowship answer is built on. [1] [3]
The Tartaglia discovery of PTPN11 mutations in Noonan syndrome in 2001 was the molecular founding event of the field, establishing the link between Noonan syndrome and the RAS/MAPK pathway and opening the door to the molecular dissection of the entire RASopathy family. Subsequent discoveries — SOS1, RAF1, KRAS, BRAF, HRAS, NRAS, LZTR1, RIT1 — expanded the pathway map and refined the genotype-phenotype correlations that guide surveillance today. [2] [4]
The ClinGen RASopathy Expert Panel has produced standardised, evidence-based specifications for the interpretation of variants in RASopathy genes, addressing the challenge that many pathway variants are rare, that the same variant may be found in different syndromes, and that the distinction between a pathogenic gain-of-function variant and a benign polymorphism requires careful assessment. The specifications are the operational document that clinical genetics services use when interpreting a multigene panel result, and a fellowship candidate should be aware of their existence and their role in ensuring consistent, evidence-based variant classification. [5]
Regional differences are practical rather than scientific. Australia and New Zealand apply the international surveillance frameworks with access to multigene panel testing through clinical genetics services, structured cardiac surveillance through paediatric cardiology, and telehealth and outreach to extend the schedule into rural and remote communities. Indigenous-health considerations intensify the need for early respiratory and cardiac surveillance, and the funding and organisation of subspecialty access shape how the checklist is delivered. The principles are constant; the delivery adapts to the setting. [8] [1]
In Australia and New Zealand, every infant with a suspected or confirmed RASopathy receives an echocardiogram by six weeks, a multigene RASopathy panel through a clinical genetics service, and a coagulation screen before any invasive procedure. Structured cardiac surveillance is delivered through paediatric cardiology services in the major centres, and telehealth extends the genetics and developmental follow-up into rural and remote communities. Growth hormone therapy for Noonan syndrome is available through the Pharmaceutical Benefits Scheme under specialist authorisation, and transition to adult care is increasingly structured through transition clinics. Indigenous-health considerations prompt earlier and more intensive cardiac and respiratory surveillance in affected children from remote communities. [1] [7]
Exam Pearls
A fellowship candidate answering on Noonan syndrome and the RASopathies should land five anchor points and avoid four classic traps. The anchors are the framework examiners listen for; the traps are where candidates lose easy marks. [1] [3]
Anchor one: the RAS/MAPK pathway unifies the family. Noonan syndrome, Noonan syndrome with multiple lentigines, Costello syndrome, cardio-facio-cutaneous syndrome, neurofibromatosis type 1, and Legius syndrome are all caused by germline variants in the same signal transduction cascade. The shared phenotype — characteristic facies, short stature, congenital heart disease, variable developmental delay — is explained by the shared pathway, and the discriminating features are explained by the specific gene and its level in the cascade. [2] [4]
Anchor two: the heart comes first. Pulmonary valve stenosis and hypertrophic cardiomyopathy are the two cardiac lesions that define the RASopathies, and an echocardiogram by six weeks in every infant is the non-negotiable safeguard. Hypertrophic cardiomyopathy can be silent yet lethal, and RAF1 variants carry the highest risk. [7] [8]
Anchor three: the gene determines the tumour risk. Costello syndrome carries the highest tumour predisposition, followed by NF1, and the surveillance schedule must be tailored accordingly. A child with an unrecognised Costello syndrome who does not receive tumour surveillance is at risk of a late-presenting, preventable malignancy. [9]
Anchor four: the bleeding diathesis is real and perioperative. One third to one half of children with Noonan syndrome have a coagulation abnormality, and a pre-procedure coagulation screen and factor XI level are mandatory. The safeguard is simple, and its omission is an avoidable harm. [1] [3]
Anchor five: confirm with a multigene panel, not a single gene. The clinical phenotype cannot reliably predict the mutated gene, and single-gene testing misses the substantial minority of cases caused by genes other than PTPN11. A panel interpreted with the ClinGen specifications is the standard approach, and it should be ordered early rather than after a period of single-gene testing. [5]
The four traps to avoid are ordering PTPN11 alone, reassuring a well newborn without an echocardiogram, proceeding with surgery without a coagulation screen, and failing to recognise the different tumour risks across the RASopathy family. Avoid these and the rest of the answer falls into place. [1] [3]
References
- [1]Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet, 2013.PMID 23312968
- [2]Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet, 2001.PMID 11704759
- [3]Romano AA, Allanson JE, Dahlgren J, et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics, 2010.PMID 20876176
- [4]Sarkozy A, Carta C, Moretti S, et al. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular diversity and associated phenotypic spectrum. Hum Mutat, 2009.PMID 19206169
- [5]Gelb BD, Cave H, Dillon MW, et al. ClinGen's RASopathy Expert Panel consensus methods for variant interpretation. Genet Med, 2018.PMID 29493581
- [6]Romano AA. Growth and growth hormone treatment in Noonan syndrome. Pediatr Endocrinol Rev, 2019.PMID 31115197
- [7]Zenker M, Dikow N, Faul C, et al. Cardiovascular aspects of Noonan syndrome and related disorders. Med Genet, 2025.PMID 40207038
- [8]Ichikawa Y, Kono K, Kotani T, et al. Cardiac features of Noonan syndrome in Japanese patients. Cardiol Young, 2023.PMID 35475426
- [9]Niihori T, Aoki Y, Narumi Y, et al. HRAS mutants identified in Costello syndrome patients can induce cellular senescence: possible implications for the pathogenesis of benign tumours. J Hum Genet, 2011.PMID 21850009