Paeds · genetics-dysmorphology-and-metabolism
Organic acidaemias
Also known as Organic acidurias · Propionic acidaemia · Methylmalonic acidaemia · Isovaleric acidaemia · Glutaric aciduria type 1 · Branched-chain organic acidaemias
A fellowship approach to the organic acidaemias: recognise the high-anion-gap metabolic acidosis with ketosis that distinguishes them from the urea cycle disorders, treat on suspicion with calorie loading, carnitine and toxin removal before the enzyme diagnosis returns, distinguish the cofactor-responsive subtypes, and lock in long-term protein-restricted diet, transplantation, and an emergency sick-day plan.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who holds three ideas at once. The first is the acid-base discriminator: a urea cycle disorder gives a normal anion gap with a respiratory alkalosis, while an organic acidaemia gives a high anion-gap metabolic acidosis with ketosis — this single difference rewrites the differential and the diet. The second is the cofactor trial: methylmalonic acidaemia may respond to vitamin B12, and multiple carboxylase deficiency responds to biotin, so a labelled "mild" or "treatment-responsive" variant can still hide behind a dangerous first presentation. The third is the chronic burden: these are multisystem diseases of basal-ganglia injury, cardiomyopathy and renal failure that persist between crises, and the emergency sick-day plan is what keeps the child alive. [1] [7]
Overview & Definition
The organic acidaemias are a family of autosomal recessive inherited metabolic diseases caused by deficiency of an enzyme in the catabolism of the branched-chain amino acids (isoleucine, leucine and valine) and related substrates. A block at any step halts the pathway, and the intermediate immediately upstream accumulates as a toxic organic acid that circulates and injures tissues. The classic disorders are propionic acidaemia (PA), methylmalonic acidaemia (MMA), isovaleric acidaemia (IVA) and glutaric aciduria type 1 (GA1), with multiple carboxylase deficiency a treatable cousin. [1] [7]
Clinically, the organic acidaemias are the archetype of the intoxication-type inborn errors of metabolism: the disease is the accumulating toxin, and the priority is toxin removal and source control, not enzyme replacement. The toxic acids inhibit the urea cycle, producing secondary hyperammonaemia; they impair mitochondrial oxidative phosphorylation, producing lactic acidosis; and they are selectively neurotoxic to the basal ganglia, producing the choreoathetosis, dystonia and intellectual disability that dominate long-term morbidity. This framing dictates the whole management. [1] [6]
Classification
The classification that matters clinically is the enzyme block, because the position of the block in branched-chain amino-acid catabolism determines the accumulating toxic acid, the diagnostic acylcarnitine and organic-acid pattern, the cofactor responsiveness, and the chronic organ burden. Propionic and methylmalonic acidaemia share the early pathway (isoleucine, valine, methionine, threonine and odd-chain fatty acids converge on propionyl-CoA), while isovaleric acidaemia sits on the leucine arm and glutaric aciduria type 1 on the lysine, hydroxylysine and tryptophan arm. [1] [7]

Propionyl-CoA is the gateway: it is carboxylated to D-methylmalonyl-CoA by propionyl-CoA carboxylase (PCC, deficient in propionic acidaemia), then isomerised to L-methylmalonyl-CoA and converted to succinyl-CoA by methylmalonyl-CoA mutase (MUT, or its cobalamin cofactor pathway, deficient in methylmalonic acidaemia). On the leucine arm, isovaleryl-CoA dehydrogenase (IVD, deficient in isovaleric acidaemia) converts isovaleryl-CoA, while on the lysine arm glutaryl-CoA dehydrogenase (GCDH, deficient in glutaric aciduria type 1) converts glutaryl-CoA. Multiple carboxylase deficiency — from holocarboxylase synthetase or biotinidase defects — impairs all biotin-dependent carboxylases and is treatable with biotin. [1] [10]
[1] [4]Epidemiology & Risk Factors
The organic acidaemias are individually rare but collectively significant, and newborn screening has changed their epidemiology by detecting presymptomatic cases. Propionic acidaemia affects roughly one in 30,000 to 100,000 live births, methylmalonic acidaemia around one in 50,000 (with the cobalamin-responsive forms a sizeable fraction), isovaleric acidaemia around one in 40,000, and glutaric aciduria type 1 roughly one in 100,000, though rates vary by population and are higher in communities with consanguinity. The Reischl-Hajiabadi newborn-screening outcome study from 2024 shows that screened children with PA and MMA still suffer significant morbidity, confirming that early detection reduces but does not abolish the burden. [1] [2]
The Nizon cohort of 80 patients with classical organic acidurias is one of the most informative long-term datasets, demonstrating that neurological outcome is dominated by the number and severity of acute decompensations — children who escape crises do better, and those who suffer recurrent ketoacidemic episodes accumulate basal-ganglia injury and intellectual disability. This is the epidemiological engine behind the emergency sick-day plan: the disease is rare, but the consequence of each untreated crisis is permanent and large. [3]
The major risk factor for any presentation is a catabolic trigger — intercurrent infection, fasting, surgery, immunisation, or a high-protein load — because catabolism floods the blocked pathway with substrate. A family history of neonatal or unexplained death, consanguinity, or a sibling with a similar pattern raises the pre-test probability further. Newborn screening detects most PA, MMA, IVA and GA1 cases on the bloodspot, but a normal screen in a sick neonate does not exclude an organic acidaemia — some variants and secondary causes are not captured, and the clinical picture must drive investigation. [1] [11]
Pathophysiology
The molecular story begins with the blocked enzyme. When a step in branched-chain amino-acid catabolism is deficient, the acyl-CoA intermediate immediately upstream accumulates, and the corresponding organic acid floods the mitochondrion and the circulation. These organic acids are the disease: they are the toxin that produces the metabolic acidosis, the secondary hyperammonaemia and the chronic neurodegeneration. [1] [6]
Three mechanisms drive the acute crisis. First, the accumulating organic acids (propionic acid in PA, methylmalonic acid in MMA) are acids, so they consume bicarbonate and produce a high-anion-gap metabolic acidosis; the body's response is hyperventilation, and the accumulating ketones from impaired energy metabolism add to the acid load. Second, the organic acids inhibit the urea cycle — propionyl-CoA depletes the intracellular CoA pool and methylmalonic acid directly inhibits carbamoyl phosphate synthetase — producing secondary hyperammonaemia that further injures the brain. Third, the organic acids impair mitochondrial oxidative phosphorylation, depleting ATP and raising lactate, which compounds the acidosis and the energy failure. [6] [10]
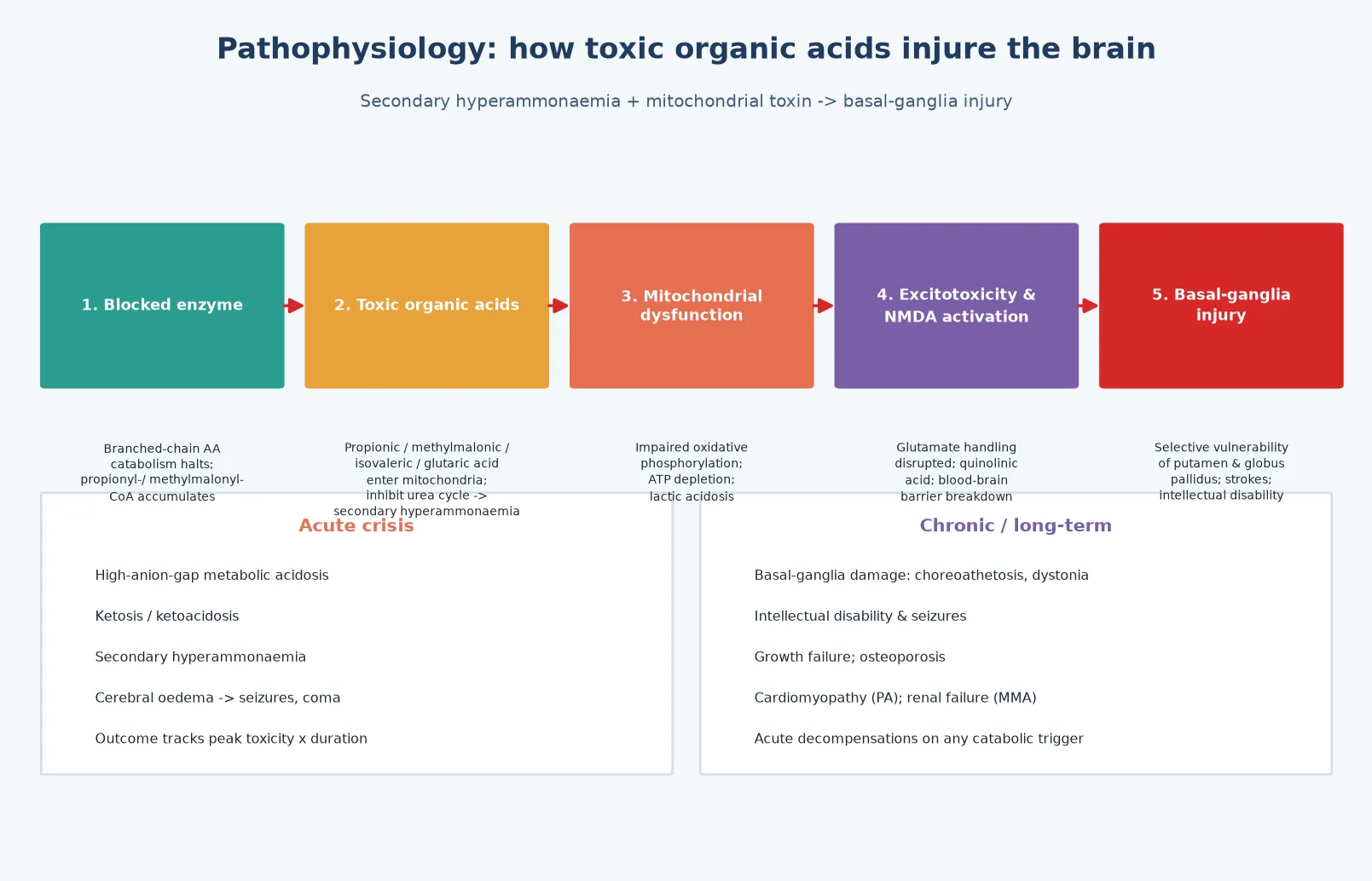
The chronic neurodegeneration is driven by selective vulnerability of the basal ganglia. Propionic and methylmalonic acid are directly toxic to the putamen and globus pallidus, producing the acute dystonia and choreoathetosis of a metabolic stroke that occurs during decompensation; glutaric acid is neurotoxic to the striatum, and the acute striatal necrosis of GA1 produces severe irreversible dystonia after a single febrile illness. The injury is proportional to both the peak toxic-acid exposure and its duration, which is why speed of toxin removal is the dominant determinant of outcome. [3] [4]
The mitochondrial connection extends beyond the brain. Schumann and colleagues demonstrated that propionyl-CoA directly damages renal tubular epithelial mitochondria, which is the molecular basis of the tubulointerstitial nephropathy and progressive renal failure of propionic acidaemia. This mechanism — toxic organic acids damaging mitochondria in the most metabolically active organs — explains why the organic acidaemias are multisystem diseases of brain, heart, kidney and bone, not simply metabolic crises. [10]
Clinical Presentation
The presentation is best framed in three patterns. Neonatal / early-infantile presentation is the classic, dramatic form: a term baby, well at birth, who deteriorates 24 to 72 hours into feeds with poor feeding, vomiting, lethargy, hypotonia and seizures, and a blood gas showing a high-anion-gap metabolic acidosis with ketosis. This picture looks like sepsis, and the discriminating clue is the metabolic panel — the acid-base and anion gap set the organic acidaemia differential apart from sepsis and from a urea cycle disorder. [1] [3]
Intermittent late-onset presentation is increasingly recognised and is the form most often missed. It occurs in older infants and children with partial enzyme deficiency, precipitated by a catabolic stressor — infection, fasting, surgery, or a high-protein meal. The picture is episodic ketoacidosis: vomiting, dehydration, altered consciousness, and ataxia or dystonia, often labelled initially as a gastroenteritis, a diabetic ketoacidosis, or a toxin. A low threshold to send a metabolic panel in any child with unexplained metabolic acidosis is the lesson. [1] [12]
Chronic presentation reflects the cumulative multisystem burden. A child with undiagnosed or poorly controlled PA or MMA may present with growth failure, intellectual disability, recurrent dystonia or choreoathetosis from basal-ganglia injury, cardiomyopathy (characteristic of PA), or progressive renal impairment (characteristic of MMA). GA1 has a distinct chronic prodrome — macrocephaly and hypotonia in infancy — followed by acute striatal necrosis after an intercurrent illness that produces severe, irreversible dystonia. [3] [4]
Differential Diagnosis
The differential is the differential of the high-anion-gap metabolic acidosis with ketosis, and the organic acidaemias are one — important but not the only — cause. The first task is to separate the organic acidaemias from the urea cycle disorders, because the management differs: a urea cycle disorder gives a normal anion gap with a respiratory alkalosis and raised glutamine, whereas the organic acidaemias give a high anion-gap metabolic acidosis with ketosis, raised lactate and secondary hyperammonaemia. This single distinction changes the diet, the scavenger choice, and the urgency of cofactor trial. [1]
Within the high-anion-gap acidosis differential, the organic acidaemias must be separated from lactic acidaemia (primary mitochondrial disease or mitochondrial encephalopathy), diabetic ketoacidosis, salicylate or other toxin ingestion, and renal failure. The discriminating tests are the acylcarnitine profile and the urinary organic acids: a raised propionylcarnitine (C3) with methylcitrate points to PA or MMA, a raised isovalerylcarnitine (C5) points to IVA, and glutaric and 3-hydroxyglutaric acids point to GA1. The metabolic profile — not the label — sets the answer. [1] [7]
Clinical & Bedside Assessment
The bedside assessment has two speeds. In the acute presentation, the question is resuscitation and the metabolic panel, not the enzyme diagnosis. Take a focused history — onset relative to feeds, the presence of a catabolic trigger, and the family history — while simultaneously drawing the blood gas, glucose, ammonia (free-flowing, on ice, to the laboratory urgently), lactate, ketones, and the metabolic first-tier tests. Do not delay resuscitation for a complete history. [1]
In the stable or recovering child, build a structured picture. A three-generation pedigree asks explicitly about neonatal deaths, unexplained developmental delay or intellectual disability, consanguinity, and reactions to protein or fasting. The examination looks for the stigmata of chronic disease in the late-onset case — growth failure, developmental delay, hepatomegaly, an abnormal odour, the macrocephaly of GA1, and the movement disorder of basal-ganglia injury — while the neonate is examined for the signs of dehydration, acidosis and encephalopathy that drive the urgency. [1] [3]
The bedside assessment converts directly into the investigation plan: acylcarnitines and urinary organic acids to identify the accumulating acid, plasma amino acids and quantitative carnitine to characterise the profile, and molecular genetic testing for confirmation and family counselling. In a neonatal crisis the enzyme diagnosis will not return before treatment decisions, so the acute plan is driven by the acid-base picture, the ammonia trajectory and the acylcarnitine pattern. [1] [7]
Investigations
The first-tier investigation is a coordinated metabolic panel interpreted together. Blood gas defines the acid-base picture and the anion gap — the single most important discriminator between an organic acidaemia and a urea cycle disorder. Glucose, lactate and ketones characterise the metabolic state: organic acidaemias typically show ketosis with a raised lactate, whereas fatty-acid oxidation defects show inappropriately low ketones for the hypoglycaemia. Ammonia is measured free-flowing, on ice and urgently, because the organic acidaemias produce secondary hyperammonaemia that must be treated. [1]
The acylcarnitine profile identifies the accumulating acyl-CoA ester: a raised propionylcarnitine (C3) with methylcitrate suggests PA or MMA, a raised isovalerylcarnitine (C5) suggests IVA, and a raised glutarylcarnitine (C5DC) suggests GA1. Urinary organic acids confirm and quantify: methylmalonic acid (MMA), 3-hydroxypropionate and methylcitrate (PA), isovalerylglycine and 3-hydroxyisovalerate (IVA), and glutaric and 3-hydroxyglutaric acid (GA1). Quantitative plasma carnitine typically shows low free carnitine and a high esterified-to-free ratio, confirming carnitine depletion and the rationale for supplementation. [1] [11]
Why a B12-responsive methylmalonic acidaemia changes the prognosis
Methylmalonic acidaemia has a spectrum of causes: a defect in the methylmalonyl-CoA mutase enzyme itself (mut-type, B12-nonresponsive) or a defect in the cobalamin processing pathway (cblA–cblD, some B12-responsive). The B12-responsive subtypes are milder and may be substantially improved by pharmacological vitamin B12, so a cobalamin responsiveness trial — measuring methylmalonic acid excretion before and after intramuscular hydroxocobalamin — is mandatory in every newly diagnosed MMA patient, because it defines the prognosis and the treatment. [1] [6]
Molecular confirmation is then undertaken by sequencing the candidate gene suggested by the biochemical pattern (PCCA/PCCB for PA, MUT/MMAB/MMACH for MMA, IVD for IVA, GCDH for GA1) or by a metabolic gene panel or exome when the pattern is ambiguous. Confirming the molecular defect defines the exact diagnosis, enables cascade carrier testing and prenatal or preimplantation diagnosis for the family, and — for MMA — distinguishes the B12-responsive cobalamin pathway defects from the non-responsive mut-type. Newborn screening detects most cases on the bloodspot, but confirmatory testing is always required. [1] [2]
Management — Resuscitation
Resuscitation is the section that earns or loses the most marks, because outcome is determined here. The principle is to treat on suspicion before the enzyme diagnosis returns: the moment a high-anion-gap metabolic acidosis with ketosis is identified in an encephalopathic child, start the emergency protocol. The first moves are to stop all protein intake, establish intravenous access, and begin aggressive calorie provision — 10 percent glucose with intralipid, with insulin as needed to drive anabolism — because switching the child from catabolism to anabolism is the single most important physiological intervention to reduce endogenous toxic-acid generation. [1]
L-carnitine is the specific detoxifier: it conjugates with the accumulating acyl-CoA esters to form acylcarnitines, which are renally excreted, removing the toxin and replenishing the depleted CoA pool. Carnitine is given intravenously in the acute crisis and orally for maintenance, and it is a cornerstone of organic acidaemia management from the first hour. Bicarbonate corrects the metabolic acidosis when severe, and N-carbamylglutamate may be used in propionic acidaemia to augment residual urea-cycle flux and lower the secondary hyperammonaemia. [1]
The decision to use extracorporeal removal — haemofiltration or haemodialysis — is made on the severity and trajectory of the acidosis and hyperammonaemia. Refractory metabolic acidosis, a very high or rising ammonia despite medical therapy, or deepening encephalopathy are the triggers, and the objective is to clear the toxic organic acids and ammonia within hours. Continuous kidney replacement therapy is preferred in the haemodynamically unstable neonate. Do not wait for the enzyme or molecular diagnosis before starting treatment — outcome tracks peak toxic-acid exposure multiplied by duration. [1]
Management — Definitive & Stepwise
Once the acute crisis is controlled, definitive management is a long-term, multidisciplinary framework built around five pillars: a protein-restricted (low branched-chain amino acid) diet, cofactor therapy where responsive, L-carnitine maintenance, prevention of catabolism during intercurrent illness, and transplantation for severe disease. The diet is the cornerstone: natural protein restricted to tolerance, with a special amino-acid formula depleted of the offending amino acid (isoleucine, leucine, valine for PA and MMA; leucine for IVA; lysine and tryptophan restriction for GA1), and adequate non-protein calories to maintain anabolism. [1] [4]
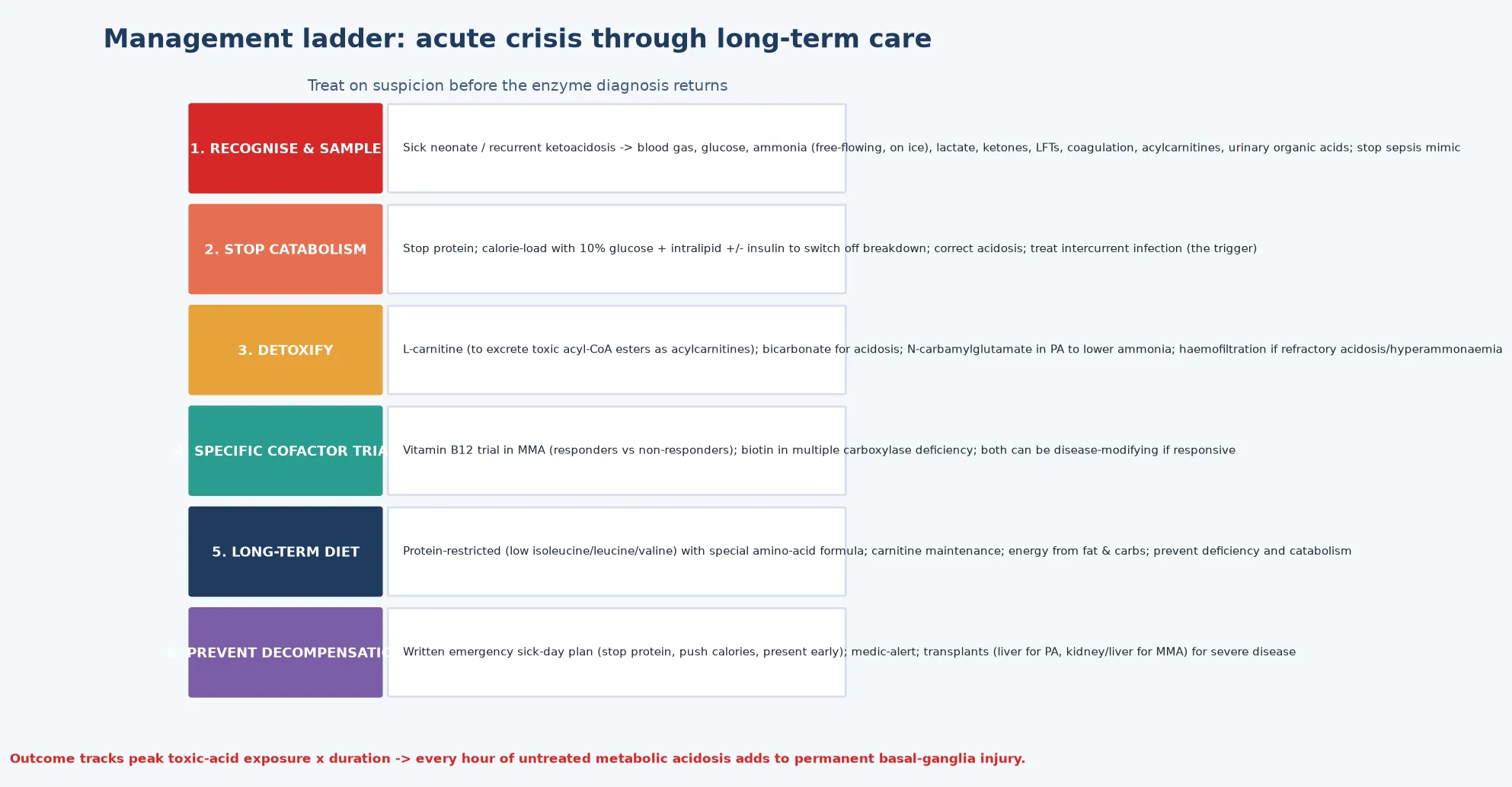
Cofactor therapy is the second pillar and the one that can be disease-modifying. Vitamin B12 responsiveness is assessed in every newly diagnosed MMA patient by the cobalamin trial; responders receive lifelong pharmacological hydroxocobalamin or cyanocobalamin, which substantially lowers methylmalonic acid excretion and improves outcome. Biotin is the specific therapy for multiple carboxylase deficiency (from holocarboxylase synthetase or biotinidase deficiency), and biotinidase activity is screened on the newborn bloodspot in many programmes. Carnitine maintenance is given orally to all patients to sustain detoxification and correct the chronic carnitine depletion. [1] [6]
Prevention of catabolism is taught to every family as an emergency sick-day plan: at the first sign of illness, stop natural protein, increase calories from glucose and the special formula, and present early for intravenous management. A medic alert and a written emergency letter travel with the child. This plan is the single most important intervention for preventing recurrent decompensation and the cumulative basal-ganglia injury that dominates long-term morbidity, and every transition — into school, into adolescence, and into adult metabolic services — is a high-risk point for its loss. [1] [12]
Transplantation is the definitive therapy for the severe forms. Liver transplantation corrects the hepatic enzyme defect in propionic acidaemia, restoring metabolic stability and liberating the child from strict dietary restriction and the constant threat of decompensation, though it does not reverse established neurological injury. The Sen ACMG points-to-consider statement and the Molema European transplant overview guide the indications: liver for PA (with the cardiomyopathy often resolving), and kidney or liver (or both) for MMA, because MMA causes progressive renal failure that kidney transplantation addresses while liver transplantation corrects the metabolic defect. Transplantation is complementary to, not a substitute for, excellent emergency care. [8] [9]
S.C.A.L.P. the toxic acid \u2014 the acute protocol
Specific Subtypes & Scenarios
Propionic acidaemia is often the most severe of the branched-chain organic acidaemias. Neonatal presentation with profound metabolic acidosis and hyperammonaemia is typical, and chronic complications include cardiomyopathy (which can be the presenting or life-threatening feature), prolonged QTc, recurrent pancreatitis, basal-ganglia injury, intellectual disability, and growth failure. Management combines the standard protocol with attention to the cardiac risk, and liver transplantation is increasingly the standard for severe disease because it corrects the metabolic defect and often resolves the cardiomyopathy. [1] [8]
Methylmalonic acidaemia deserves special mention for its renal and cofactor dimensions. It causes a progressive tubulointerstitial nephropathy leading to chronic renal failure — the molecular basis for which Schumann and colleagues traced to propionyl-CoA-induced mitochondrial damage in renal epithelial cells — so renal surveillance and transplantation are integral to management. The cobalamin-responsive subtypes are milder and may be substantially improved by vitamin B12, which is why a responsiveness trial is mandatory at diagnosis. [6] [10]
Isovaleric acidaemia is often the mildest of the classic organic acidaemias. The characteristic sweaty-feet odour during decompensation is a high-yield bedside clue, and the Thimm systematic review confirms that many newborn-screened patients remain asymptomatic, though an acute neonatal crisis can be severe. Management combines the standard protocol with glycine supplementation (glycine conjugates isovaleryl-CoA to form isovalerylglycine, an alternative detoxification pathway) alongside carnitine. [11]
Glutaric aciduria type 1 is the striatal-necrosis scenario. Its hallmarks are macrocephaly and hypotonia in infancy, followed by an acute encephalopathic crisis — typically after a febrile illness in the first two to three years — that produces irreversible striatal necrosis and severe dystonia. The Boy third-revision guidelines (building on the Kölker first revision) make prevention of the first crisis the central goal: aggressive emergency management of any intercurrent illness, with lysine and tryptophan restriction and carnitine, because once striatal necrosis occurs the neurological damage is permanent. Early diagnosis through newborn screening and rigorous emergency management can allow near-normal development. [4] [5]
Complications & Pitfalls
The complications divide into the acute metabolic injuries, the chronic multisystem burden, and the cognitive traps that cost marks. The acute injuries are metabolic acidosis, cerebral oedema, seizures, metabolic stroke (acute dystonia or choreoathetosis from basal-ganglia injury during decompensation), coma and death. The Nizon cohort demonstrates that the number and severity of acute decompensations is the dominant predictor of long-term neurological outcome, which is the evidence base that underpins the urgency of treatment and the centrality of the emergency sick-day plan. [3]
The chief cognitive trap is delay, which takes three forms: failure to send a metabolic panel in a sick neonate because sepsis seems more likely; failure to start treatment before the diagnosis is confirmed; and failure to use haemofiltration because the acidosis "might come down with medication". The second trap is misclassification — confusing an organic acidaemia with a urea cycle disorder or a fatty-acid oxidation defect, which changes the diet, the scavenger choice and the cofactor trial. The third is missing the family — the autosomal recessive inheritance obliges carrier testing and reproductive counselling, and an affected sibling may be presymptomatic. [1] [3]
Prognosis & Disposition
Prognosis is determined by three factors: the specific defect (with PA typically the most severe, IVA often the mildest), the number and severity of acute decompensations, and the quality and consistency of long-term metabolic management. The Reischl-Hajiabadi newborn-screening outcome data confirm that even with early detection, children with PA and MMA carry a significant burden of morbidity, but rigorous emergency management and transplantation improve the trajectory substantially. A child who escapes crises and receives consistent metabolic care can achieve a far better outcome than one who suffers recurrent decompensations. [2] [3]
Transplantation changes the trajectory for severe disease. Liver transplantation for PA corrects the metabolic defect and often resolves the cardiomyopathy, restoring metabolic stability and liberating the child from strict dietary restriction; for MMA, kidney transplantation addresses the renal failure and liver transplantation corrects the metabolic defect, and combined transplantation may be considered. Transplantation cannot reverse established neurological injury, which is why it is best performed in a metabolically stable window rather than as a rescue. [8] [9]
Disposition is shared, lifelong, multidisciplinary care: a specialist metabolic service owns the diet, the cofactor regimen, and the transplantation pathway; the general paediatrician owns coordination, immunisation, and the emergency sick-day plan; and the family owns the day-to-day vigilance that prevents decompensation. The transition to adult metabolic services is a high-risk point — dietary adherence slips, the emergency plan may not transfer, and pregnancy becomes a concern — so the plan must travel with the patient. The Tuncel adult-cohort data show that late complications — renal failure, neurological deterioration, psychiatric disturbance — accumulate, underscoring the need for lifelong surveillance. [12]
Special Populations
The same organic acidaemia behaves differently across populations because access, recognition, and service models are unevenly distributed. In remote and Indigenous communities, later presentation, distance from a metabolic service, and the need for aeromedical retrieval during a neonatal crisis mean that the window between onset and treatment is longer and outcomes are worse — so a written, location-specific emergency plan and a low threshold to send a metabolic panel in any sick neonate are disproportionately important. In migrant, refugee, and asylum-seeking families, consanguinity raises the pre-test probability of autosomal recessive defects, language barriers complicate the emergency teaching of a sick-day plan, and an interpreter must be used at every key consultation. [1]
In adolescents transitioning to adult metabolic care, the move is a high-risk point: dietary adherence often slips, the emergency plan may not transfer, and the cumulative burden of basal-ganglia injury and organ damage becomes apparent. In families managing complex chronic metabolic disease, fragmentation of care is the chief threat, and a written, shared, reconciled care plan is the intervention that matters most. Pregnancy in a woman with an organic acidaemia requires a metabolic obstetric plan, because the catabolic stress of labour and the postpartum period can precipitate decompensation. [12] [1]
Evidence, Guidelines & Regional Differences
The evidence base rests on three pillars: consensus clinical guidelines, longitudinal cohort outcome data, and emerging molecular and therapeutic evidence. The Baumgartner guidelines (2014) are the international standard for the diagnosis and management of methylmalonic and propionic acidaemia, setting the structure of acute management, long-term dietary and pharmacological therapy, cofactor responsiveness, and transplantation that most national programmes adopt. The Boy third-revision guidelines (2023), building on the Kölker first revision (2011), are the equivalent standard for glutaric aciduria type 1, and the Forny organic-aciduria guideline-development review contextualises how evidence has informed practice. [1] [4] [5] [7]
The longitudinal cohort data rest largely on nationwide registries and single-centre series. The Nizon cohort of 80 patients with classical organic acidurias is among the most informative long-term datasets, demonstrating that neurological outcome is dominated by the number and severity of acute decompensations. The Reischl-Hajiabadi newborn-screening outcome study (2024) shows that screened children with PA and MMA still suffer significant morbidity despite early detection, and the Tuncel adult cohort (2018) documents the late complications that accumulate over a lifetime. [2] [3] [12]
The molecular and therapeutic evidence is moving fast. The Head pathophysiology review grounds MMA neurotoxicity in mitochondrial mechanisms, the Schumann renal study traces the nephropathy of PA to propionyl-CoA-induced mitochondrial damage, the Sen ACMG transplantation statement and the Molema European transplant overview refine who benefits from solid-organ transplantation and when, and the Thimm IVA systematic review consolidates the practical management of isovaleric acidaemia. Together these define the modern, severity-adjusted, multidisciplinary approach. [6] [8] [9] [10] [11]
In Australia and New Zealand, the organic acidaemias are managed through the state-based metabolic services (most coordinated through the major children's hospitals in each state and Starship in New Zealand), with newborn screening programmes detecting propionic, methylmalonic, isovaleric and glutaric aciduria type 1 on the bloodspot through tandem mass spectrometry — though a normal screen does not exclude an organic acidaemia in a sick neonate, because some variants and secondary causes are not captured. Aeromedical retrieval to a tertiary metabolic and intensive-care centre is the expected pathway for a neonatal crisis, and continuous kidney replacement therapy is available at the paediatric intensive care units that receive these patients. The cobalamin responsiveness trial is performed at the specialist metabolic service, and liver and kidney transplantation are coordinated through the national transplant programmes. Genetic counselling, carrier testing, and prenatal or preimplantation genetic diagnosis are coordinated through clinical genetics services, and the NDIS (Australia) and equivalent disability support (New Zealand) fund the allied health, dietetic, and developmental supports. Always confirm the current local retrieval pathway and metabolic service contact, as these vary by state.
[1][4]Exam Pearls
A fellowship candidate answering on the organic acidaemias should land six anchor points and avoid three classic traps. The anchors are the acid-base discriminator (high-anion-gap metabolic acidosis with ketosis, not the normal-anion-gap respiratory alkalosis of a urea cycle disorder), the five major subtypes and their accumulating toxic acids, the S.C.A.L.P. emergency protocol (stop protein and catabolism, carnitine, acidosis correction, cofactor trial, purify with haemofiltration), the cofactor responsiveness of MMA (B12) and multiple carboxylase deficiency (biotin), the role of transplantation (liver for PA, kidney or liver for MMA), and the chronic multisystem burden of basal-ganglia injury, cardiomyopathy and renal failure. The traps are delay (waiting for the diagnosis), misclassification (confusing an organic acidaemia with a urea cycle disorder), and missing GA1 until the first striatal necrosis. The candidate who names the sweaty-feet odour of IVA, the macrocephaly prodrome of GA1, and the disease-modifying potential of the cobalamin trial in MMA will stand out. [1] [3]
References
- [1]Baumgartner MR, Hörster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis, 2014.PMID 25205257
- [2]Reischl-Hajiabadi AT, Schnabel E, Gleich F, Mengler K, Lindner M, Burgard P, et al. Outcomes after newborn screening for propionic and methylmalonic acidemia and homocystinurias. J Inherit Metab Dis, 2024.PMID 38563533
- [3]Nizon M, Ottolenghi C, Valayannopoulos V, Arnoux JB, Barbier V, Habarou F, et al. Long-term neurological outcome of a cohort of 80 patients with classical organic acidurias. Orphanet J Rare Dis, 2013.PMID 24059531
- [4]Boy N, Mühlhausen C, Maier EM, Ballhausen D, Baumgartner MR, Beblo S, et al. Recommendations for diagnosing and managing individuals with glutaric aciduria type 1: Third revision. J Inherit Metab Dis, 2023.PMID 36221165
- [5]Kölker S, Christensen E, Leonard JV, Greenberg CR, Boneh A, Burlina AB, et al. Diagnosis and management of glutaric aciduria type I--revised recommendations. J Inherit Metab Dis, 2011.PMID 21431622
- [6]Head PE, Meier JL, Venditti CP. New insights into the pathophysiology of methylmalonic acidemia. J Inherit Metab Dis, 2023.PMID 37078237
- [7]Forny P, Hörster F, Baumgartner MR, Kölker S, Boy N. How guideline development has informed clinical research for organic acidurias (et vice versa). J Inherit Metab Dis, 2023.PMID 36591944
- [8]Sen K, Burrage LC, Chapman KA, Ginevic I, Mazariegos GV, Graham BH, et al. Solid organ transplantation in methylmalonic acidemia and propionic acidemia: A points to consider statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med, 2023.PMID 36534118
- [9]Molema F, Martinelli D, Hörster F, Kölker S, Tangeraas T, de Koning B, et al. Liver and/or kidney transplantation in amino and organic acid-related inborn errors of metabolism: An overview on European data. J Inherit Metab Dis, 2021.PMID 32996606
- [10]Schumann A, Belche V, Schaller K, Grünert SC, Kaech A, Baumgartner MR, et al. Mitochondrial damage in renal epithelial cells is potentiated by protein exposure in propionic aciduria. J Inherit Metab Dis, 2021.PMID 34297429
- [11]Thimm E, Riederer A, Vockley J, Dobbelaere D, Williams M, MacDonald A, et al. Practical Considerations for the Diagnosis and Management of Isovaleryl-CoA-Dehydrogenase Deficiency (Isovaleric Acidemia): Systematic Search and Review and Expert Opinions. Int J Neonatal Screen, 2025.PMID 41133704
- [12]Tuncel AT, Boy N, Morath MA, Hörster F, Mütze U, Kölker S. Organic acidurias in adults: late complications and management. J Inherit Metab Dis, 2018.PMID 29335813