Paeds · genetics-dysmorphology-and-metabolism
Prader-Willi and Angelman syndromes
Also known as 15q11-q13 imprinting disorders · PWS · Angelman syndrome · Happy puppet syndrome · Genomic imprinting disorders of chromosome 15
A fellowship approach to Prader-Willi and Angelman syndromes: recognise the reciprocal imprinting errors at 15q11-q13, confirm the diagnosis with methylation analysis, and build syndrome-specific multidisciplinary management — growth hormone and behavioural support for PWS, anticonvulsant and communication strategies for AS — combined with imprinting-aware cascade genetic counselling.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who thinks in three layers at once. The first layer is the child in front of you: the hypotonic neonate failing to feed (PWS) or the ataxic toddler with seizures and a happy demeanour (AS). The second is the molecular mechanism: a parent-of-origin-specific imprinting error at 15q11-q13 that silences one parental allele and leaves the other unable to compensate. The third is the family: the subtype (deletion, uniparental disomy, imprinting defect, or UBE3A mutation) sets the recurrence risk, and counselling must be imprinting-aware — because imprinting-centre defects and UBE3A mutations carry up to 50 per cent recurrence, while typical deletions and UPD are sporadic. [1] [2] [4]
Overview & Definition
Prader-Willi syndrome and Angelman syndrome are the prototypical disorders of genomic imprinting in humans, and they arise from reciprocal errors at the same chromosomal locus — 15q11-q13. Genomic imprinting is the parent-of-origin-specific expression of certain genes, where only the maternal or paternal copy is active in specific tissues. In the 15q11-q13 region, paternally expressed genes (including SNRPN, MAGEL2, NDN, and the SNORD116 snoRNA cluster) are active on the paternal chromosome, while UBE3A, a ubiquitin-protein ligase, is active primarily on the maternal chromosome in the brain. [3] [5]
Loss of the paternal contribution produces Prader-Willi syndrome: the child has no functional paternally expressed genes, and the maternal copies are silenced by imprinting. The neonate presents with profound hypotonia, poor feeding, and failure to thrive, and the phenotype shifts after infancy to hyperphagia, obesity, intellectual disability, short stature, and hypogonadism. Loss of the maternal contribution at the same locus — specifically of UBE3A — produces Angelman syndrome: the child lacks functional UBE3A protein in the brain because the paternal allele is silenced in neurons, and the result is severe intellectual disability, ataxia, a characteristic happy demeanour, seizures, and microcephaly. [1] [2]
Classification
The clinical classification that matters most is molecular, because the molecular subtype determines recurrence risk and, in some cases, phenotype severity. Both PWS and AS have three or four molecular subtypes, and the distribution differs between them. [3] [4]

For Prader-Willi syndrome, approximately 70 per cent of cases carry a paternal deletion of the 15q11-q13 critical region (Type I or Type II, differing in deletion size), approximately 25 per cent carry maternal uniparental disomy (two maternal copies and no paternal copy), and approximately 5 per cent carry an imprinting-centre defect that silences the paternal allele without a deletion or UPD. Deletion and UPD are sporadic events with low recurrence risk (less than 1 per cent), but imprinting-centre defects can carry a recurrence risk up to 50 per cent if a parent carries the defect. [1] [4]
For Angelman syndrome, approximately 70 per cent carry a maternal deletion of 15q11-q13, approximately 10 per cent carry a UBE3A mutation, approximately 3 to 5 per cent carry paternal uniparental disomy, and approximately 3 to 5 per cent carry an imprinting-centre defect. The deletion subtype tends to produce the most severe phenotype (seizures, absent speech, microcephaly, fair skin and hair), while the UPD and imprinting-defect subtypes are often milder. Critically, the UBE3A mutation form has a normal methylation result, so methylation analysis alone will miss it — UBE3A sequencing is required when the phenotype is classic but methylation is normal. The UBE3A mutation subtype carries a 50 per cent recurrence risk if the mother is a carrier. [2] [4]
Epidemiology & Risk Factors
Prader-Willi syndrome and Angelman syndrome are rare, with broadly similar birth prevalences: PWS at approximately 1 in 15,000 to 25,000 live births and AS at approximately 1 in 12,000 to 20,000. The similarity reflects the fact that both arise from errors at the same locus and that the major mechanism — a de novo deletion — is the same type of event in both, differing only in which parental chromosome is affected. [3] [5]
The major epidemiological determinant of subtype is parental origin and chromosomal event. The deletion subtype is a de novo interstitial deletion that occurs during gametogenesis or early embryogenesis, and its risk is not strongly related to parental age. Uniparental disomy, however, is associated with advanced maternal age for the maternal UPD form of AS, because the meiotic nondisjunction that produces a trisomic zygote (which then undergoes trisomy rescue, losing the paternal chromosome and leaving two maternal copies) is more likely with increasing maternal age. [3]
The practical consequence is that recurrence-risk counselling is subtype-dependent and requires molecular subtype determination after a positive methylation result. Imprinting-centre defects and UBE3A mutations can be inherited and carry high recurrence risk (up to 50 per cent), while deletions and UPD are sporadic with very low recurrence. This is why determining the molecular subtype is not an academic exercise — it is the counselling answer. [4]
Pathophysiology
The molecular story begins with the concept of genomic imprinting — the parent-of-origin-specific silencing of certain genes through DNA methylation and histone modification. At the 15q11-q13 locus, an imprinting centre near the SNRPN gene controls the methylation state of the entire region. On the paternal chromosome, the imprinting centre is unmethylated and active, allowing expression of paternally expressed genes including SNRPN, MAGEL2, NDN, and the SNORD116 snoRNA cluster. On the maternal chromosome, the imprinting centre is methylated and silenced, but the maternal UBE3A allele is active in neurons. [3] [5]
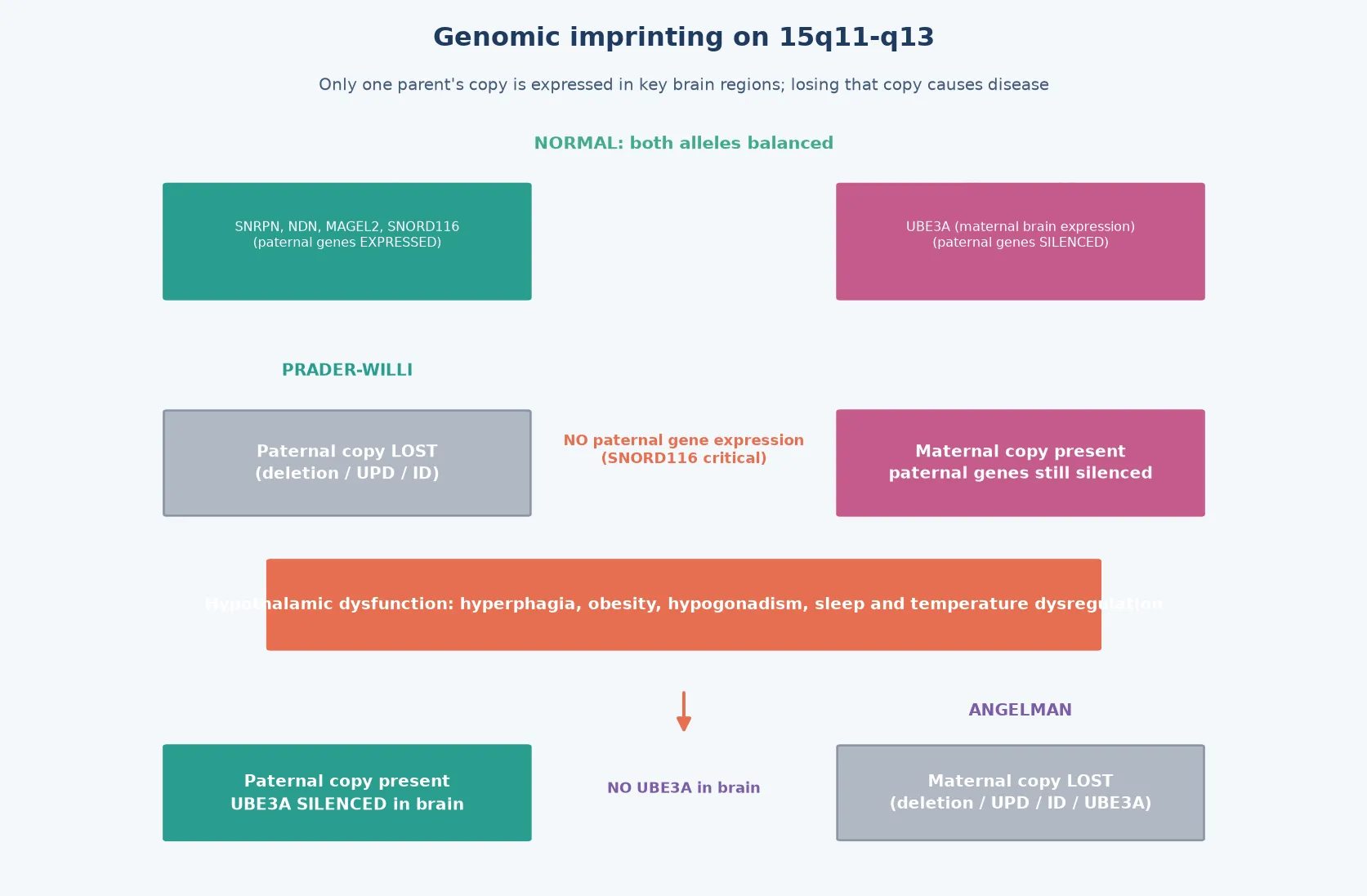
In Prader-Willi syndrome, loss of the paternal contribution — whether by deletion, maternal UPD, or imprinting defect — eliminates expression of all paternally expressed genes. The absence of the SNORD116 snoRNA cluster is now considered a key driver of the hyperphagia and growth dysregulation, because animal models show that SNORD116 knockout mice develop hyperphagia and growth failure. The absence of other paternally expressed genes (MAGEL2, NDN) contributes to the neonatal hypotonia, endocrine dysfunction, and behavioural phenotype. [1] [5]
In Angelman syndrome, the critical gene is UBE3A, which encodes a ubiquitin-protein ligase (UBE3A, also called E6-AP). In most tissues, both the maternal and paternal UBE3A alleles are expressed, but in neurons the paternal allele is silenced by an antisense transcript (UBE3A-ATS) expressed from the paternal SNRPN locus. This means that loss of the maternal UBE3A allele eliminates functional UBE3A protein in the brain, because the paternal allele is already silenced. UBE3A is a key regulator of synaptic protein turnover via the ubiquitin-proteasome pathway, and its loss disrupts synaptic plasticity, long-term potentiation, and dendritic spine development — producing the severe neurodevelopmental phenotype. [2] [10]
This brain-specific maternal-only expression of UBE3A is the therapeutic rationale for emerging gene therapy strategies in Angelman syndrome. Because the paternal UBE3A allele is intact but silenced by UBE3A-ATS, unsilencing the paternal allele — through antisense oligonucleotides that block UBE3A-ATS — could restore functional UBE3A protein in the brain. This approach is in active clinical development and represents one of the most promising precision medicine strategies in any neurogenetic disorder. [9] [10]
Clinical Presentation
The presentation of PWS is dramatically age-dependent and passes through two distinct phases that a fellowship candidate must know cold. Phase one (neonatal to early infancy) is dominated by profound hypotonia, poor feeding, weak cry, hyporeflexia, and failure to thrive. The baby often requires nasogastric or gastrostomy feeding for months, and the differential at this stage is the floppy infant — spinal muscular atrophy, congenital myotonic dystrophy, and metabolic disease. Facial features may be subtle but include a narrow bifrontal diameter, almond-shaped palpebral fissures, a thin upper lip, and a down-turned mouth. Cryptorchidism and hypogonadism are common in male infants. [1] [5]
Phase two (typically beginning around 18 months to 3 years) is the transition to hyperphagia and obesity. The child develops an insatiable appetite, food-seeking behaviour, and rapid weight gain if intake is not strictly controlled. This hyperphagia is neurologically driven — the child genuinely does not feel satiated — and it is accompanied by intellectual disability (typically mild to moderate), short stature, small hands and feet, delayed motor milestones, sleep disturbance, and characteristic behavioural features including temper outbursts, skin-picking, and rigid thinking. [1]
Angelman syndrome presents differently and is usually recognised in infancy or early childhood. The key features are severe intellectual disability with absent or minimal speech, ataxic or puppet-like gait with uplifted flexed arms, a happy demeanour with frequent, often inappropriate laughter and excitability, seizures that are often multiple-type and treatment-resistant, microcephaly (usually postnatal), and prognathism. Children with the deletion subtype may also have fair skin and hair relative to their family, and seizures tend to be more severe and medication-resistant in this subtype. Sleep disturbance is very common. [2] [6]
Differential Diagnosis
The differential of PWS in the neonate is the differential of neonatal hypotonia, and it includes spinal muscular atrophy (posterior column weakness, tongue fasciculations, normal cognition), congenital myotonic dystrophy (maternal history, facial weakness, arthrogryposis), congenital myopathies, metabolic disease (mitochondrial, peroxisomal), and benign congenital hypotonia. PWS is distinguished by the combination of hypotonia with poor feeding, characteristic facial features, hypogonadism, and the molecular test. In older children, the differential of obesity with intellectual disability includes Bardet-Biedl syndrome (retinitis pigmentosa, polydactyly, renal anomalies), Cohen syndrome, and leptin or melanocortin pathway defects. [1] [5]
The differential of Angelman syndrome includes Rett syndrome (a regression in a girl after normal early months, with MECP2 mutation), Mowat-Wilson syndrome (ZEB2, Hirschsprung disease, distinctive face), Christianson syndrome (SLC9A6, X-linked), and cerebral palsy with ataxia. The happy demeanour, seizure pattern, and severe expressive language impairment with relatively better receptive language are distinctive for AS. The critical point is that methylation testing of 15q11-q13 will identify the deletion, UPD, and imprinting-defect forms of AS in a single assay, but UBE3A sequencing is needed for the mutation form when methylation is normal. [2] [4]
Clinical & Bedside Assessment
The bedside assessment of suspected PWS begins in the neonatal period with structured observation of tone, feeding, reflexes, and facial gestalt. Quantify the hypotonia (central vs peripheral, using the hypothesis that PWS is central hypotonia with preserved cognition early), document the feeding pattern (poor suck, weak cry, failure to thrive), and examine for dysmorphic features (narrow forehead, almond-shaped palpebral fissures, thin upper lip, down-turned mouth), cryptorchidism, small hands and feet, and acromicria. Draw a three-generation pedigree asking about neonatal hypotonia, obesity, intellectual disability, and consanguinity. [1]
For suspected Angelman syndrome, the assessment centres on the developmental trajectory (often normal early, then plateau or regression around 6-12 months), the seizure history (onset typically 1-3 years, multiple types), the communication profile (minimal or absent speech, but relatively good receptive language), the gait (ataxic, wide-based, uplifted arms), the facial features (microcephaly, prognathism, deep-set eyes, fair skin and hair in deletion subtype), and the behavioural phenotype (happy demeanour, excitability, hand-flapping, fascination with water). The happy demeanour and the seizure pattern are the two discriminators that should trigger methylation testing. [2] [6]
Investigations
The molecular investigation strategy for both syndromes is tiered and starts with methylation analysis. Methylation-specific PCR (MS-PCR) or methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) detects the parent-of-origin methylation imprint at the SNRPN locus and can diagnose both PWS and AS in a single assay. In PWS, only the maternal methylation pattern is seen (absent paternal pattern), confirming loss of paternal expression. In AS (deletion, UPD, and imprinting-defect forms), only the paternal methylation pattern is seen (absent maternal pattern), confirming loss of maternal expression. [1] [4]
Why a normal methylation result does not always exclude Angelman syndrome
Approximately 10 per cent of Angelman syndrome cases are caused by a UBE3A mutation, not a deletion, UPD, or imprinting defect. These children have a normal methylation pattern at SNRPN, because the imprinting marks are intact and both parental copies are present. The reflex is to sequence UBE3A when the clinical phenotype is classic AS (happy demeanour, ataxia, severe ID, seizures) but the methylation result is normal. Missing the UBE3A mutation form is a serious pitfall because it carries a 50 per cent recurrence risk if the mother is a carrier. [2] [4]
A positive methylation result must be followed by subtype determination, because the subtype sets recurrence risk and may modify phenotype severity. Fluorescence in situ hybridisation (FISH) or chromosomal microarray distinguishes deletion from UPD and imprinting defect. If a deletion is confirmed, the case is a de novo event with low recurrence risk (less than 1 per cent). If no deletion is found, UPD testing by microsatellite polymorphism analysis of both parents determines whether the child has uniparental disomy. If neither deletion nor UPD is found, an imprinting-centre defect is likely, and parental testing determines whether a parent carries the defect — because this subtype carries up to 50 per cent recurrence risk. For the UBE3A mutation form of AS, maternal carrier testing determines whether the mother carries the mutation. [4]
Baseline assessments that complete the diagnostic work-up include polysomnography (for sleep-disordered breathing, particularly before growth hormone initiation in PWS), body composition analysis, endocrine panel (IGF-1, thyroid function, gonadal hormones for hypogonadism), EEG (for seizure characterisation in AS), vision and hearing assessment, developmental or cognitive assessment, and scoliosis screening. [1] [6]
Management — Resuscitation
Resuscitation in PWS rarely involves an acute physiological collapse in the neonatal period, but two scenarios demand urgent action. The first is severe feeding failure in the neonate, where nasogastric or gastrostomy feeding is required to prevent aspiration and failure to thrive. The second is an acute metabolic or gastrointestinal complication of obesity in older children and adults — type 2 diabetes, respiratory compromise, obstructive sleep apnoea, and the life-threatening complication of gastric dilation or rupture from binge eating. Any PWS patient presenting with abdominal pain, vomiting, or distension after suspected binge eating must be assessed urgently for gastric dilation or necrosis, because this carries a high mortality. [1]
In Angelman syndrome, the acute scenario is status epilepticus or severe seizure clusters. AS seizures are often multiple-type (atypical absence, myoclonic, tonic-clonic, atonic) and can be medication-resistant. Standard paediatric protocols apply — benzodiazepines first, then loading with appropriate anticonvulsants — but the child may require multiple agents for adequate control. The underlying principle in both syndromes is that the imprinting disorder does not change the resuscitation algorithm, but it changes the threshold to act and the comorbidities to anticipate. [2] [6]
Management — Definitive & Stepwise
Definitive management of PWS is a multi-domain framework that adapts across the lifespan. The neonatal and infant phase is dominated by feeding support (nasogastric or gastrostomy feeding, caloric supplementation). From early childhood, the focus shifts to strict dietary and environmental control of hyperphagia: locked food access, structured meal plans with visual supports, and behavioural strategies, because there is currently no approved pharmacotherapy for the hyperphagia itself. Growth hormone therapy is standard of care and is supported by consensus guidelines, improving body composition, height, lean muscle mass, and cognitive outcomes — but it requires surveillance (sleep study before initiation, scoliosis monitoring, IGF-1 levels) because of the risk of sleep-disordered breathing and scoliosis progression. Endocrine management includes monitoring and treatment of hypogonadism, hypothyroidism, and osteoporosis. [7] [1]
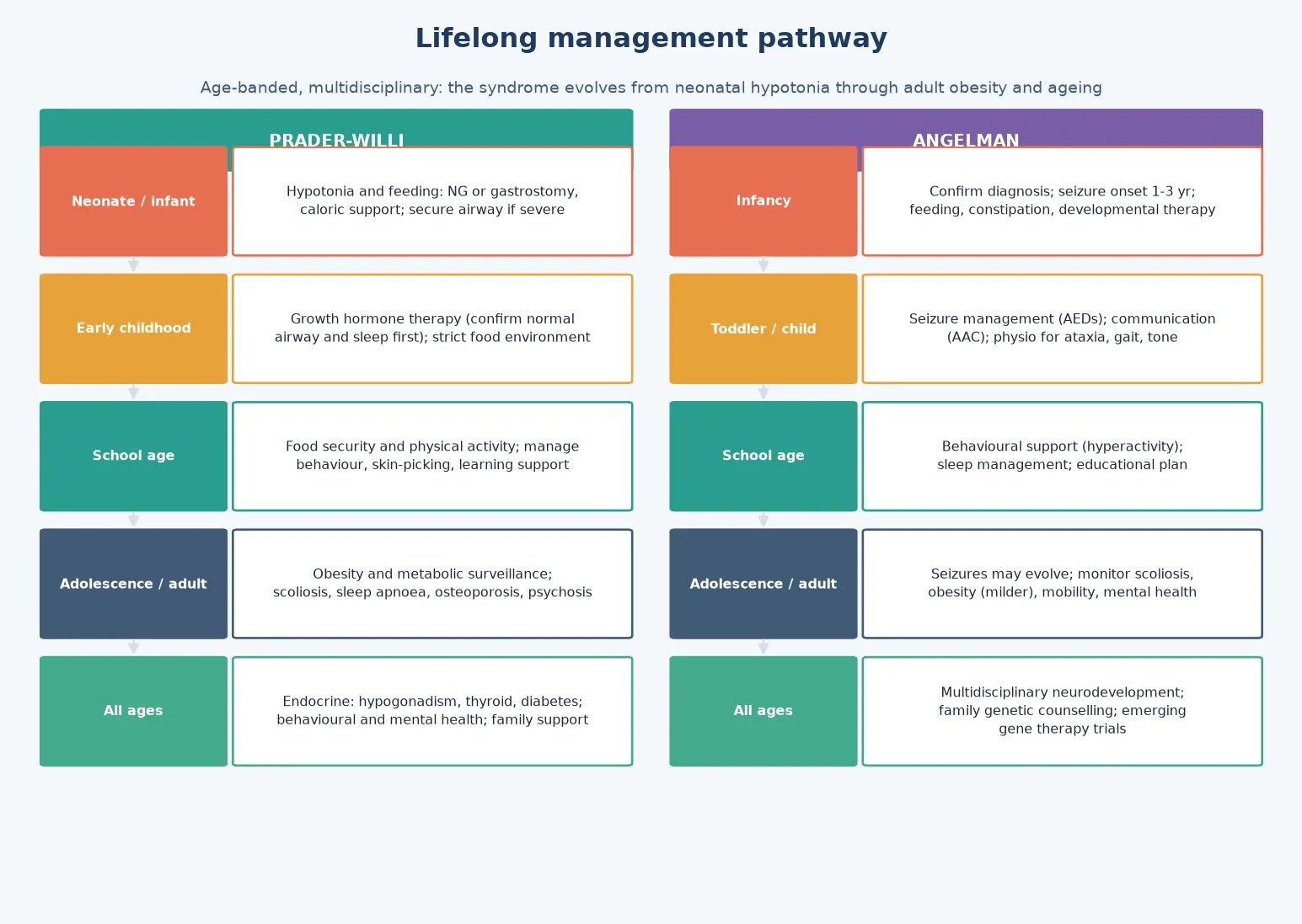
Definitive management of Angelman syndrome is anchored by seizure control, communication, and motor support. Anticonvulsant optimisation is central: valproate, levetiracetam, and clonazepam are commonly used, and some centres prefer ethosuximide or a ketogenic diet for atypical absence seizures. Carbamazepine can worsen myoclonic seizures in some patients and should be used with caution. Communication strategies are critical, because expressive language is typically absent or minimal while receptive language is relatively preserved — augmentative and alternative communication (AAC), sign language, and picture-based systems unlock the child's communicative potential. Motor and physical therapy addresses the ataxia, hypotonia, and gait abnormality, and sleep management — which may include melatonin, clonidine, or behavioural sleep strategies — addresses the very common sleep disturbance. [6] [2]
P.W.S. \u2014 the management anchors
Specific Subtypes & Scenarios
The neonate with PWS is the scenario where early diagnosis changes everything. The hypotonic, poorly feeding neonate needs nasogastric feeding, careful monitoring for aspiration, and early methylation testing to confirm the diagnosis before discharge. Parents must be counselled about the trajectory from failure to thrive to hyperphagia, the role of growth hormone, and the lifelong dietary vigilance that will be required. Early diagnosis also allows early enrolment in early intervention services and proactive developmental support. [1]
The AS child with refractory epilepsy is a scenario requiring a systematic anticonvulsant strategy. Multiple seizure types may coexist, and sequential trials of valproate, levetiracetam, topiramate, clonazepam, or a ketogenic diet may be needed. The communication barrier makes seizure detection harder — parents and carers must be trained to recognise subtle seizure patterns. The goal is functional seizure freedom with minimal medication burden, balancing seizure control against sedation and behavioural side effects. [6]
The imprinting-centre-defect family is the scenario where recurrence risk is highest. If a parent carries an imprinting-centre defect, the recurrence risk for subsequent children can approach 50 per cent, and prenatal or preimplantation genetic diagnosis must be offered. This is why subtype determination after a positive methylation result is not optional — it is the counselling answer that shapes the family's reproductive decisions. [4]
The PWS adolescent and young adult faces transition challenges: worsening behavioural rigidity, risk of psychotic illness (particularly in the UPD subtype), increasing obesity burden if dietary control falters, hypogonadism requiring endocrine management, and the need for supported living arrangements. The transition to adult care must explicitly hand over the care plan, the genetic record, and the behavioural and dietary strategies, because loss of structured support is a high-risk point. [5] [8]
The UBE3A-mutation AS child is the scenario where methylation testing is normal. The clinical phenotype is classic AS (happy demeanour, ataxia, seizures, severe ID), but the methylation result shows both parental patterns. The reflex is UBE3A sequencing, and if a mutation is found, the mother's carrier status must be determined, because a carrier mother has a 50 per cent recurrence risk in each pregnancy. [2] [9]
Complications & Pitfalls
The complications of PWS accumulate over a lifetime and are the main determinants of morbidity and mortality. Morbid obesity drives type 2 diabetes, obstructive sleep apnoea, obesity-hypoventilation syndrome, and cardiovascular disease. Temperature dysregulation and a reduced pain response can mask serious illness. Scoliosis is common and may be exacerbated by growth hormone therapy. Osteoporosis results from hypogonadism and low muscle mass. Psychiatric complications — including temper outbursts, obsessive-compulsive behaviours, and psychosis in young adults (particularly the UPD subtype) — are major sources of morbidity. Early death is most commonly related to respiratory and cardiopulmonary complications of obesity. [1] [8]
The complications of AS centre on refractory epilepsy, communication barriers that mask medical problems, scoliosis and contractures from abnormal tone, aspiration risk, and the lifelong dependency that severe intellectual disability produces. Life expectancy is relatively normal with good seizure control and supportive care, but quality of life depends on communication access, seizure management, and family support. [2] [6]
Prognosis & Disposition
Prognosis in PWS is determined by the severity of hyperphagia and obesity control, the comorbidity burden (sleep apnoea, diabetes, scoliosis), and the quality and timing of growth hormone and behavioural support. With modern multidisciplinary care including growth hormone therapy, strict dietary control, and proactive comorbidity management, life expectancy has improved substantially, though it remains reduced relative to the general population. The leading modifiers of outcome are weight management, sleep-disordered breathing treatment, and behavioural and psychiatric support through adolescence and adulthood. [1] [5]
Prognosis in AS is determined by seizure control, communication access, motor function, and the quality of multidisciplinary support. Life expectancy is relatively normal with good supportive care, and most individuals achieve some degree of ambulation and functional communication with AAC. The leading modifiers of outcome are seizure management, physical and communication therapy, and family resilience. [2] [6]
Disposition is shared, lifelong, medical-home care in both syndromes: the general paediatrician or GP owns coordination and preventive care, the genetics service owns the counselling and cascade testing, endocrinology owns growth hormone and gonadal management (PWS), neurology owns seizure management (AS), and allied health and education own the developmental, behavioural, and communication support. Every transition — into school, into adolescence, and into adult services — is a point at which support can be lost, so the plan must travel with the child. [1] [2]
Special Populations
The same imprinting diagnosis behaves differently across populations because access, recognition, and service models are unevenly distributed. In remote and Indigenous communities, later presentation, limited genetic service access, and lower rates of cascade testing mean that affected children and their relatives are diagnosed late, if at all, and culturally safe genetic counselling is essential. In migrant, refugee, and asylum-seeking families, language barriers, incomplete family histories, and different prior test records complicate the pedigree and the counselling, and an interpreter must be used at every key consultation. [1]
In families managing complex disability, fragmentation of care is the chief threat to support; a written, shared care plan reconciled at every visit is the intervention that matters. In adolescents transitioning to adult care, the move is a high-risk point for loss of behavioural, dietary, and endocrine support (PWS) or seizure and communication support (AS), so the transition plan must explicitly hand over the care plan and the genetic record. The PWS adolescent with emerging psychiatric features and the AS adult with increasing seizure burden are the two highest-risk transition scenarios. [5] [6]
Evidence, Guidelines & Regional Differences
The evidence base rests on four pillars: GeneReviews clinical guidelines, the EMQN/ACGS molecular testing standards, the Duis Angelman consensus on standards of care, and the PWS growth hormone consensus. The GeneReviews entries for PWS (Driscoll et al., PMID 20301505) and AS (Dagli et al., PMID 20301323) set the structure of clinical features, diagnosis, and management that most national programmes adopt. The Cassidy imprinting review (PMID 9556704) and the Cassidy and Driscoll Genetics in Medicine review (PMID 22237428) frame the reciprocal relationship between the two syndromes as the paradigm of genomic imprinting. [1] [2] [3] [5]
The EMQN/ACGS molecular testing guidelines (Beygo et al., PMID 31235867) define the testing algorithm: methylation analysis as first-tier, subtype determination by deletion/UPD testing, and UBE3A sequencing for methylation-normal cases. The Duis Angelman consensus (PMID 35150089) establishes multidisciplinary standards of care for AS, and the PWS growth hormone consensus (PMID 23609333) anchors the endocrine management framework. Emerging precision medicine approaches — antisense oligonucleotides to unsilence paternal UBE3A (PMID 39168152, PMID 38248358) and investigational hyperphagia pharmacotherapy for PWS — represent the next frontier but are not yet standard of care. [4] [6] [9]
In Australia and New Zealand, methylation testing for PWS and AS is publicly funded when clinical criteria are met, and genetic counselling is delivered through state clinical genetics services with access in metropolitan, regional, and (by outreach or telehealth) remote settings. Cascade testing of at-risk relatives is coordinated through the genetics service once an index case is confirmed and the molecular subtype is determined. Growth hormone therapy for PWS is publicly funded under the Pharmaceutical Benefits Scheme when criteria are met, and disability support frameworks (the NDIS in Australia, and the equivalent in New Zealand) fund the allied health, educational, and behavioural supports that anchor long-term care. Always confirm the current local eligibility criteria for genetic testing, growth hormone, and disability funding, as these change.
[1][6]Exam Pearls
A fellowship candidate answering on PWS or AS should land five anchor points and avoid three classic traps. The anchors are the imprinting definition (paternal loss = PWS, maternal UBE3A loss = AS), the molecular subtype distribution and its recurrence-risk implications, the methylation-based diagnostic strategy (with UBE3A sequencing for methylation-normal AS), the syndrome-specific management framework (growth hormone and dietary control for PWS; anticonvulsants and communication for AS), and the cascade-testing obligation with imprinting-aware counselling. The traps are failing to test (waiting for the classic face or attributing the happy child to simple delay), failing to determine the subtype after a positive methylation result, and missing the UBE3A-mutation form of AS because methylation is normal. [1] [2] [4]
References
- [1]Driscoll DJ, Miller JL, Schwartz S, Cassidy SB. Prader-Willi Syndrome. GeneReviews, 1993.PMID 20301505
- [2]Dagli AI, Mueller J, Williams CA. Angelman Syndrome. GeneReviews, 1993.PMID 20301323
- [3]Cassidy SB. Prader-Willi and Angelman syndromes. Disorders of genomic imprinting. Medicine, 1998.PMID 9556704
- [4]Beygo J, Buiting K, Ramsden SC, Ellis R, Clayton-Smith J, Kurth I, et al. Update of the EMQN/ACGS best practice guidelines for molecular analysis of Prader-Willi and Angelman syndromes. Eur J Hum Genet, 2019.PMID 31235867
- [5]Cassidy SB, Driscoll DJ. Prader-Willi syndrome. Eur J Hum Genet, 2012.PMID 22237428
- [6]Duis J, van Bon L, Bunt C, Hoffmann L, Wegner K, Johnson S, et al. A multidisciplinary approach and consensus statement to establish standards of care for Angelman syndrome. Mol Genet Genomic Med, 2022.PMID 35150089
- [7]Koch L. Growth hormone in health and disease: Consensus guidelines for GH therapy in Prader-Willi syndrome--this way forward? Nat Rev Endocrinol, 2013.PMID 23609333
- [8]Pellikaan K, Widdershoven J, Csabai L, Khanna M, Nicol C, Theunissen TEJ, et al. Malignancies in Prader-Willi Syndrome: Results From a Large International Cohort and Literature Review. J Clin Endocrinol Metab, 2023.PMID 37267430
- [9]Manssen L, Wirths O, Bhatti MFM. Precision Medicine in Angelman Syndrome. Neuropediatrics, 2025.PMID 39168152
- [10]Roy B, DunAW, Patel P, Pahan K. UBE3A: The Role in Autism Spectrum Disorders (ASDs) and a Potential Candidate for Biomarker Studies and Designing Therapeutic Strategies. Diseases, 2023.PMID 38248358