Paeds · genetics-dysmorphology-and-metabolism
Skeletal dysplasias
Also known as Skeletal dysplasias · Osteochondrodysplasias · Constitutional disorders of bone · Bone dysplasias · Chondrodysplasias
A fellowship approach to the skeletal dysplasias: recognise the disproportionate child and the lethal short-limbed newborn, group the disorders by molecular pathway (FGFR3, type II collagen, type I collagen, skeletal ciliopathies), confirm with a skeletal survey and targeted molecular testing, and match the therapy — vosoritide for achondroplasia, bisphosphonates for osteogenesis imperfecta — to the diagnosis while running surveillance for the lethal complications of foramen magnum compression and recurrent fracture.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A baby born at term with a very narrow chest and short limbs develops grunting respiratory distress in the delivery room and is intubated with difficulty; the chest radiograph shows short horizontal ribs and a small thorax, and the question is whether this is a lethal dysplasia. Down the corridor, a two-year-old with rhizomelic short limbs, a large head, and trident hands attends clinic for growth and developmental surveillance, and her parents ask whether any treatment can increase her adult height. In both rooms the unifying task is to recognise a skeletal dysplasia, group it by its molecular pathway, and match the management to whether it is lethal or compatible with long life. [1] [10]
B · O · N · E · S
Overview & Definition
A skeletal dysplasia is an inherited disorder in which a gene that governs cartilage or bone development is altered, so that the skeleton grows abnormally in shape, length, or density from early fetal life onward. The skeleton forms by two routes — endochondral ossification, where a cartilage template is replaced by bone at the growth plate, and intramembranous ossification, where bone is laid down directly — and most dysplasias disturb endochondral ossification at the growth plate. The result is disproportionate short stature, meaning the limbs are shortened relative to the trunk or one segment of the limb is shortened relative to another, and the disproportion distinguishes a skeletal dysplasia from the proportionate short stature of endocrine, nutritional, or constitutional causes. [2] [5]
The 2019 revision of the international nosology lists four hundred and sixty-one distinct skeletal disorders in forty-two groups, defined by their causative gene and their clinical and radiographic phenotype. The number is daunting, but a small cluster of diagnoses accounts for most clinical encounters: achondroplasia is the commonest non-lethal dysplasia, osteogenesis imperfecta is the commonest dysplasia seen in general paediatric fracture practice, and thanatophoric dysplasia is the commonest lethal dysplasia recognised at birth. Mastering these three, together with the principle that the molecular pathway predicts the management, equips the candidate to reason through almost any skeletal dysplasia vignette. [3] [4]
The single most useful clinical concept is the lethal versus non-lethal dichotomy, because it sets the entire management trajectory. A lethal dysplasia — thanatophoric dysplasia, achondrogenesis, osteogenesis imperfecta type II, and the short-rib polydactyly syndromes — causes death from respiratory failure in the perinatal period because the small thorax cannot support lung inflation, and the management shifts to palliation, accurate molecular diagnosis for recurrence counselling, and prenatal diagnosis in future pregnancies. A non-lethal dysplasia such as achondroplasia or osteogenesis imperfecta types I, III, and IV is compatible with a normal lifespan and intellect, and the task becomes surveillance, therapy, and psychosocial support. Naming this dichotomy at the first consultation frames every later decision. [2] [10]
Classification
Clinicians classify the skeletal dysplasias by the molecular pathway that is disrupted, because the pathway predicts the phenotype and increasingly the therapy. The figure lays out the four high-yield pathway groups alongside their representative diagnoses and the gene involved; the groups most likely to appear in an examination are the FGFR3 group, the type II collagenopathies, osteogenesis imperfecta, and the skeletal ciliopathies. [2] [3]
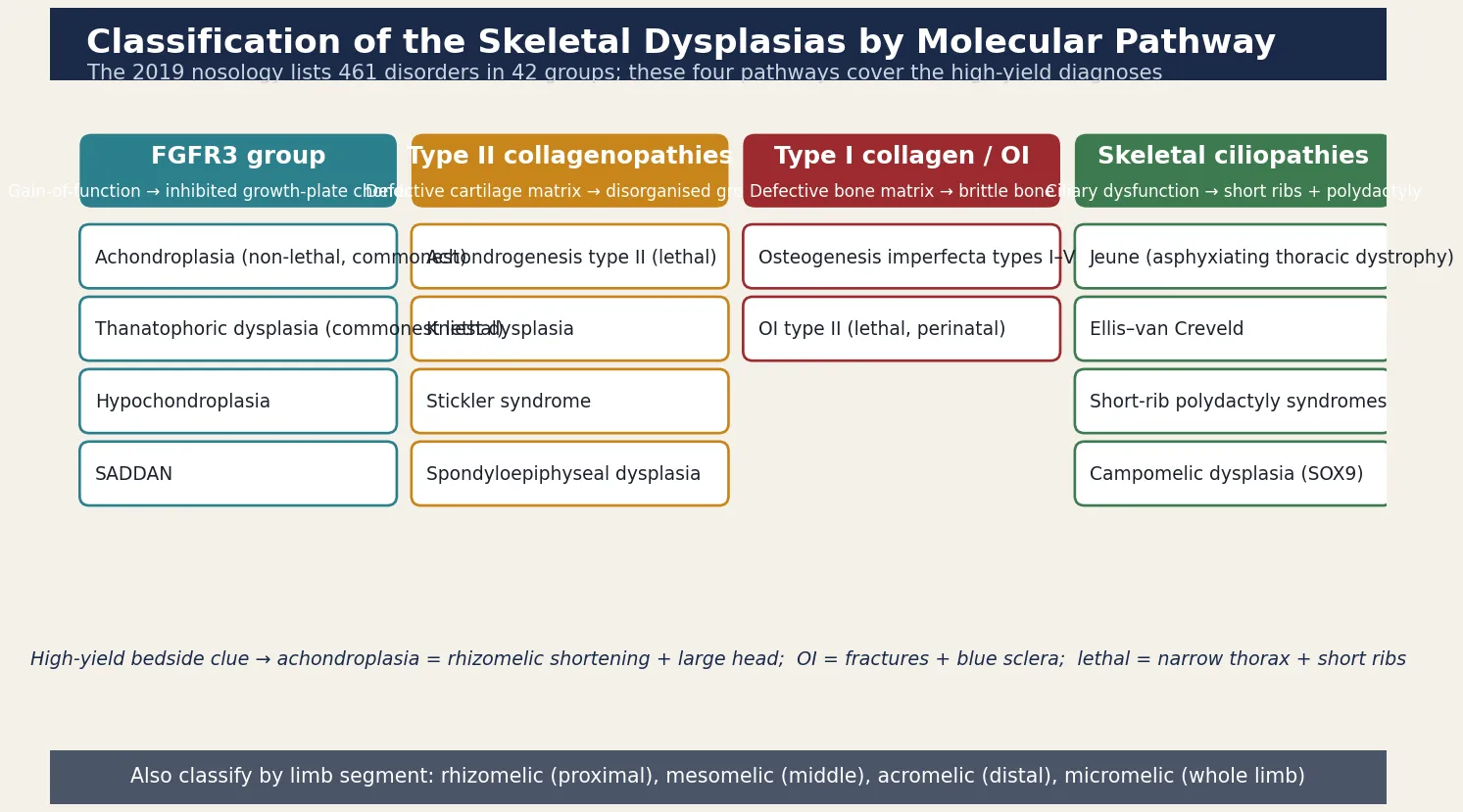
A second, more practical axis separates the disorders by which limb segment is shortened, because the pattern is obvious at the bedside and narrows the differential quickly. Rhizomelic shortening, in which the proximal segment (femur and humerus) is most affected, is the hallmark of achondroplasia and the thanatophoric dysplasias. Mesomelic shortening, affecting the middle segment (radius, ulna, tibia, fibula), points to the mesomelic dysplasias and is rarer. Acromelic shortening, affecting the hands and feet, occurs in the peripheral dysostoses. Micromelic shortening, in which the whole limb is uniformly short, marks the severe and often lethal dysplasias such as achondrogenesis. Reading the segment first converts a vague "short limbs" into a focused differential before the radiograph is taken. [2] [13]
Lethal versus non-lethal — the trajectory-defining split
- Lethal in the perinatal period (small thorax, pulmonary hypoplasia): thanatophoric dysplasia (FGFR3), achondrogenesis (types I and II), osteogenesis imperfecta type II, short-rib polydactyly syndromes, campomelic dysplasia. Management is palliation, molecular confirmation, and recurrence counselling.
- Non-lethal, compatible with normal lifespan and intellect: achondroplasia, hypochondroplasia, osteogenesis imperfecta types I, III, and IV, the spondyloepiphyseal dysplasias. Management is surveillance, syndrome-specific therapy, and multidisciplinary support.
- The discriminator: thoracic size and rib shortening on the chest radiograph drive respiratory outcome — a narrow chest with short horizontal ribs is the single best predictor of lethality. [2] [10]
Epidemiology & Risk Factors
Individually each skeletal dysplasia is rare, but collectively they are encountered regularly in paediatric practice. Pooled estimates put the overall birth prevalence of skeletal dysplasias at roughly one in five thousand live births, with achondroplasia the commonest non-lethal form at about one in twenty-five thousand live births, osteogenesis imperfecta at one in fifteen thousand, and thanatophoric dysplasia the commonest lethal form at one in thirty to fifty thousand. The birth prevalence of lethal skeletal dysplasias detected at prenatal ultrasound is higher again, because many are recognised and managed before delivery. These figures mean that a busy general paediatrician will care for several children with achondroplasia and osteogenesis imperfecta over a career. [2] [3]
Most skeletal dysplasias follow autosomal dominant or autosomal recessive inheritance, and the inheritance matters because it changes the recurrence risk and the counselling. Achondroplasia is autosomal dominant, but around eighty per cent of cases arise from a new variant in the father's germ line, so most affected infants are born to average-statured parents of advanced paternal age — a fact that reshapes recurrence risk from the twenty-five per cent of a recessive disorder to the small gonadal mosaicism risk of a new dominant variant. Thanatophoric dysplasia shares the same new-variant FGFR3 mechanism. The recessive ciliopathies — Jeune and Ellis–van Creveld — cluster in consanguineous or founder populations and carry a one-in-four recurrence risk that makes prenatal diagnosis central to future pregnancies. [5] [6]
Achondroplasia carries a specific genetic counselling nuance that examiners test. Because the FGFR3 variant is dominant, a child who inherits the variant from one affected parent has heterozygous (typical) achondroplasia, but a child of two affected parents has a one-in-four chance of inheriting two pathogenic copies. Homozygous achondroplasia is a severe, lethal disorder with an extremely small thorax, and the reproductive counselling of two affected adults must address this risk directly through prenatal or preimplantation genetic testing. [1] [6]
Pathophysiology
The skeleton lengthens at the growth plate, a cartilaginous template where resting chondrocytes proliferate, hypertrophy, and are replaced by bone through endochondral ossification. Most skeletal dysplasias disturb this growth plate, and the specific gene change dictates whether the chondrocytes fail to proliferate, fail to mature, or lay down a defective matrix. The figure traces the cascade from gene variant to growth-plate defect to skeletal phenotype, and overlays the therapeutic levers that exploit the biology. [5] [12]
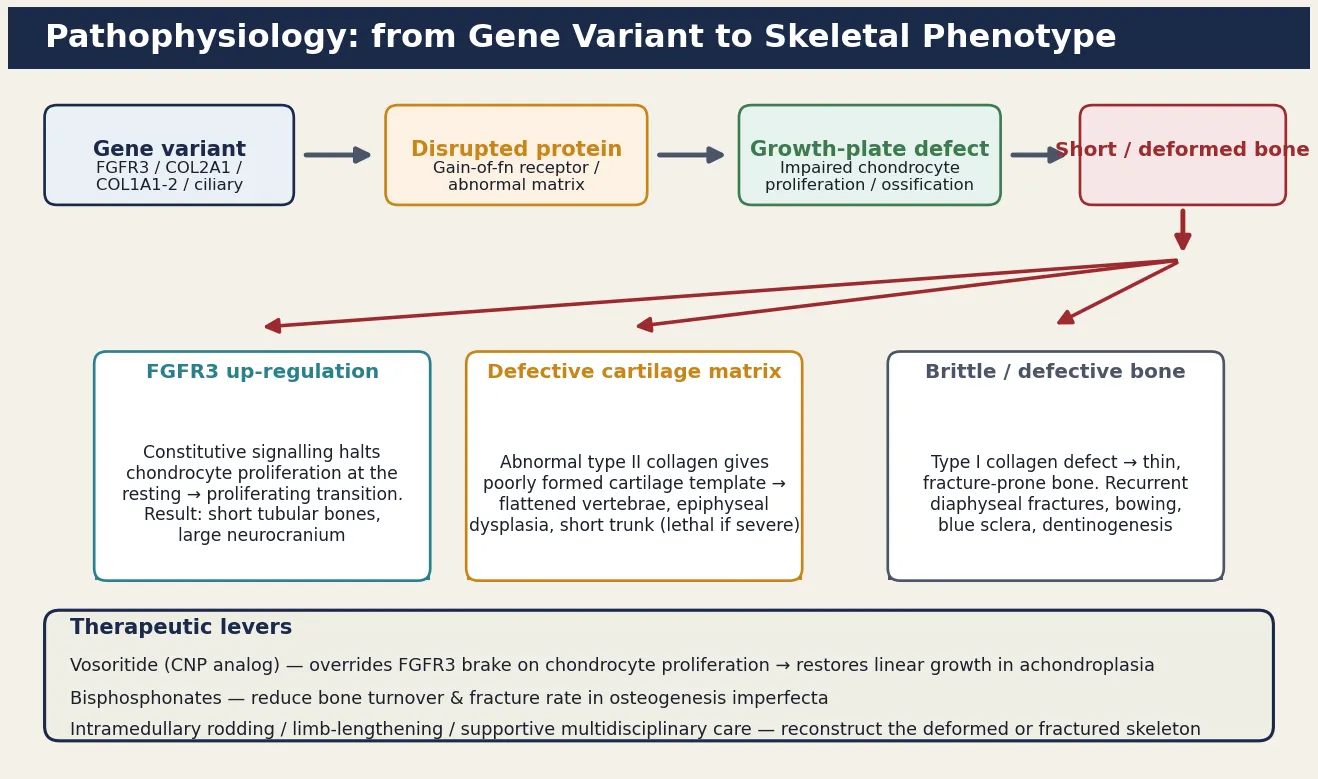
The FGFR3 family is the highest-yield mechanism because it explains both the commonest non-lethal and the commonest lethal dysplasia. Fibroblast growth factor receptor 3 is a brake on chondrocyte proliferation: in normal growth it restrains the growth plate, but a gain-of-function variant removes the restraint's off-switch, so the receptor fires constitutively and halts the chondrocytes at the resting-to-proliferating transition. The result is shortened tubular bones with a large neurocranium, because the membranous bone of the skull (which grows by intramembranous, not endochondral, ossification) is unaffected and grows normally while the endochondral bones are arrested. The severity of the gain-of-function determines the phenotype: a mild variant gives achondroplasia, a stronger one gives the lethal thanatophoric dysplasia. [5] [12]
Osteogenesis imperfecta works by a different mechanism entirely. Type I collagen, the major structural protein of bone, is a triple helix assembled from two alpha-1 chains and one alpha-2 chain encoded by COL1A1 and COL1A2. A pathogenic variant in either gene either reduces the amount of normal collagen (the milder type I, giving a "quantitative" defect) or produces structurally abnormal collagen that disrupts the triple helix (the more severe types II, III, and IV, giving a "qualitative" defect). The bone laid down is thin and disorganised, so it fractures after minimal or no trauma, bowing of long bones and vertebral compression follow, and in type II the in-utero fractures produce a severely malformed, lethal skeleton. Recognising that osteogenesis imperfecta is a matrix disorder rather than a growth-plate disorder explains why stature may be reduced by fracture and deformity rather than by primary growth arrest. [8] [9]
Clinical Presentation
The presentation depends on whether the dysplasia is lethal or non-lethal, and on whether it declares itself before or after birth. The first pattern is the lethal short-limbed newborn. A baby born with a very narrow chest, short limbs, and soft skull bones develops severe respiratory distress from pulmonary hypoplasia in the delivery room, and the examination shows a small bell-shaped thorax, short ribs, and marked micromelic shortening. This pattern points to thanatophoric dysplasia (with its characteristic telephone-receiver femurs and cloverleaf skull in some cases), achondrogenesis (with absent or minimal ossification of the spine and skull), osteogenesis imperfecta type II (with in-utero fractures, blue sclerae, and a soft cranium), or a short-rib polydactyly syndrome. The recognition that this is a lethal skeletal dysplasia, not a treatable respiratory disorder, is the resuscitation-level decision. [10] [11]
The second pattern is the disproportionate infant or child. A child with achondroplasia looks normal at birth apart from a slightly large head and short limbs, and the phenotype becomes obvious over the first year as the limbs fail to lengthen proportionately. The examination is distinctive: rhizomelic shortening of the proximal limbs, a large head with frontal bossing and midface hypoplasia, a small nasal bridge, a trident configuration of the hands (short fingers separated into two groups with a gap at the base of the middle finger), genu varum, lumbar lordosis, and a normal trunk. Intellect is normal, and the short stature is disproportionate rather than global. The combination of rhizomelic shortening and a large head in a child with normal development is the classic achondroplasia presentation. [1] [6]
The third pattern is the fracturing child. A child with osteogenesis imperfecta presents with recurrent fractures after minimal trauma, often beginning in infancy or the preschool years, with blue or grey sclerae, early-onset hearing loss in some types, joint hypermobility, and dentinogenesis imperfecta (opalescent, fragile teeth). The fractures may raise the spectre of non-accidental injury, and the active distinction between the two — by clinical phenotype, family history, and the radiographic pattern of osteopenia and healed fractures — is one of the most important tasks in paediatric practice. Severity ranges from mild type I, with a near-normal life and occasional fractures, through progressively severe types III and IV, to the lethal perinatal type II. [8] [9]
Differential Diagnosis
When a skeletal dysplasia is suspected, the differential depends on the presenting feature. For the disproportionate child, the key distinction is between a skeletal dysplasia and the many causes of proportionate short stature. Growth hormone deficiency, hypothyroidism, Turner syndrome in a girl, chronic systemic disease, and constitutional delay of growth all produce short stature, but the child is proportionate and the limbs are not selectively shortened. The next layer distinguishes achondroplasia from the other rhizomelic dysplasias (hypochondroplasia, pseudoachondroplasia, the type II collagenopathies) and from the short-trunk dysplasias such as the spondyloepiphyseal dysplasias, in which the spine is short relative to the limbs. [2] [6]
For the fracturing child, the differential is the most fraught in paediatrics. Non-accidental injury produces fractures, but the fractures of osteogenesis imperfecta occur in the setting of a connective tissue phenotype (blue sclerae, joint laxity, dentinogenesis imperfecta, family history), generalised osteopenia, and a characteristic radiographic pattern. The discriminating features are the presence of osteopenia and wormian skull bones in osteogenesis imperfecta, the pattern of the fractures, and the family history; when the phenotype is uncertain, molecular testing of COL1A1 and COL1A2 and a specialist skeletal review resolve the question, because wrongly labelling a family either way has devastating consequences. Rickets, prematurity-related osteopenia, and other metabolic bone diseases enter the differential of the fracturing infant and are excluded by biochemistry. [8] [9]
For the lethal short-limbed fetus or newborn, the differential is among the lethal dysplasias themselves, and it is resolved by the skeletal survey. Thanatophoric dysplasia shows short curved femurs and a narrow thorax; achondrogenesis shows severely deficient ossification of the spine and skull; osteogenesis imperfecta type II shows in-utero fractures and a soft skull; the short-rib polydactyly syndromes add polydactyly and visceral malformations. A small number of non-dysplastic conditions — severe hydrops, congenital neuromuscular disorder with arthrogryposis, and lethal skeletal malformation syndromes — can mimic a dysplasia, but the radiographic pattern of a skeletal dysplasia is usually distinctive enough to make the call at birth. [10] [11]
Clinical & Bedside Assessment
The bedside assessment is built around three questions. First, is this dysplasia lethal or non-lethal — because this sets the urgency and the whole management pathway. Second, which limb segment is shortened and what does the radiograph show — because this narrows the molecular pathway. Third, is there a family history of short stature, fractures, or consanguinity — because this sets the inheritance and the recurrence risk. A focused history and examination answer all three in a single consultation and direct the imaging and genetic workup. [2] [13]
The growth and proportion assessment is the single most informative bedside step. Measure the weight, length, and head circumference on the appropriate syndrome-specific chart — achondroplasia growth charts exist and must be used, because plotting an achondroplastic child on a standard chart produces false alarm about head circumference and growth failure. Then measure the upper-to-lower body segment ratio and the arm span, and inspect for rhizomelic, mesomelic, acromelic, or micromelic shortening. Document the head shape, the facial features (frontal bossing, midface hypoplasia, nasal bridge), the hands (trident configuration, brachydactyly, polydactyly), the chest (thoracic size and rib shortening), the spine (gibbus, scoliosis, lordosis), and the lower limbs (genu varum, bowing, fractures). A thorough dysmorphology examination often identifies the syndrome at the bedside. [1] [6]
The neurological and respiratory examination is essential because the complications of the dysplasias are life-threatening. In achondroplasia, examine for signs of cervicomedullary compression — increased tone, hyperreflexia, clonus, asymmetric movements, or apnoea — because foramen magnum stenosis can cause sudden unexpected death in infancy. Ask specifically about snoring, witnessed apnoea, and restless sleep, because sleep-disordered breathing from midface hypoplasia and adenoidal hypertrophy is common. Check the head circumference trend and fontanelle, because hydrocephalus complicates achondroplasia. In osteogenesis imperfecta, assess the hearing (early-onset conductive and sensorineural hearing loss develops), the dentition, and the range of movement of fractured or bowed limbs. [1] [5]
Investigations
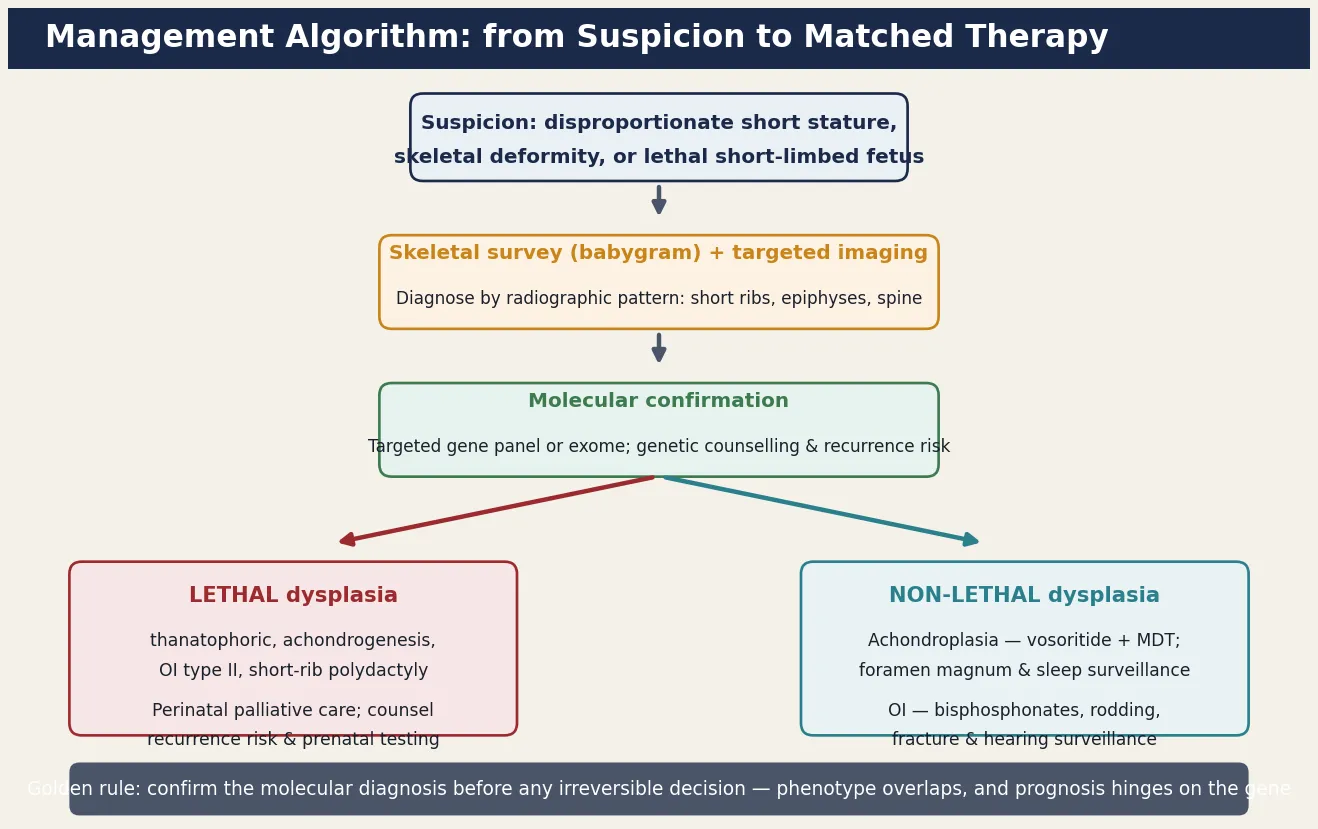
The investigation strategy is radiographic first and molecular second, because the skeletal survey is the diagnostic cornerstone and the molecular test confirms. The first investigation is a full skeletal survey — a babygram in the newborn or a complete set of skull, spine, chest, pelvis, and long-bone radiographs in the older child — interpreted by a radiologist experienced in skeletal dysplasia. The radiograph answers four questions: which limb segment is shortened, whether the ribs and thorax are small (the lethality clue), whether the spine and skull are ossified normally (excluded achondrogenesis and severe osteogenesis imperfecta type II), and whether there is a pathognomonic pattern such as the telephone-receiver femur of thanatophoric dysplasia or the in-utero fractures of osteogenesis imperfecta type II. [2] [13]
The molecular test confirms the diagnosis and defines the variant. Targeted testing of the recurrent FGFR3 variant (the glycine-to-arginine substitution at position 380, p.Gly380Arg) confirms around ninety-nine per cent of achondroplasia cases, while a skeletal dysplasia gene panel or exome sequencing is used when the phenotype does not fit a single recurrent variant. Molecular confirmation matters for three reasons: it confirms the diagnosis before irreversible decisions, it refines the prognosis where the phenotype is borderline, and it enables accurate recurrence-risk counselling and prenatal or preimplantation genetic testing in future pregnancies. In the lethal dysplasias, molecular confirmation from a post-mortem skin or cartilage sample is the basis for the recurrence counselling that follows. [3] [11]
Supportive investigations stage the complications and set the surveillance baseline. A cranial ultrasound or MRI checks for ventriculomegaly and cervicomedullary compression in achondroplasia. A sleep study quantifies sleep-disordered breathing. Echocardiography screens for the congenital heart defects that accompany some ciliopathies (Ellis–van Creveld, common atrium). Audiometry and a dental review are part of osteogenesis imperfecta care. Bone densitometry, where available, quantifies the osteopenia of osteogenesis imperfecta and tracks the response to bisphosphonate therapy. The goal is to stage the skeleton and its complications precisely so that therapy and surveillance are matched to the individual child. [1] [8]
Management — Resuscitation
Most children with a non-lethal dysplasia do not present in collapse, but the lethal short-limbed newborn is a genuine resuscitation emergency. A baby born with a narrow chest and respiratory distress is managed with a focus on comfort rather than escalation when the dysplasia is confirmed lethal, because intubation and ventilation cannot overcome the fixed pulmonary hypoplasia and only prolong the dying process. The resuscitation-level decision is to confirm the diagnosis rapidly by chest and skeletal radiographs at the bedside, to involve the neonatal and genetic teams and the parents in a shared decision about the limits of intervention, and to move to palliation with the diagnosis established in time for accurate molecular confirmation and recurrence counselling. [10] [11]
The non-lethal dysplasias carry their own emergencies. An achondroplastic infant with new apnoea, hypotonia, or signs of myelopathy may have acute cervicomedullary compression from foramen magnum stenosis, and this is managed with urgent MRI and neurosurgical decompression, because untreated compression causes sudden death and irreversible neurological injury. A child with osteogenesis imperfecta who sustains a fracture is managed with gentle handling, splinting, and orthopaedic assessment for reduction or intramedullary rodding, recognising that these children are prone to fracture with minimal handling and that pain control and fracture stabilisation run in parallel. Hydrocephalus in achondroplasia and severe kyphoscoliosis with respiratory compromise in the type II collagenopathies are other scenarios that need prompt recognition. [1] [8]
Management — Definitive & Stepwise
The definitive management matches the therapy to the molecular pathway and to whether the dysplasia is lethal or non-lethal. The figure lays out the stepwise pathway from suspicion through skeletal survey and molecular confirmation to the matched therapy, and the guiding principle is that the non-lethal dysplasias now have disease-modifying therapy and surveillance that transforms the child's trajectory. [7] [12]
For achondroplasia, the landmark advance is vosoritide, a C-type natriuretic peptide analogue that overrides the FGFR3 brake and restores chondrocyte proliferation at the growth plate. A randomised, double-blind, phase three trial showed that once-daily subcutaneous vosoritide produces a sustained increase in annualised growth velocity in children with achondroplasia, and it is now approved for use from early childhood. Vosoritide does not address the complications of achondroplasia, so it runs in parallel with — not instead of — the multidisciplinary surveillance that defines modern achondroplasia care. Growth hormone has been used with modest effect in some regions; limb-lengthening surgery remains controversial and is reserved for carefully selected adolescents. [7] [12]
For osteogenesis imperfecta, bisphosphonate therapy is the cornerstone. Intravenous bisphosphonates such as pamidronate, given in cyclic infusions, reduce bone resorption, increase bone density, reduce fracture rates, relieve bone pain, and improve mobility in the moderate and severe forms. Intramedullary rodding of long bones stabilises the deformed or repeatedly fracturing bone and is often combined with bisphosphonates in the child with type III or severe type IV disease. Physiotherapy, hydrotherapy, and occupational therapy maximise mobility and function; hearing surveillance and dental care run throughout life; and emerging therapies including sclerostin inhibition and gene-based approaches are under investigation. [8] [9]
Therapy for the non-lethal skeletal dysplasias (specialist-initiated; confirm current dosing)
Symptom-directed multidisciplinary care runs in parallel with disease-modifying therapy and is often what determines quality of life. Orthopaedic surgery manages fractures, limb deformity, and scoliosis; ENT and respiratory manage sleep-disordered breathing, with adenotonsillectomy and continuous positive airway pressure where indicated; neurosurgery manages foramen magnum compression and hydrocephalus; audiology monitors hearing; dentistry manages dentinogenesis imperfecta; and developmental, educational, and psychosocial services support the child and family. A coordinated medical home with access to a specialist skeletal dysplasia service is the model of care, because these are lifelong conditions that cross every organ system. [1] [8]
Specific Subtypes & Scenarios
Achondroplasia is the prototypical non-lethal dysplasia and the one most likely to appear in an examination. An affected child has rhizomelic shortening, a large head with frontal bossing and midface hypoplasia, trident hands, genu varum, and lumbar lordosis, with normal intellect. The complications are the heart of the management: foramen magnum stenosis with cervicomedullary compression and sudden death risk in infancy, sleep-disordered breathing from midface hypoplasia, recurrent otitis media with conductive hearing loss and speech delay, hydrocephalus, and genu varum. Modern management combines syndrome-specific growth surveillance, vosoritide where available, and structured multidisciplinary follow-up, and the prognosis is a normal lifespan and intellect with a final adult height in the range of one hundred and twenty to one hundred and thirty-five centimetres. [1] [6]
Thanatophoric dysplasia is the prototypical lethal dysplasia and the commonest cause of lethal short-limbed dwarfism recognised at birth. A gain-of-function FGFR3 variant, more severe than that of achondroplasia, produces extreme micromelia, a narrow bell-shaped thorax with short horizontal ribs, a large head with frontal bossing, and — in some cases — a cloverleaf skull (craniosynostosis). Pulmonary hypoplasia from the small thorax causes death from respiratory failure within hours to days of birth, and the radiograph shows the characteristic curved telephone-receiver femurs and flattened vertebral bodies. Management is palliative, with the diagnosis confirmed at birth by the radiograph and by molecular FGFR3 testing, and the subsequent counselling addresses the small gonadal mosaicism recurrence risk and the option of prenatal diagnosis in future pregnancies. [5] [12]
Osteogenesis imperfecta is the dysplasia most likely to present to the general paediatrician because of fractures. The Sillence classification remains useful: type I is mild with blue sclerae and few fractures, type II is lethal in the perinatal period, type III is severely deforming with in-utero and perinatal fractures progressing to a short, bowed skeleton, and type IV is moderately severe with variable scleral colour. The diagnosis is clinical and molecular, based on the fracture pattern, the connective tissue phenotype, and COL1A1 or COL1A2 testing, and the active distinction from non-accidental injury is essential. Bisphosphonates, intramedullary rodding, and multidisciplinary rehabilitation have transformed the outlook for the moderate and severe forms, and many children now achieve far greater mobility than was historically possible. [8] [9]
Numbers worth carrying into the exam
Complications & Pitfalls
The complications of the skeletal dysplasias cluster by organ and are the reason these conditions need lifelong surveillance. The bones fracture and bow, producing the recurrent fractures and skeletal deformity that dominate the life of a child with osteogenesis imperfecta. The foramen magnum narrows in achondroplasia, producing cervicomedullary compression and the sudden unexpected death that haunts the first two years of life. The thorax is small in the lethal and some non-lethal dysplasias, producing restrictive lung disease and sleep-disordered breathing. The spine curves, producing the kyphoscoliosis that compromises respiration in the type II collagenopathies. Recognising that the same growth-plate or matrix defect produces a predictable set of complications is the conceptual hinge of surveillance. [1] [8]
The most consequential pitfall is failing to recognise a lethal dysplasia. Treating a thanatophoric or achondrogenesis infant as a treatable respiratory disorder subjects the family and child to invasive, futile intervention and forfeits the chance for dignified palliation and accurate recurrence counselling. A second pitfall is confusing osteogenesis imperfecta with non-accidental injury: wrongly attributing the fractures of a mild osteogenesis imperfecta to abuse tears a family apart, while missing abuse by attributing it to a dysplasia endangers the child. The resolution lies in the phenotype, the family history, the radiographic pattern of osteopenia and wormian bones, and molecular testing of COL1A1 and COL1A2, with specialist review when there is genuine uncertainty. [8] [9]
A third pitfall is neglecting surveillance in a non-lethal dysplasia that looks well. An achondroplastic infant may look comfortable and still have significant foramen magnum stenosis or sleep-disordered breathing that declares itself as an arrest; routine sleep studies and a low threshold for craniovertebral MRI are the safeguards. Plotting an achondroplastic child on a standard growth chart rather than a syndrome-specific chart generates false alarms about head size and growth failure, and using the wrong chart is a frequent source of over-investigation. Finally, failing to offer prenatal or preimplantation genetic testing to a couple with a previously affected child with a lethal dysplasia forfeits a genuine reproductive option in a high-recurrence-risk setting. [1] [11]
Prognosis & Disposition
Prognosis is determined by the specific dysplasia, the genotype, and the quality of the surveillance, and the lethal versus non-lethal dichotomy is the dominant determinant. A child with achondroplasia has a normal lifespan and normal intellect, with a final adult height around one hundred and twenty to one hundred and thirty-five centimetres, and the prognosis is transformed by modern multidisciplinary care and vosoritide. A child with mild osteogenesis imperfecta type I has a near-normal life with occasional fractures and late-onset hearing loss; a child with severe type III disease has a substantially shortened, deforming course but, with bisphosphonates and surgery, now achieves far greater function and survival than was historically possible. [1] [8]
The lethal dysplasias carry a perinatal prognosis. Thanatophoric dysplasia, achondrogenesis, osteogenesis imperfecta type II, and the severe short-rib polydactyly syndromes cause death from respiratory failure within hours to days of birth, and the task after a lethal diagnosis is palliative care, bereavement support, and accurate recurrence-risk counselling. The small gonadal mosaicism recurrence risk for a new dominant FGFR3 variant, the one-in-four recurrence risk for a recessive ciliopathy, and the availability of prenatal and preimplantation genetic testing are the elements of that counselling, and the genetic service should be involved before the next pregnancy rather than after it. [5] [11]
Disposition is to a specialist skeletal dysplasia service with a structured transition to adult care, because these are lifelong conditions that cross orthopaedics, respiratory, neurosurgery, ENT, audiology, dentistry, and genetics. The medical home coordinates surveillance — growth and development on syndrome-specific charts, craniovertebral and sleep surveillance in achondroplasia, fracture and hearing and dental surveillance in osteogenesis imperfecta, and cardiac screening in the ciliopathies — and integrates the family's psychosocial, educational, and genetic-counselling needs. Early involvement of palliative care, alongside disease-directed therapy, supports the family through the lethal dysplasias without precluding continuing care. [2] [9]
Special Populations
Prenatal diagnosis has created a distinct population — the fetus with a suspected skeletal dysplasia on routine ultrasound — whose management differs from the postnatal child. The skeletal dysplasia is suspected when the second-trimester scan shows short long bones, a small thorax, or bone bowering and fracture, and the task is to determine whether the dysplasia is lethal, because that decision governs the option of termination and the planning of perinatal management. Guidelines recommend a structured sonographic assessment of long-bone length, thoracic dimensions, skull shape, and mineralisation, supplemented by fetal MRI and molecular testing where available, with the prognosis hinging on the chest circumference and the presence of a lethal pattern. The parents need honest, calibrated counselling about the prognosis and the option of post-mortem molecular confirmation for recurrence counselling. [10] [11]
Access to vosoritide and to specialist skeletal dysplasia services varies between jurisdictions and is evolving, so the examining clinician should state the local pathway rather than assume a universal standard. In Australia and New Zealand, children with achondroplasia are managed through coordinated paediatric and genetic services with access to multidisciplinary clinics, sleep studies, and craniovertebral imaging; vosoritide is accessed through specialist centres where funded. The principles are constant: use syndrome-specific growth charts, run structured foramen magnum and sleep surveillance in the first two years, refer to a specialist skeletal dysplasia service, and confirm the molecular diagnosis before any irreversible decision. [1] [7]
Consanguineous communities and founder populations carry a higher burden of the recessive skeletal dysplasias — the ciliopathies, some of the type II collagenopathies, and rare recessive forms — and a previously affected child reshapes the reproductive risk for every subsequent pregnancy. In these families carrier testing, prenatal diagnosis, and preimplantation genetic testing are central to management. Indigenous, migrant, refugee, and remote populations face additional barriers — distance from specialist centres, language and cultural considerations, and inequitable access to expensive therapies — and the medical home must actively coordinate equitable, culturally safe care, because the complications of these conditions do not wait for geography to be overcome. [2] [11]
Evidence, Guidelines & Regional Differences
The evidence base for the skeletal dysplasias has been transformed by molecular genetics and by disease-modifying therapy. The international nosology, revised in 2010 and again in 2019, organises the disorders by causative gene and provides the framework for diagnosis and counselling, and the genotype-first approach has clarified the overlap between phenotypes that were once considered distinct. The strongest treatment evidence is for vosoritide in achondroplasia, where a randomised, double-blind, phase three placebo-controlled trial demonstrated a sustained increase in annualised growth velocity, establishing the first disease-modifying therapy directed at the underlying FGFR3 mechanism. Bisphosphonate therapy for osteogenesis imperfecta rests on a substantial body of cohort and registry evidence showing improved bone density, reduced fracture rates, and improved mobility. [3] [7]
Guidelines are pathway- and syndrome-specific. The prenatal diagnosis guidelines set out the structured sonographic approach to the fetus with suspected skeletal dysplasia and the role of molecular testing in refining the prognosis. The achondroplasia clinical reviews define the surveillance schedule — foramen magnum and sleep assessment in infancy, growth on syndrome-specific charts, ENT and audiology surveillance, and orthopaedic management of limb deformity — that constitutes modern achondroplasia care. The osteogenesis imperfecta guidelines address bisphosphonate use, rodding, and multidisciplinary rehabilitation. [1] [8]
Regional differences persist in what is funded, how specialist services are accessed, and how prenatal and perinatal care is delivered. Vosoritide availability, the structure of skeletal dysplasia clinics, access to prenatal molecular testing, and the funding of bisphosphonate regimens all vary between jurisdictions and change over time, so the clinician should ground recommendations in local policy while applying the universal principles: recognise disproportion early, confirm with a skeletal survey and molecular testing, run structured surveillance for the lethal complications, and offer recurrence-risk counselling and prenatal diagnosis in the high-risk family. [7] [11]
Exam Pearls
In a viva, lead with the clinical pattern, name the limb segment shortened and the molecular pathway, and then state the confirmatory test and the therapy — that four-step structure answers almost any skeletal dysplasia question. For a short case, the rhizomelic shortening, trident hands, and large head of achondroplasia, or the blue sclerae and healed fractures of osteogenesis imperfecta, are the findings that earn marks; for a long case, the management plan that matches the therapy to the pathway (vosoritide for achondroplasia, bisphosphonates for osteogenesis imperfecta) and sets out surveillance for foramen magnum compression, sleep-disordered breathing, and fractures is what distinguishes a pass from a commendation. Remember the lethal versus non-lethal dichotomy and the FGFR3 mechanism, because together they tie the whole topic together. [1] [8]
The single most testable principle is that a gain-of-function FGFR3 variant brakes chondrocyte proliferation at the growth plate, producing the achondroplasia family from the non-lethal commonest dysplasia to the lethal commonest, and that vosoritide overrides that brake to restore linear growth. Couple that with the recognition that osteogenesis imperfecta is a type I collagen matrix disorder producing brittle, fracturing bone that must be distinguished from non-accidental injury, and you have the framework for the whole topic. Every skeletal dysplasia question reduces to: is it lethal, which limb segment is shortened, which pathway is disrupted, and what surveillance and therapy does that pathway demand. [5] [12]
References
- [1]Pauli RM. Achondroplasia: a comprehensive clinical review. Orphanet J Rare Dis, 2019.PMID 30606190
- [2]Krakow D, Rimoin DL. The skeletal dysplasias. Genet Med, 2010.PMID 20556869
- [3]Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A, 2019.PMID 31633310
- [4]Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A, 2011.PMID 21438135
- [5]Horton WA, Hall JG, Hecht JT. Achondroplasia. Lancet, 2007.PMID 17630040
- [6]Baujat G, Legeai-Mallet L, Finidori G, Cormier-Daire V, Le Merrer M. Achondroplasia. Best Pract Res Clin Rheumatol, 2008.PMID 18328977
- [7]Savarirayan R, Ireland P, Irving J, Jones J, Thompson B, Bellus G, et al. Once-daily, subcutaneous vosoritide therapy in children with achondroplasia: a randomised, double-blind, phase 3, placebo-controlled, multicentre trial. Lancet, 2020.PMID 32891212
- [8]Forlino A, Marini JC. Osteogenesis imperfecta. Lancet, 2016.PMID 26542481
- [9]van Dijk FS, Pals G, van Rijn RR, Nikkels PG, Cobben JM. Osteogenesis Imperfecta: A Review with Clinical Examples. Mol Syndromol, 2011.PMID 22570641
- [10]Krakow D. Skeletal dysplasias. Clin Perinatol, 2015.PMID 26042906
- [11]Krakow D, Lachman RS, Rimoin DL. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet Med, 2009.PMID 19265753
- [12]Kim HY, Ko JM. Clinical management and emerging therapies of FGFR3-related skeletal dysplasia in childhood. Ann Pediatr Endocrinol Metab, 2022.PMID 35793999
- [13]Offiah AC, Hall CM. Radiological diagnosis of the constitutional disorders of bone. As easy as a, b, c? Pediatr Radiol, 2003.PMID 12612812