Paeds · genetics-dysmorphology-and-metabolism
Peroxisomal disorders
Also known as Peroxisomal disorders · Peroxisomal biogenesis disorders · Zellweger spectrum disorders · X-linked adrenoleukodystrophy · Zellweger syndrome
A fellowship approach to the peroxisomal disorders: split them at the bedside into the peroxisome biogenesis disorders (the Zellweger spectrum — a dysmorphic, hypotonic neonate with hepatomegaly, seizures and chondrodysplasia punctata) and the single-enzyme deficiencies (X-linked adrenoleukodystrophy, Refsum disease, rhizomelic chondrodysplasia punctata), confirm with a plasma very-long-chain fatty acid and phytanic acid panel, and match the therapy — haemopoietic stem cell transplant for cerebral X-ALD, cholic acid for the Zellweger spectrum, phytanic-acid restriction for Refsum — to the disorder.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A neonate is born with a high forehead, a large fontanelle, profound hypotonia, and hepatomegaly, and within days develops seizures that do not respond. Down the corridor, a healthy-looking six-year-old boy who had been doing well at school becomes withdrawn, clumsy, and hard of hearing, and his brain MRI lights up with symmetric, gadolinium-enhancing white-matter change. In both rooms the unifying question is the same: is this a peroxisomal disorder, which one, and how fast does it move. The fellowship task is to split the bedside pattern into the biogenesis group or the single-enzyme group, confirm with the right biochemistry, and act before an irreversible window closes. [2] [4]
P · E · R · O · X · I · S · O · M · E
Overview & Definition
The peroxisome is a single-membrane organelle that carries out a small set of oxidative reactions the mitochondrion cannot manage: beta-oxidation of very-long-chain fatty acids, alpha-oxidation of phytanic acid, the early steps of plasmalogen synthesis, and steps of bile-acid synthesis. A peroxisomal disorder is what happens when one of these functions is lost — either because the whole organelle fails to assemble, or because a single enzyme or transporter within it is defective. The missing function, not the missing gene, drives the phenotype, which is why a child without any peroxisomes looks completely different from a boy who simply cannot import very-long-chain fatty acids into them. [1] [6]
Two mechanistic groups organise the entire topic. The first is the peroxisome biogenesis disorders — collectively the Zellweger spectrum — caused by recessive mutations in any of roughly fourteen PEX genes that encode the peroxins responsible for importing matrix proteins into the organelle. When peroxins fail, the organelle is empty or absent and every peroxisomal pathway collapses at once, producing the most severe phenotype in metabolic medicine. The second group is the single-enzyme or single-transporter deficiencies, in which the peroxisome is intact but one reaction is blocked; X-linked adrenoleukodystrophy, Refsum disease, and rhizomelic chondrodysplasia punctata sit here, and each behaves like its own disease. [1] [3]
The inheritance pattern matters because it changes counselling and who is at risk. The Zellweger spectrum, Refsum disease, and rhizomelic chondrodysplasia punctata are autosomal recessive, so a previously affected child reshapes the reproductive risk for every later pregnancy. X-linked adrenoleukodystrophy is the exception that dominates the exam: affected boys carry the full burden, carrier mothers may show a milder myelopathy in adulthood, and a single affected male sends ripples through every female relative. Naming the inheritance at the first consultation frames every later conversation about siblings, future children, and the wider family. [4] [5]
Classification
Clinicians classify the peroxisomal disorders by mechanism, because the mechanism predicts the biochemistry, the inheritance, and the organ involvement. The figure lays out the two groups alongside their deficient genes and the biochemical signature of each; the groups most likely to appear in an examination are the Zellweger spectrum and X-linked adrenoleukodystrophy. [1] [6]
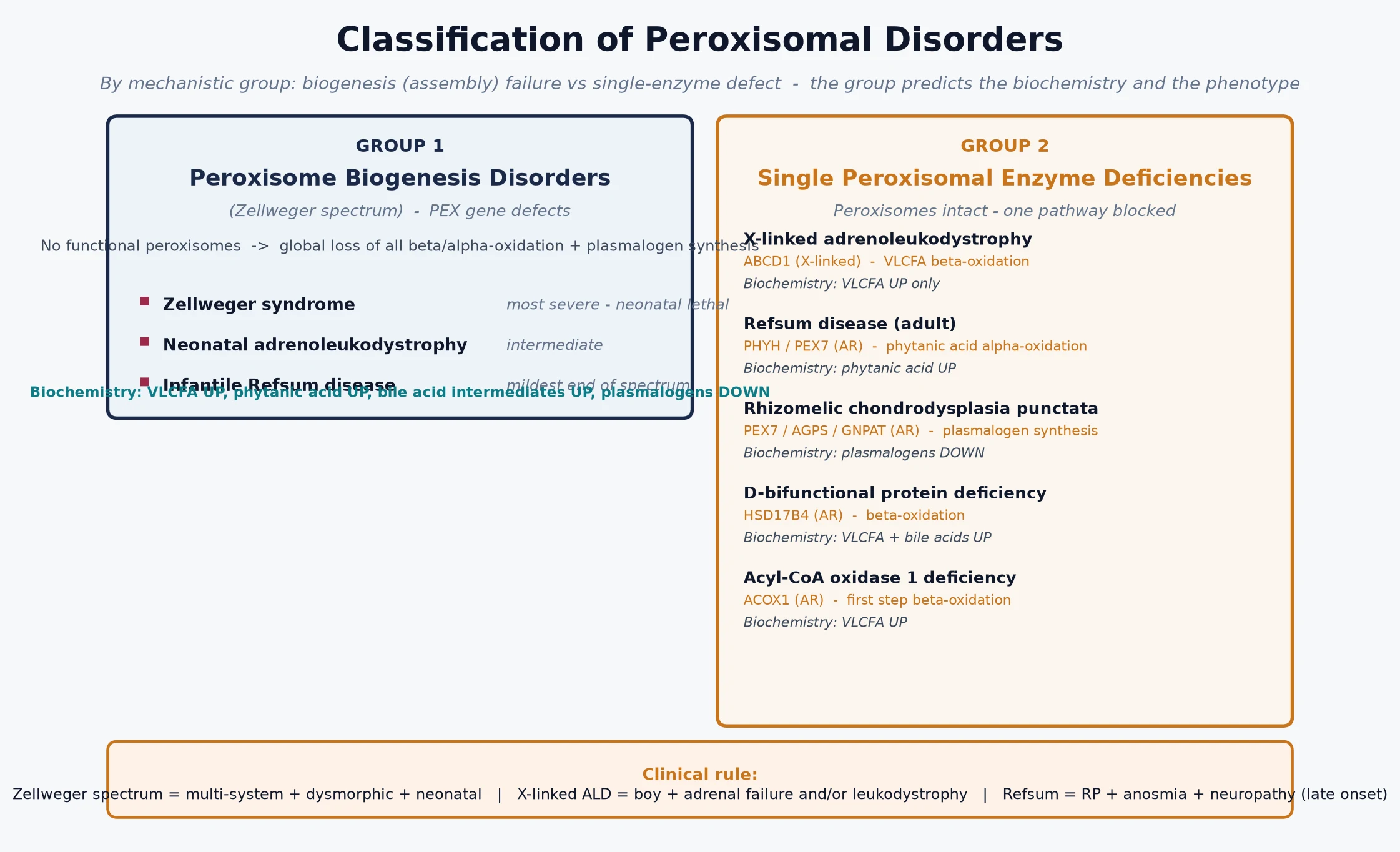
A second, more practical axis separates the disorders by the dominant organ and the pace of decline, because that distinction governs the urgency of treatment. The Zellweger spectrum declares itself in the neonate with multi-system failure and is managed supportively. Cerebral X-linked adrenoleukodystrophy declares itself in a previously well boy aged four to ten and is the one peroxisomal disorder where a treatment decision is measured in weeks. Refsum disease declares itself later still, with retinitis pigmentosa, neuropathy, and ataxia, and is managed with diet. Holding the pace in mind prevents the two common errors: reassuring a family when a transplant window is closing, or over-investigating a stable adult phenotype. [2] [4]
Biogenesis disorder versus single-enzyme deficiency — the defining split
- Peroxisome biogenesis disorder (Zellweger spectrum, PEX genes, autosomal recessive): no functional peroxisomes, so very-long-chain fatty acids rise, phytanic acid rises, bile-acid intermediates rise, and plasmalogens fall. Presents in the neonate with dysmorphism, profound hypotonia, seizures, hepatomegaly, chondrodysplasia punctata, retinal and hearing impairment.
- X-linked adrenoleukodystrophy (ABCD1, X-linked): peroxisomes intact but the very-long-chain fatty acid transporter is broken, so only VLCFA accumulate. Presents in a boy with adrenal failure and inflammatory cerebral demyelination, or in a young man with adrenomyeloneuropathy.
- Refsum disease (PHYH, autosomal recessive): only phytanic acid accumulates. Presents after infancy with retinitis pigmentosa, anosmia, ataxia, polyneuropathy, and deafness. [1] [3]
Epidemiology & Risk Factors
Individually each peroxisomal disorder is rare, but together they are a meaningful burden in paediatric metabolic practice. The Zellweger spectrum affects roughly one in fifty thousand live births, and X-linked adrenoleukodystrophy — the most common single peroxisomal disorder — affects about one in seventeen thousand males and one in seventeen thousand female carriers. Founder effects concentrate both groups: higher Zellweger rates are reported in certain French-Canadian and consanguineous communities, and adrenoleukodystrophy is well described across all populations. Consanguinity and a previously affected sibling are the strongest risk factors for the autosomal recessive forms and should lower the threshold for testing any child with a compatible phenotype. [2] [6]
The epidemiology is shifting rapidly because of newborn screening. X-linked adrenoleukodystrophy has been added to bloodspot panels in a growing number of regions, detected by an elevated C26:0 lysophosphatidylcholine, and that has changed the presenting population from a symptomatic school-age boy to an asymptomatic neonate. This presymptomatic detection is the whole point: it opens the surveillance window so that the first sign of cerebral disease is a subtle MRI change rather than a failing child, and it allows adrenal insufficiency to be found and treated before a crisis. The trade-off is that many screen-positive infants carry variants of uncertain significance or will never develop cerebral disease, so calibrated follow-up has become its own clinical skill. [9] [5]
Pathophysiology
The peroxisome maintains a oxidative interior in which a handful of enzymes break down substrates the mitochondrion cannot handle. Very-long-chain fatty acids — chains of twenty-four and twenty-six carbons — enter the peroxisome through a transporter encoded by ABCD1 and are shortened by beta-oxidation. Phytanic acid, a branched-chain fatty acid from the diet, is shortened by alpha-oxidation. Plasmalogens, essential membrane lipids particularly abundant in brain and heart, are made in the peroxisome before being exported. When a PEX gene is defective the organelle cannot import its enzymes and none of these reactions occur; when ABCD1 is defective only the very-long-chain fatty acid import fails. The figure traces both cascades from gene to multi-organ disease and overlays the therapeutic levers. [1] [8]
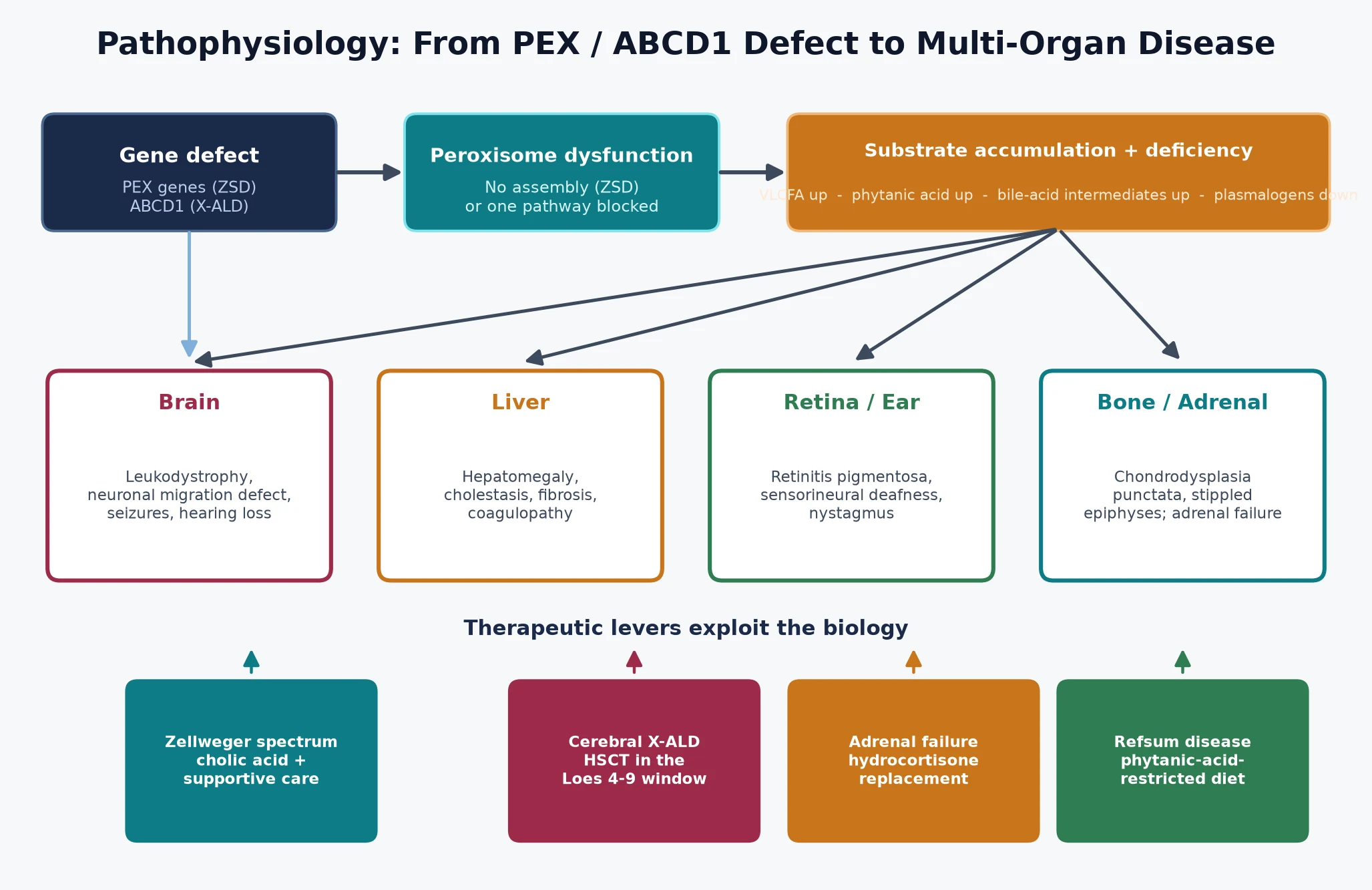
The organ phenotype follows where the substrate lands. In the Zellweger spectrum, plasmalogen deficiency disrupts neuronal migration and brain development, giving the structural brain malformations and profound hypotonia, while accumulated very-long-chain fatty acids and bile-acid intermediates injure the liver and retina. In X-linked adrenoleukodystrophy, very-long-chain fatty acids accumulate in the adrenal cortex — causing primary adrenal insufficiency — and in the white matter, where they trigger an inflammatory cascade that strips myelin from the occipito-parietal regions outward. The inflammation, not the storage alone, is what makes cerebral disease so fast and so destructive, and it is the reason a boy can look well one month and fail the next. [4] [3]
Treatment is built on this same biology. Haemopoietic stem cell transplant works for cerebral adrenoleukodystrophy not by replacing a gene but by supplying donor-derived microglia that carry a functional transporter and calm the inflammatory cascade — which is why it works only while the demyelination is still early and inflammatory, and is futile once it is advanced. Cholic acid therapy for the Zellweger spectrum supplies the missing end-product of bile-acid synthesis and reduces the toxic bile-acid intermediates. Phytanic-acid restriction in Refsum disease attacks the problem from the input side, slowing the accumulation of the substrate that drives the neuropathy. [12] [4]
Clinical Presentation
The presentation depends on the group, the specific gene, and the age at onset, but a small number of patterns account for most encounters. The first is the dysmorphic neonate. A baby is born with a high forehead, a large anterior fontanelle, a broad nasal bridge, and hypertelorism, and is profoundly hypotonic with poor feeding, seizures that are hard to control, and hepatomegaly with cholestasis. Cataracts or pigmentary retinopathy, sensorineural deafness, and stippled epiphyses on the skeletal survey complete the picture. This is the severe end of the Zellweger spectrum, and survival beyond infancy is uncommon. [2] [10]
The second pattern is the previously well boy who fails. Between the ages of four and ten, a boy develops behavioural change, declining school performance, subtle visual or hearing loss, and progressive clumsiness, and his brain MRI shows symmetric, confluent, gadolinium-enhancing demyelination in the occipito-parietal white matter. This is childhood cerebral X-linked adrenoleukodystrophy, and it progresses to a vegetative state and death within two to five years if untreated. The same ABCD1 defect can instead declare itself as adrenomyeloneuropathy in a young man, with progressive spastic paraparesis and peripheral neuropathy, or as isolated adrenal insufficiency with no neurological involvement at all. Female carriers develop an adrenomyeloneuropathy-like myelopathy in middle age in roughly half of cases. [4] [5]
Differential Diagnosis
When a peroxisomal disorder is suspected, the differential depends on which presentation brought the child to attention. The dysmorphic, hypotonic neonate with seizures and hepatomegaly must be separated from the other causes of severe neonatal hypotonia and multi-system disease: Zellweger spectrum from the lysosomal storage disorders, mitochondrial disease (which brings lactic acidosis and multi-organ failure), the congenital disorders of glycosylation, and spinal muscular atrophy (where the face and the liver are spared). Prader–Willi and benign congenital hypotonia do not have hepatomegaly, seizures, and chondrodysplasia punctata. The discriminating test is the plasma very-long-chain fatty acid panel, which is abnormal across the whole Zellweger spectrum. [7] [6]
The failing school-age boy raises a different list. Acquired demyelination, a brain tumour, and the leukodystrophies of other origin — metachromatic leukodystrophy, Krabbe disease, and the late-onset neuronal ceroid lipofuscinoses — can all produce subacute neurological decline in a child. The discriminating features are the symmetric occipito-parietal distribution with gadolinium enhancement on MRI, which is highly characteristic of cerebral adrenoleukodystrophy, and the adrenal involvement. A morning cortisol and a plasma very-long-chain fatty acid level should be sent in any boy with an unexplained leukodystrophy, because adrenal insufficiency may precede the neurological disease by years. [4] [8]
For the late-onset retinitis pigmentosa and neuropathy pattern of Refsum disease, the differential includes the other causes of syndromic retinitis pigmentosa — Usher syndrome, mitochondrial disease, and the neuronal ceroid lipofuscinoses. The combination of retinitis pigmentosa with anosmia is striking and under-recognised; anosmia is one of the earliest findings in adult Refsum disease and should prompt a phytanic acid level. [1] [6]
Clinical & Bedside Assessment
The bedside assessment is built around three questions. First, which group is this — a dysmorphic neonate pointing to the biogenesis disorders, or a previously well boy pointing to X-linked adrenoleukodystrophy. Second, is the brain involved, and how fast is it declining, because this sets the urgency of imaging and transplant referral. Third, is there adrenal involvement, because adrenal insufficiency can be silent and is treatable. A focused history and examination answer all three in a single consultation and direct the laboratory workup. [2] [5]
For the neonate, the examination records the dysmorphic features, the tone and reflexes, the liver size, and any seizures, and it seeks the ophthalmological and auditory findings. Measuring the fontanelle, documenting the facial gestalt, and performing a skeletal survey for stippled epiphyses are high-yield because the combination is specific. For the older child, the neurological examination maps the pace and pattern of decline — behavioural change, visual and hearing loss, ataxia, spasticity — and the skin is examined for hyperpigmentation that signals adrenal failure. A developmental trajectory, what the child could do three and six months ago, is more informative than any single finding. [3] [7]
The family history is non-negotiable in X-linked adrenoleukodystrophy. A maternal family history of adrenal disease, unexplained male deaths, or a male relative with a spastic paraparesis should raise the suspicion and prompt carrier testing of the mother and at-risk female relatives. In the autosomal recessive forms, consanguinity and a previously affected sibling are the key flags. Documenting the pedigree at the first consultation frames the genetic counselling and the cascade testing that will follow. [4] [5]
Investigations
The investigation strategy is layered and escalates from a single blood test to definitive molecular confirmation. The first tier is the plasma very-long-chain fatty acid panel — C26:0 and the C26-to-C22 ratio — which is abnormal in the whole Zellweger spectrum and in X-linked adrenoleukodystrophy, and is the single most useful first-line test. Adding phytanic acid, pristanic acid, bile-acid intermediates, and red-cell plasmalogens broadens the panel and distinguishes the groups: a global abnormality points to a biogenesis disorder, an isolated very-long-chain fatty acid elevation points to adrenoleukodystrophy, and an isolated phytanic acid elevation points to Refsum disease. [8] [7]
The second and confirmatory tier is molecular genetic testing. Sequencing the PEX gene panel confirms the Zellweger spectrum and, increasingly, predicts severity by the specific gene and variant; ABCD1 sequencing confirms X-linked adrenoleukodystrophy and enables cascade carrier testing of at-risk female relatives and prenatal or preimplantation testing for future pregnancies. Molecular testing is essential because the biochemical pattern alone can be misleading, and pseudodeficiency and carrier states must be excluded before any irreversible decision such as transplant. [6] [5]
Newborn bloodspot screening for X-linked adrenoleukodystrophy has reshaped this pathway in the screened regions. An elevated C26:0 lysophosphatidylcholine on the bloodspot is a screen, not a diagnosis, and must be confirmed with plasma very-long-chain fatty acids, ABCD1 sequencing, a baseline morning cortisol, and a brain MRI. The confirmatory step is what distinguishes a boy who needs urgent surveillance from one who may never develop cerebral disease, and it frames the calibrated counselling the family will need. [9]
Supportive investigations quantify organ involvement and set the surveillance baseline. A brain MRI with gadolinium is mandatory for any boy with confirmed X-linked adrenoleukodystrophy, and the Loes score quantifies the demyelination burden and guides the transplant decision. A morning cortisol and synacthen test define adrenal reserve. Ophthalmology and audiology assess vision and hearing across the whole spectrum, and echocardiography and liver function tests complete the baseline. The goal is to stage the disease precisely so that therapy and surveillance are matched to the individual child. [4] [2]
Management — Resuscitation
Most children with a peroxisomal disorder do not present in collapse, but two acute scenarios demand prompt recognition. The first is adrenal crisis in a boy with X-linked adrenoleukodystrophy: hyperpigmentation, lethargy, vomiting, hypotension, and hyponatraemia with hyperkalaemia signal primary adrenal failure, and management is parenteral hydrocortisone with fluid resuscitation followed by lifelong oral replacement with a stress-day plan. The recognition that an Addisonian presentation in a boy may be the first sign of adrenoleukodystrophy is the resuscitation-level decision, because a plasma very-long-chain fatty acid level sent at that admission can change the trajectory of the whole family. [4] [5]
The second is acute neurological decline in cerebral adrenoleukodystrophy. A boy who had been stable may deteriorate rapidly as the inflammatory demyelination accelerates, with rising intracranial pressure, seizures, or a marked drop in function. The resuscitation-level decision here is that this is a treatable disorder with a closing window — the priority is to stabilise the child, confirm the diagnosis and the Loes score, and mobilise the haemopoietic stem cell transplant pathway within days. Seizures are managed symptomatically, and raised intracranial pressure with standard measures, while the disease-modifying pathway is set in motion. [4]
In the Zellweger spectrum the resuscitation phase focuses on the complications of multi-system disease: seizure control, feeding and growth support, management of cholestasis and liver dysfunction, and respiratory support. These infants are managed in a neonatal or paediatric intensive care setting with a metabolic service involved from the outset, because the prognosis and the goals of care must be discussed openly with the family early. [2] [10]
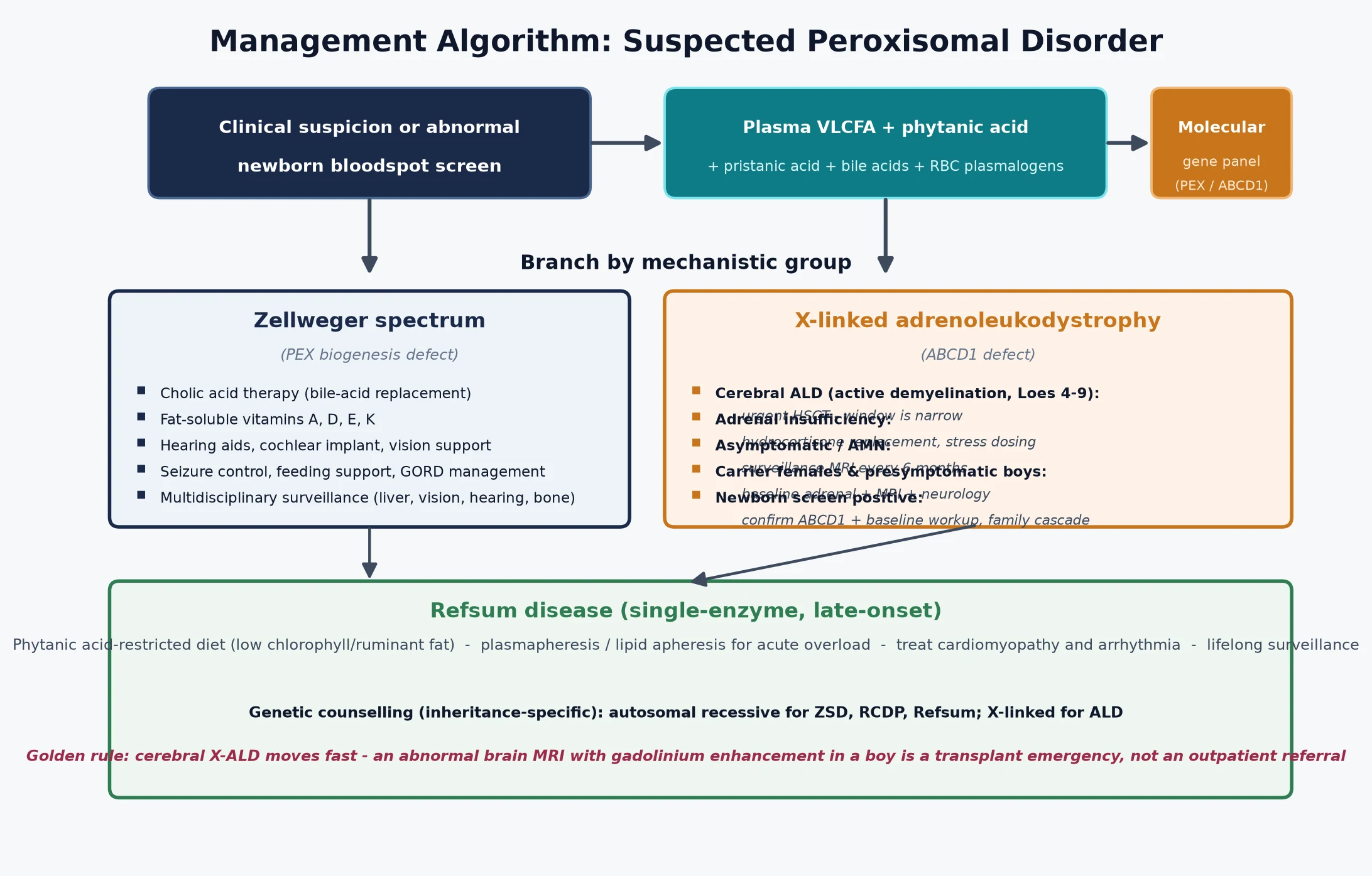
Management — Definitive & Stepwise
The definitive management matches the therapy to the disorder and, for X-linked adrenoleukodystrophy, to the stage. The figure lays out the decision pathway from suspicion or an abnormal screen to the matched treatment branch. [2] [4]
For cerebral X-linked adrenoleukodystrophy, haemopoietic stem cell transplant is the established disease-modifying therapy. Transplant works because engrafted donor microglia supply a functional transporter and modulate the inflammatory cascade that drives the demyelination, and it is effective when given inside a defined window — typically a Loes score between four and nine, with gadolinium enhancement and a still-ambulant, still-functioning child. Transplant carries significant morbidity and mortality and is offered at specialist centres, and the decision is made jointly by the metabolic, neurology, and transplant teams with the family. Adrenal insufficiency is treated in parallel with hydrocortisone replacement and a stress-day plan, because transplant does not rescue the adrenal cortex. [4] [5]
For the Zellweger spectrum, cholic acid therapy is the one disease-directed pharmacological option and is approved for reducing the toxic bile-acid intermediates that accumulate when peroxisomal bile-acid synthesis fails. Beyond cholic acid, management is supportive and multidisciplinary: fat-soluble vitamin supplementation, seizure control, feeding and growth support, hearing aids or cochlear implantation, and vision rehabilitation. For Refsum disease, a phytanic-acid-restricted diet — avoiding ruminant fat and dairy, and foods high in chlorophyll-bound phytol — is the cornerstone, with plasmapheresis or lipid apheresis reserved for acute overload that threatens the heart or the vision. Gene therapy and other investigational approaches are expanding and are accessed through clinical trials at specialist centres. [12] [2]
Cholic acid therapy — Zellweger spectrum (specialist-initiated, confirm current dosing)
Symptom-directed multidisciplinary care runs in parallel with disease-modifying therapy and is often what determines quality of life. Neurology and rehabilitation manage spasticity, weakness, and developmental support; ophthalmology and audiology manage the sensory impairments; endocrinology manages adrenal replacement; and gastroenterology manages feeding, liver function, and nutrition. For the most severe Zellweger presentations, early involvement of paediatric palliative care alongside disease-directed therapy supports the family through a progressive illness and does not preclude continuing treatment where it is available. [2] [11]
Specific Subtypes & Scenarios
The Zellweger spectrum is a continuum, and the clinical severity ranges from the lethal neonatal Zellweger syndrome through neonatal adrenoleukodystrophy to the milder infantile Refsum disease. The severe end presents at birth with the characteristic dysmorphism, profound hypotonia, seizures, hepatomegaly, and chondrodysplasia punctata, and most affected infants do not survive the first year. The milder end presents later with developmental delay, retinitis pigmentosa, sensorineural hearing loss, and liver disease, and some patients survive into adolescence and adulthood with a chronic multisystem disorder. The severity correlates broadly with the residual peroxisomal function, which the molecular result can help predict. [2] [10]
X-linked adrenoleukodystrophy is the disorder most likely to appear as a long-case vignette, and the phenotype split is the central teaching point. Childhood cerebral disease is the most devastating form, with inflammatory demyelination peaking between ages four and ten and progressing rapidly without transplant. Adrenomyeloneuropathy is the most common adult phenotype, with a slowly progressive spastic paraparesis and peripheral neuropathy that begins in the twenties. Isolated adrenal insufficiency affects a subset of boys who may never develop neurological disease, and treating the adrenal failure is mandatory regardless of the neurological status. Female carriers are not asymptomatic — roughly half develop an adrenomyeloneuropathy-like myelopathy in middle age — so surveillance of carrier women is part of lifelong care. [4] [5]
Refsum disease is the great mimic of ophthalmology clinics. An autosomal recessive defect in phytanic acid alpha-oxidation, it presents after infancy with retinitis pigmentosa, anosmia, progressive sensorineural deafness, ataxia, and a polyneuropathy, and it can be complicated by cardiac arrhythmia and ichthyosis. The phytanic acid level is the key test, and a phytanic-acid-restricted diet can halt progression and reverse some of the neuropathy. The combination of retinitis pigmentosa with anosmia is the bedside clue that should prompt the test, because anosmia is one of the earliest and most under-recognised findings. [1] [6]
Complications & Pitfalls
The complications of the peroxisomal disorders are the complications of untreated storage and progressive organ failure, and they cluster by group. In the Zellweger spectrum, the complications are the multi-system failure of a child without functional peroxisomes: intractable seizures, progressive liver disease, respiratory failure, feeding difficulty and failure to thrive, and progressive loss of vision and hearing. In X-linked adrenoleukodystrophy, the complications are the consequences of inflammatory demyelination — progressive dementia, blindness, deafness, spasticity, and a vegetative state in untreated cerebral disease — and the silent adrenal failure that can declare itself as a crisis. [2] [4]
The most consequential pitfall is delay in cerebral adrenoleukodystrophy. Treating a failing boy as a behavioural problem or a learning difficulty for months forfeits the transplant window and converts a potentially modifiable disease into a fatal one. A boy with new behavioural or cognitive change, with or without subtle neurological signs, deserves a brain MRI with gadolinium, not reassurance. A second pitfall is missing the adrenal insufficiency: a boy with adrenoleukodystrophy can present with adrenal failure years before any neurological disease, and an unexplained Addisonian picture in a boy should always trigger a very-long-chain fatty acid level. [5] [9]
A third pitfall is misreading a newborn screening result. An elevated C26:0 lysophosphatidylcholine on the bloodspot reflects a biochemical state, not a clinical destiny, and many screen-positive infants carry variants of uncertain significance or will develop a milder phenotype. Confirmatory plasma very-long-chain fatty acids, ABCD1 sequencing, and a baseline brain MRI are essential before counselling the family, and the follow-up must be calibrated — not every screen-positive boy needs a transplant, but every screen-positive boy needs structured surveillance. Finally, assuming that haemopoietic stem cell transplant will rescue a Loes-advanced brain sets up both clinician and family for avoidable disappointment, because transplant works only in the early inflammatory window. [9] [4]
Prognosis & Disposition
Prognosis is determined by the specific disorder, the age at onset, the genotype, and crucially the timing of treatment. The severe end of the Zellweger spectrum remains lethal in infancy despite supportive care and cholic acid, while the milder end allows survival into adolescence and adulthood with a chronic multisystem disorder. Childhood cerebral adrenoleukodystrophy, uniformly fatal within two to five years without treatment, is modified by haemopoietic stem cell transplant when the transplant is given inside the Loes-score window, with a meaningful proportion of boys retaining function — though transplant-related morbidity and residual disease are real. Adrenomyeloneuropathy and the carrier myelopathy pursue a slower, adult-onset course. [2] [4]
Disposition is to a specialist metabolic service with a structured transition to adult care, because these are lifelong diseases that cross every organ system and every life stage. The medical home coordinates surveillance — growth, development, vision, hearing, adrenal function, and brain MRI for the adrenoleukodystrophy group — and integrates the family's psychosocial, educational, and genetic-counselling needs. For the most severe Zellweger presentations, early involvement of paediatric palliative care alongside disease-directed therapy supports the child and the family through a progressive illness, and does not preclude continuing treatment where it is available. [11]
Special Populations
Newborn screening has created a distinct population — the asymptomatic neonate with an abnormal screen for X-linked adrenoleukodystrophy — whose management differs from the symptomatic child. The metabolic service must distinguish true disease, which demands structured surveillance and a baseline brain MRI, from variants of uncertain significance and late-onset phenotypes that may never cause cerebral disease. Families need honest, calibrated counselling: not every abnormal screen means a failing boy, but every abnormal screen demands a thorough, time-sensitive workup, because the cost of missing early cerebral disease is irreversible neurological injury. [9] [5]
The conditions screened for peroxisomal disease vary between jurisdictions and evolve over time, so the examining clinician should state the local panel rather than assume a universal list. In Australia and New Zealand, X-linked adrenoleukodystrophy is being progressively added to bloodspot panels as evidence and access to transplant and gene therapy mature. The principles are constant: an elevated C26:0 lysophosphatidylcholine is a screen, not a diagnosis; confirm with plasma very-long-chain fatty acids and ABCD1 sequencing within weeks; establish baseline adrenal function and a brain MRI; and refer to the regional metabolic service for structured surveillance. [9]
Consanguineous communities and founder populations carry a higher burden of the autosomal recessive forms, and a previously affected sibling reshapes the reproductive risk for every subsequent pregnancy. In these families carrier testing, prenatal diagnosis by chorionic villus sampling or amniocentesis, and preimplantation genetic testing are central to management, and the metabolic and genetics services should be involved before the next pregnancy rather than after it. Migrant, refugee, and remote populations face additional barriers — distance from specialist centres, language and cultural considerations, and inequitable access to expensive therapies and transplant — and the medical home must actively coordinate equitable, culturally safe care. [2]
Evidence, Guidelines & Regional Differences
The evidence base for treatment has grown substantially but remains uneven across the family of disorders. Haemopoietic stem cell transplant for cerebral adrenoleukodystrophy rests on cohort and registry evidence showing that transplant inside the early Loes-score window halts or slows the demyelination, and it is the rationale that underpins newborn screening for the disorder. Cholic acid therapy for the Zellweger spectrum is supported by extension studies showing a reduction in toxic bile-acid intermediates, though the clinical impact on survival and neurodevelopment is harder to measure in a rare and heterogeneous population. Gene therapy for cerebral adrenoleukodystrophy has shown early promise and is now approved or in advanced trials in several regions, broadening the options beyond transplant. [4] [12]
Guidelines are disorder-specific and specialist-led. The Zellweger spectrum overview sets the framework for cholic acid therapy and multidisciplinary care; the X-linked adrenoleukodystrophy guidance defines the transplant pathway, the surveillance schedule, and the adrenal management; and the newborn-screening consensus defines the confirmatory and follow-up pathway for a screen-positive infant. [2] [5] Regional differences persist in what is screened, what therapies are funded, and how transplant and gene therapy are accessed, so the clinician should ground recommendations in local policy while applying the universal principles: confirm fast, treat before the brain is injured, support the family across the lifespan. [9]
The frontier is gene therapy. Lentiviral and adeno-associated-virus-mediated approaches aim to deliver a functional copy of ABCD1 to the patient's own haemopoietic stem cells or directly to the brain, offering the prospect of disease modification without a donor and without graft-versus-host disease. These advances make early, accurate diagnosis more valuable than ever, because a boy who was transplant-ineligible a decade ago may be modifiable today — provided the cerebral disease is found before the inflammatory window closes. The fellowship answer therefore closes with an active posture: keep the peroxisomal disorders on the differential of any dysmorphic neonate or failing boy, confirm fast, and refer early. [4]
Exam Pearls
In a viva, lead with the clinical pattern, name the group (biogenesis versus single-enzyme), state the biochemical signature and the gene, and then state the confirmatory test and the treatment modality — that four-step structure answers almost any peroxisomal question. For a short case, the dysmorphic neonate with stippled epiphyses, the failing boy with occipito-parietal demyelination, and the retinitis-pigmentosa-and-anosmia adult are the findings that earn marks. For a long case, the management plan that matches the therapy to the group and sets out surveillance, adrenal replacement, and genetic counselling is what distinguishes a pass from a commendation. Remember the inheritance — X-linked for adrenoleukodystrophy — because it changes the family counselling and the cascade testing. [2] [4]
The single most testable principle is that the peroxisomal disorders split into the biogenesis disorders and the single-enzyme deficiencies, and that split predicts the biochemistry, the inheritance, and the treatment. Couple that with the recognition that cerebral X-linked adrenoleukodystrophy is the one disorder where a treatment decision is measured in weeks, and that newborn screening exists precisely to find these boys before the inflammatory window closes, and you have the framework that ties the whole topic together. Every peroxisomal disorder question ultimately reduces to: which group, which gene, is the brain involved, and what is the window for the therapy that reaches it. [1] [9]
References
- [1]Wanders RJA, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem, 2006.PMID 16756494
- [2]Klouwer FCC, Berendse K, Engelen M, Wanders RJA, Ferdinandusse S, Poll-The BT. Zellweger spectrum disorders: clinical overview and management approach. Orphanet J Rare Dis, 2015.PMID 26627182
- [3]Braverman NE, Raymond GV, Rizzo WB, Moser AB, Wilkinson ME, Stone EM, et al. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis and clinical care. Mol Genet Metab, 2016.PMID 26750748
- [4]Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol, 2007.PMID 17342190
- [5]Raymond GV, Moser AB, Fatemi A. X-linked adrenoleukodystrophy. GeneReviews, 1993.PMID 20301491
- [6]Braverman NE, Raymond GV, Rizzo WB, Moser AB. Zellweger spectrum disorder. GeneReviews, 1993.PMID 20301621
- [7]Klouwer FCC, Koot BGP, Engelen M, Wanders RJA, Ferdinandusse S, Poll-The BT. Clinical and biochemical pitfalls in the diagnosis of peroxisomal disorders. Neuropediatrics, 2016.PMID 27089543
- [8]Wanders RJA, Klouwer FCC, Ferdinandusse S, Engelen M, Poll-The BT. Clinical and laboratory diagnosis of peroxisomal disorders. Methods Mol Biol, 2017.PMID 28409475
- [9]Lee S, McCandless SE, Mallott J, Grebe T, Rinaldo P, Hedlund G, et al. Evaluation of X-linked adrenoleukodystrophy newborn screening in North Carolina. JAMA Netw Open, 2020.PMID 32003821
- [10]Bose M, Beck-Woölner N, Birch J, Bhatia K, Brown R, Cif L, et al. Characterization of severity in Zellweger spectrum disorder by clinical findings: A scoping review. Cells, 2022.PMID 35741019
- [11]Berendse K, Engelen M, Ferdinandusse S, Klootwijk L, Mooijer PW, Touraine R, et al. Zellweger spectrum disorders: clinical manifestations in patients surviving into adulthood. J Inherit Metab Dis, 2016.PMID 26287655
- [12]Klouwer FCC, Bams-Mengech A, Engelen M, van der Plaat D, Goorden SMI, van Roermund CWT, et al. The cholic acid extension study in Zellweger spectrum disorders: Results and implications for therapy. J Inherit Metab Dis, 2019.PMID 30793331