Paeds · genetics-dysmorphology-and-metabolism
Syndromic craniosynostosis and craniofacial disorders
Also known as syndromic craniosynostosis · FGFR craniosynostosis · Crouzon syndrome · Apert syndrome · Pfeiffer syndrome · Muenke syndrome · Saethre-Chotzen syndrome · craniofacial dysostosis
A fellowship approach to syndromic craniosynostosis and the genetic craniofacial disorders: recognise that an abnormal head shape with midface hypoplasia, exorbitism or a limb anomaly is syndromic until proven otherwise, name the big six syndromes by their gene (FGFR2 for Crouzon, Apert and Pfeiffer; FGFR3 p.Pro250Arg for Muenke; TWIST1/TCF12 for Saethre-Chotzen; EFNB1 for craniofrontonasal; RAB23/MEGF8 for Carpenter), confirm the diagnosis with skull imaging and targeted genetic testing, secure the airway, the exposed eye and raised intracranial pressure before any cosmetic plan, and coordinate an age-based multidisciplinary craniofacial team through to adult transition.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A baby is brought in with a tall, tower-shaped head and eyes that seem to bulge; or a toddler has a broad great toe and a flat forehead; or a newborn grunts and snores through a face that never grew forward. Each is the same biological signal — a fibroblast growth factor receptor switched on too hard — and the fellowship task is to see the head, the face and the hand as one diagnosis, to send the test that names the gene, and to build a surgical and surveillance net across the airway, the eye, the brain and the developing child before any of them comes to harm. [1] [7]
The big six by gene — the viva list
Overview & Definition
A baby's skull is not a single bone but a set of plates held together by sutures, and those sutures stay open in infancy precisely so the brain can grow. Craniosynostosis is the premature fusion of one or more of those sutures, and the immediate consequence is that the skull grows in the wrong direction — perpendicular to the fused suture — because the brain pushes outward only along the paths that remain open. The head shape is therefore a clue to which suture fused, and recognising that geometry is where the assessment begins. [5] [1]
The syndromic label is earned the moment the fused suture travels with a facial or limb feature. A child who has fused a single suture and has an otherwise normal face, hands and development has nonsyndromic disease, and that is the commoner form — around four out of five cases. A child whose fused suture is joined by midface hypoplasia, exorbitism, a cleft, syndactyly, or a broad and deviated thumb or great toe has syndromic craniosynostosis, and that packaging is the fingerprint the paediatrician is trained to see. The reason the features travel together is that the same signalling pathway — the fibroblast growth factor receptor family — builds the skull, the face and the limbs in parallel, so a single activating variant reshapes all three at once. [1] [6]
The lifespan task is no longer simply to reshape the head. With modern multidisciplinary craniofacial care, children with syndromic craniosynostosis grow into adults who attend school, work and form relationships, and the general paediatrician sits at the centre of a trajectory that runs from the neonatal airway through staged childhood surgery into adult craniofacial and psychological care. The head shape is the presenting complaint, but the airway, the eye, the intracranial pressure and the developing child are what the surveillance protects across that whole life. [8] [5]
Classification
Classification begins with a single gate that earns the syndromic label, and the gate is this: any craniosynostosis accompanied by a midface, orbital, limb or developmental feature is syndromic until proven otherwise. On the far side of the gate sit the nonsyndromic single-suture forms — sagittal scaphocephaly, metopic trigonocephaly, unicoronal plagiocephaly and the rare lambdoid — which carry no travelling feature and a much lower genetic yield. On the near side sit the syndromes, and there the gene is the organising principle, because the gene predicts the face, the limb, the inheritance and, increasingly, the surgical risk. [5] [6]
The fibroblast growth factor receptor family carries most of the syndromic load, and the big three live in FGFR2. Crouzon syndrome fuses the coronal and other sutures with exorbitism and midface hypoplasia but leaves the hands normal; Apert syndrome adds the most severe symmetric syndactyly of hands and feet — the mitten hand — alongside cleft palate and a more variable cognition; and Pfeiffer syndrome is marked by the broad, medially-deviated thumb and great toe. A recurrent variant in FGFR3, p.Pro250Arg, defines Muenke syndrome, which is the commonest single-gene cause and the one most often mistaken for nonsyndromic disease. TWIST1 — and its basic helix-loop-helix partner TCF12 — defines Saethre-Chotzen, with its low frontal hairline, ptosis and small ears. [6] [2] [4]
A handful of syndromes fall outside the FGFR axis and round out the differential. Craniofrontonasal syndrome, from EFNB1, is X-linked and carries the paradox that heterozygous girls are more severely affected than the hemizygous boys, with a coronal synostosis, a bifid nasal tip and often developmental difference. Carpenter syndrome, from RAB23 or MEGF8, is the autosomal-recessive exception, presenting with polysyndactyly, congenital heart defects and short stature; its recessive inheritance changes the recurrence counselling from a 50 per cent autosomal-dominant risk to a 25 per cent sibling risk. Naming the recessive and the X-linked forms is what marks a complete classification answer. [5] [6]
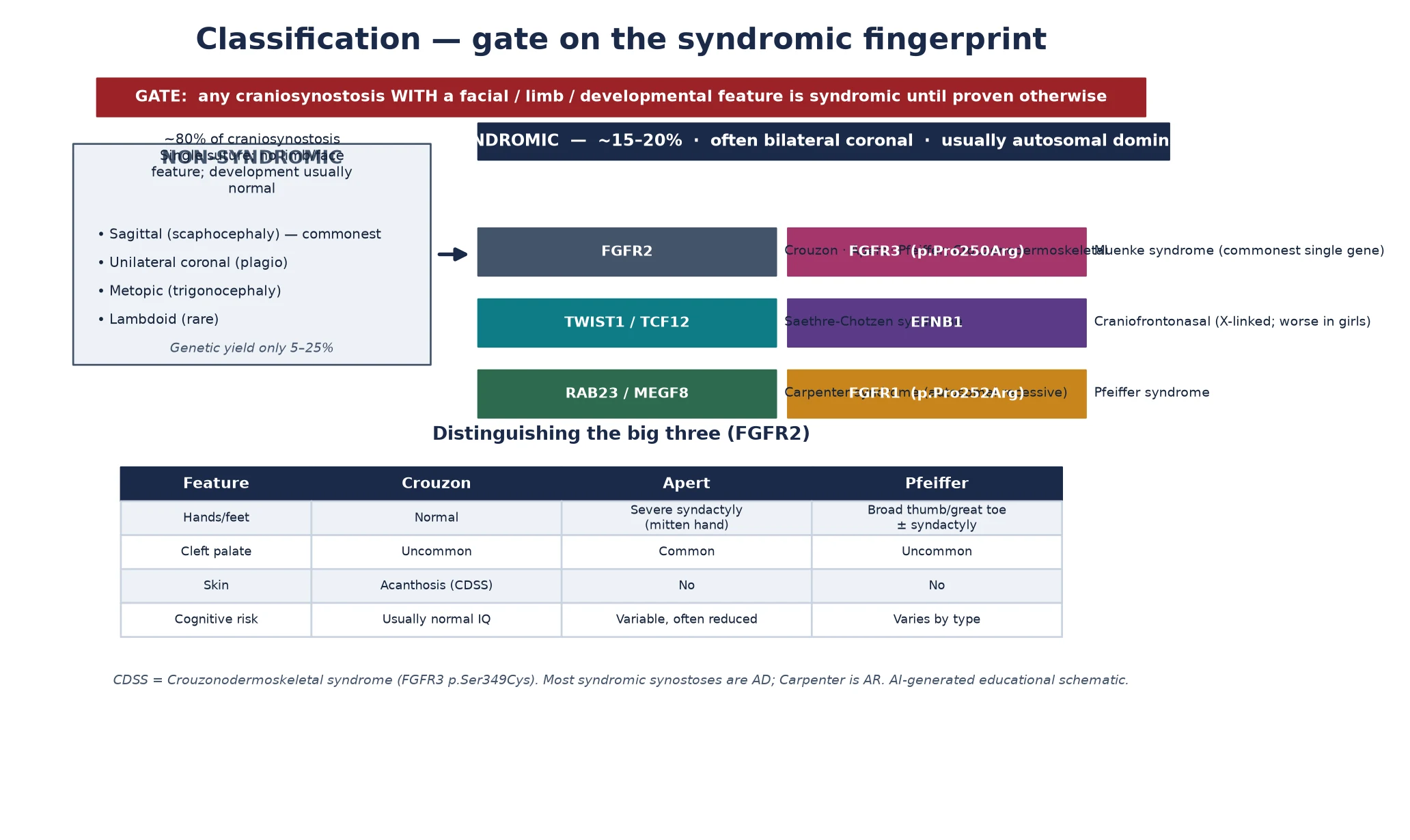
| Feature | Crouzon | Apert | Pfeiffer |
|---|---|---|---|
| Hands and feet | Normal | Severe symmetric syndactyly (mitten hand) | Broad, medially-deviated thumb and great toe |
| Cleft palate | Uncommon | Common | Uncommon |
| Skin | Acanthosis in the FGFR3-related form | No | No |
| Cognitive outcome | Usually normal intelligence | Variable, often reduced | Varies with type and severity |
Epidemiology & Risk Factors
Craniosynostosis as a whole occurs in roughly one in every 2000 to 2500 live births, and the syndromic forms make up about 15 to 20 per cent of that total, which places them among the more recognisable congenital malformations a general paediatrician will meet. The population-level burden of birth defects more broadly has been mapped by national surveillance programmes, which frame craniosynostosis within the wider congenital-anomaly picture that drives screening and service planning, and against which the rarer syndromic fraction is measured. [5] [10]
The reason the syndromic forms are stable, recognisable entities across populations is that they are driven by recurrent, specific point variants. The FGFR2 variants that cause Apert and the FGFR3 p.Pro250Arg that causes Muenke arise repeatedly and independently, and the paternal-age effect — in which activating FGFR2 variants confer a selective growth advantage on the spermatogonial stem cell, so that the variant accumulates with the father's age — explains why Apert syndrome in particular shows a paternal-age association even though most cases are de novo. Recurrent variant plus recurrent mechanism equals a stable prevalence. [1] [6]
Most syndromic craniosynostoses are autosomal dominant, and that is why the family history matters even when a case appears sporadic. A parent who carries the variant — often with a milder expression that was never diagnosed — has a 50 per cent recurrence risk in each pregnancy, and variable expressivity and incomplete penetrance mean the same variant can sit silently in one family member and announce itself severely in the next. Identifying the proband therefore opens a cascade of parental and sibling testing that catches the mildly affected adults whose own airway, hearing and ICP surveillance has been missed. [4] [7]
Pathophysiology
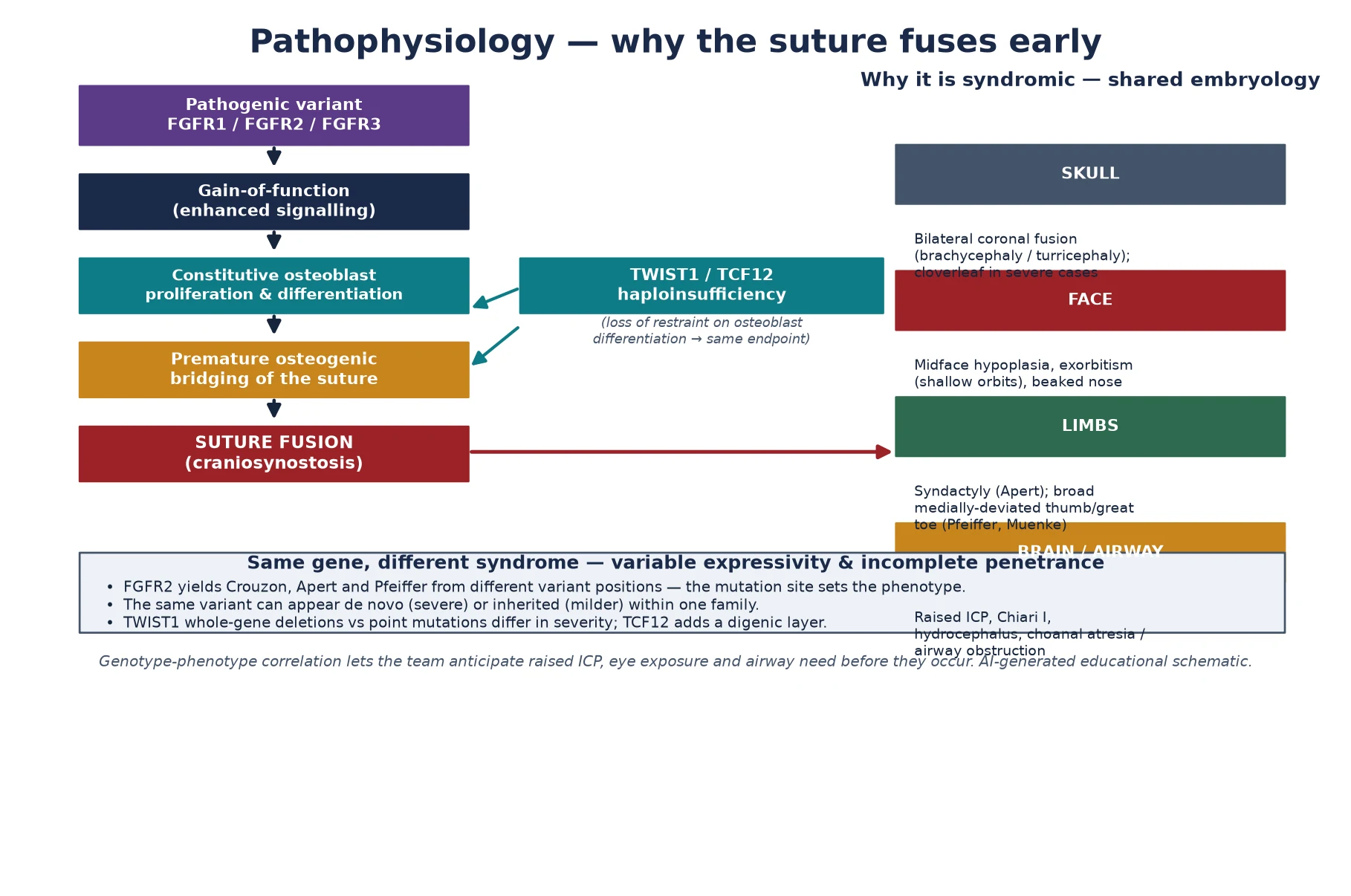
The mechanism that fuses the suture is a gain-of-function variant in a fibroblast growth factor receptor, and understanding it is what makes the phenotype intelligible rather than a list of associations. The fibroblast growth factor receptors sit on the surface of osteoblasts and their precursors, and when their ligand binds they switch on a signalling cascade that tells the cell to divide and to lay down bone. A pathogenic variant in FGFR1, FGFR2 or FGFR3 locks that receptor into an over-active state, so the osteoblasts at the sutural edge proliferate and differentiate beyond their cue and bridge the gap with bone before the brain has finished growing. [1] [6]
The elegance of the system is that the position of the variant within the gene sets the syndrome. A single gene, FGFR2, yields Crouzon, Apert and Pfeiffer from different variant positions, because each position perturbs a slightly different ligand-binding or disulfide-bonding domain, and the downstream signal lands at a slightly different strength on skull, face and limb. That genotype-phenotype correlation is what lets the team anticipate the airway, the eye and the intracranial pressure before they declare themselves, and it is the reason modern surgical planning has become genotype-aware rather than purely anatomical. [9] [1]
Not every syndromic synostosis is an over-active FGFR. Saethre-Chotzen syndrome arises from loss of TWIST1, a transcription factor that restrains osteoblast differentiation, and the loss of that brake produces the same endpoint of premature sutural bridging; TCF12, which encodes a binding partner of TWIST1, is a frequent cause of coronal craniosynostosis in its own right and adds a digenic layer when the two act together. The fellowship point is that the suture can be fused either because the accelerator is stuck on or because the brake is off, and both converge on the same clinical need to secure the airway, the eye and the brain. [4] [2]
Why the package travels together is a lesson in shared embryology. The skull vault, the midface, the orbital walls and the limb buds are all shaped by the same fibroblast-growth-factor and neural-crest signals, so a variant that distorts one distorts them all, producing the recognisable combination of bilateral coronal fusion, shallow orbits with exorbitism, midface hypoplasia and a limb anomaly. Variable expressivity and incomplete penetrance — shaped by modifier genes, mosaicism and stochastic factors — explain why the same variant can produce a severely affected neonate in one family member and a subtly affected adult in another, and why the surveillance plan must be staged to the individual rather than assumed from the label. [1] [7]
Clinical Presentation
The presentation is a head, a face and a hand, and a fellowship answer earns depth by separating them and then reuniting them. The head declares the fused suture through its shape: bilateral coronal fusion produces the short, broad brachycephaly or the tall turricephaly, and the most severe FGFR2 variants produce the trilobed kleeblattschädel, or cloverleaf skull, that marks the dangerous end of the spectrum. The shape is the presenting complaint, and photographing and measuring it from above is the first clinical act. [5] [1]
The face carries the danger. Exorbitism — the protrusion of the eyes because the orbits never grew deep enough to hold them — is the signature of the FGFR2 craniofacial dysostoses and is the feature that converts a cosmetic concern into an ophthalmic emergency, because the exposed cornea dries, ulcerates and can perforate. Midface hypoplasia gives the beaked nose, the class three malocclusion and, critically, the small nasopharynx that narrows the airway; choanal atresia or stenosis compounds the obstruction in the severe forms. The face is where the sight-threatening and airway-threatening complications live, and examining it actively is what catches them. [3] [7]
The limb names the syndrome. Severe symmetric syndactyly of the hands and feet — the mitten hand, where the digits are fused into a single mass — is the Apert signature and is visible from birth. A broad, short and medially-deviated thumb and great toe, sometimes with cutaneous syndactyly, marks Pfeiffer. Broad great toes with carpal and tarsal fusion and a sensorineural hearing loss mark Muenke. Brachydactyly with a low frontal hairline, ptosis and small ears marks Saethre-Chotzen, and polysyndactyly with congenital heart disease points to Carpenter. The limb is the single most efficient discriminator among the FGFR2 syndromes, and examining the hands and feet is non-negotiable. [6] [4]
The brain and the airway declare the complications rather than the diagnosis. Raised intracranial pressure — from multi-suture restriction of the vault, from venous outflow obstruction, or from a Chiari I herniation through a small foramen magnum — presents as irritability, sleep disturbance, headache in the verbal child, vomiting, a developmental plateau, and papilloedema on fundoscopy. Airway obstruction presents as snoring, stertor, feeding difficulty, failure to thrive and, on a sleep study, obstructive sleep apnoea. The well-looking child with a broad toe and an odd head shape is the presentation that must not close the search, because the silent raised pressure and the silent apnoea are what harm the developing brain. [7] [3]
Differential Diagnosis
The differential splits into three questions: is the head shape a fused suture at all, is the fused suture syndromic or nonsyndromic, and which syndrome is it. The first question is the one that prevents unnecessary surgery, because the commonest mimic of craniosynostosis is a deformational or positional head shape — the parallelogram plagiocephaly of a baby who lies on one side — which fuses no suture, improves with repositioning and physiotherapy, and needs no operation. The suture is felt along its length; a fused suture has a hard bony ridge, a deformational head does not. [5]
Once a suture is genuinely fused, the syndromic gate applies. Nonsyndromic single-suture disease — sagittal, metopic, unicoronal, lambdoid — carries no travelling facial or limb feature, a much lower genetic yield, and a far lower rate of raised intracranial pressure, and it is managed with a single operation and surveillance. The syndromic forms carry the package, the multi-suture involvement, the airway and eye and ICP burden, and a 50 per cent recurrence risk for the dominant genes. Mistaking one for the other either subjects a child to needless lifelong surveillance or, more dangerously, strips a syndromic child of the airway, eye and pressure protection that the surveillance exists to provide. [1] [6]
Among the syndromes, the limb is the great separator, and the comparison table in the classification section is the tool. The trap within the syndromic group is Muenke syndrome, whose recurrent FGFR3 p.Pro250Arg variant produces a coronal synostosis that can look isolated, so the child is labelled nonsyndromic unicoronal or bicoronal disease and the diagnosis is missed until a broad great toe, a hearing loss, or a reoperation for recurrent fusion surfaces the gene. Testing the apparently nonsyndromic coronal case for FGFR3 p.Pro250Arg is the safeguard, because a positive result restores the recurrence counselling and the hearing and ICP surveillance that the miss would have forfeited. [12] [1]
A broader set of genetic syndromes produce craniofacial disproportion without fusing a suture and enter the differential by their face. Treacher Collins syndrome, a mandibulofacial dysostosis from TCOF1 or POLR1D, produces the down-slanting palpebral fissures, malar hypoplasia and micrognathia of a child whose sutures are open. Pierre Robin sequence produces the small jaw and cleft palate that compromise the airway but again fuses no suture. Noonan and the RASopathies carry short stature and a characteristic face, and 22q11.2 deletion carries the conotruncal heart and cleft palate. The discriminating question is always the suture: fuse the suture and the syndromic synostosis family comes into view; keep the suture open and the craniofacial differential shifts to the dysostoses and sequences. [4] [5]
Clinical & Bedside Assessment
The recognition move is to look at the head from above, then the face front-on, then the hands and feet — and to do it in that order, because the head sets the question, the face finds the danger, and the limb names the syndrome. The head shape is assessed from the vertex for brachycephaly or turricephaly, the sutures are palpated for a bony ridge, the fontanelles are felt, and the head circumference is measured and plotted, because a head crossing centiles upward or, more subtly, faltering in its growth against a fused vault is a clue to rising pressure. [5]
The history gathers the discriminators that drive the syndromic label and the safety triad. Ask about the onset and progression of the head shape, about snoring, noisy breathing and observed apnoea, about eye symptoms of exposure such as redness or watering, about feeding difficulty and growth, and about developmental concern. Ask about a family history of craniofacial or limb difference, of early hearing loss, or of a relative who had a head operation, because the autosomal-dominant inheritance means a mildly affected parent is a real and recurrent finding. Growth, feeding and the developmental trajectory function as vital signs here, because faltering or a plateau often signals an unrecognised airway, eye or pressure problem. [7]
Examination then moves to the danger zones. Inspect the eyes for exorbitism, measure the degree of protrusion, check the cornea for exposure, and look for strabismus; the exophthalmos of Crouzon and Apert is quantifiable and is the sign that converts the visit into an ophthalmic referral. Examine the airway for stertor and work of breathing, and arrange a sleep study rather than relying on the bedside impression, because obstructive sleep apnoea in midface hypoplasia is frequently silent to history. Examine the hands and feet fully — every digit, every web, every nail — because the limb sign is the single most efficient gene predictor and is easy to under-examine in a busy clinic. [3] [6]
The first visit also assesses the family's practical capacity for a lifelong, staged, surgical condition, because a surveillance plan that demands repeated travel to a distant craniofacial centre is only as good as the family's ability to attend it. Map the access to the centre, the travel and accommodation support, the schooling and learning support, the psychosocial and peer-support needs, and the interpreter and cultural requirements, and identify a named coordinator early. Combining the clinical gestalt with the genetic result — and holding both when they seem to disagree — is what protects against both over-diagnosis of an incidental variant and under-diagnosis of a classical syndrome. [8] [1]
Investigations
Imaging maps every fused suture and the brain behind it, and it is the indispensable first investigation. A skull radiograph often suffices to show the fused suture as a bony bridge with loss of the suture line, but a low-dose three-dimensional computed tomography of the cranial vault is obtained when the pattern is complex or syndromic, because it maps every suture, defines the skull-base anatomy including the foramen magnum for Chiari, and assesses the venous sinuses. Magnetic resonance imaging is added when a brain malformation, a Chiari I herniation, or a venous outflow stenosis is suspected, because those findings change the surgical plan. [5] [1]
Polysomnography is not an optional extra; it is an essential investigation in the syndromic child, because obstructive sleep apnoea from midface hypoplasia is common, frequently under-reported by families, and a driver of pulmonary hypertension and failure to thrive when missed. A baseline sleep study is obtained at diagnosis and repeated as the child grows and around any airway or midface surgery. Ophthalmology assessment is equally mandatory: the cornea is examined for exposure, the optic discs for papilloedema as a sign of raised intracranial pressure, and the refraction and squint for the strabismus that accompanies the shallow orbits. [3] [7]
Genetic testing confirms the syndrome and unlocks the counselling, and it is now targeted and efficient. A panel covering FGFR1, FGFR2, FGFR3, TWIST1 and TCF12 — or a targeted test for the recurrent FGFR3 p.Pro250Arg variant when the pattern is coronal — names the gene in the majority of syndromic cases, and a chromosome microarray is added when a chromosomal mimic is in play. The gene predicts the face, the limb, the inheritance and, increasingly, the surgical risk, so the result is not a label to file but a tool to plan with. Audiology completes the baseline, because hearing loss — conductive from middle-ear effusion, or sensorineural in Muenke — is common and treatable, and an unrecognised loss compounds any developmental concern. [12] [2]
Raised intracranial pressure is the complication that imaging and clinical signs together screen for, and it is evaluated actively. Fundoscopy looks for papilloedema, optical coherence tomography quantifies the nerve-fibre layer in the cooperative child, and, in selected cases, overnight intracranial pressure monitoring confirms the borderline suspicion that neither fundoscopy nor imaging has resolved. When pressure is confirmed, the surgical plan accelerates toward vault expansion, because unrecognised raised pressure harms the visual system and the developing brain. Prenatal detection closes the loop: an abnormal head shape or limb anomaly on the anatomy scan triggers prenatal genetic testing and delivery planning at a craniofacial centre. [7] [9]
Management — Resuscitation
Resuscitation in syndromic craniosynostosis means securing the three things that harm before any cosmetic plan for the head, and the order is airway, eye, and intracranial pressure. A newborn with severe midface hypoplasia or choanal atresia may present in airway distress, and the immediate management is airway positioning, a nasopharyngeal airway, continuous positive airway pressure, and, in the most severe, a tracheostomy to secure the airway while the midface is planned. Treating the head shape first, in a child whose airway is compromised, is the cardinal resuscitation error. [7] [5]
The exposed eye is the second emergency. Exorbitism from shallow orbits dries and ulcerates the cornea, and the immediate management is copious lubrication, a moisture chamber, and — when exposure is severe and threatens perforation — urgent tarsorrhaphy to protect the globe until definitive orbital reconstruction can be performed. The fellowship point is that protecting the eye is not a phase of the cosmetic plan but a parallel emergency that runs from the first clinic visit, and the ophthalmology team is involved at diagnosis rather than after a complication. [3]
Raised intracranial pressure is the third resuscitation-grade threat, and it is a neurosurgical emergency when papilloedema, a clear developmental plateau, or confirmed pressure is present. The immediate management is escalation toward vault expansion to create room for the growing brain, with a shunt for hydrocephalus and foramen-magnum decompression for a symptomatic Chiari I in the relevant syndromes. The trap is to attribute the headache or the developmental plateau to the syndrome and to wait, because the pressure is treatable and the harm of waiting is visual and cognitive. [7] [9]
Feeding and growth are resuscitation concerns in their own right, because the airway obstruction that compromises breathing also compromises feeding, and a child who burns calories working to breathe fails to thrive. Nutritional support, feeding augmentation, and treatment of the airway run together, and the family is given an honest early explanation of the genetic, lifelong, multidisciplinary nature of the diagnosis, because a family that understands the staged plan from the start adheres to it. A severe presentation — a kleeblattschädel with choanal atresia, the Pfeiffer type 2 or 3 picture with airway and cerebral compromise — demands urgent transfer to a specialist craniofacial centre where the staged pathway can be executed. [11] [1]
Management — Definitive & Stepwise
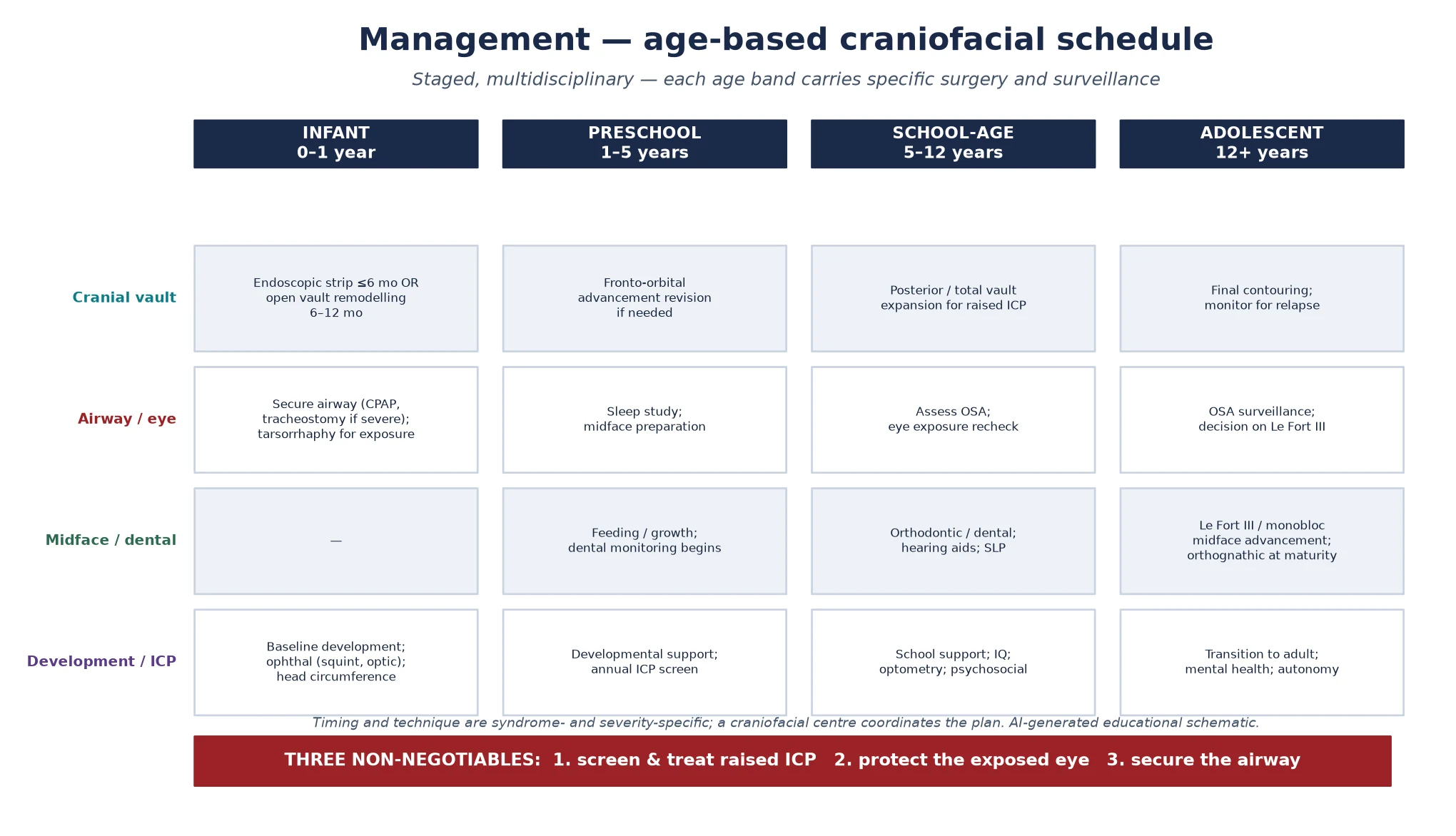
Definitive management is an age-based surgical and surveillance schedule that moves with the person from infancy into adult care, and the craniofacial team — neurosurgery, plastic and craniofacial surgery, genetics, ophthalmology, ear-nose-throat and audiology, speech and language, dental and orthodontics, and psychology — coordinates it. The head is reshaped early, the face is advanced later, and the airway, eye, pressure and development are surveilled throughout, because the cranial operation is one event in a lifelong plan rather than its conclusion. [5] [8]
In infancy, the priority is the cranial vault. The fused sutures are released and the vault is remodelled, either by an endoscopic strip craniectomy followed by helmet moulding in the very young infant — typically under six months — or by an open vault and fronto-orbital remodelling in the older infant. The choice of technique is centre-, age- and syndrome-specific, and the goal is to create room for the growing brain and to reshape the head while the bone is still malleable. The airway, the eye and the pressure are managed in parallel, because an infant whose vault is expanded but whose airway is unaddressed has exchanged one problem for another. [9] [7]
Across the preschool and school years, the plan layers fronto-orbital advancement revision when the forehead retrudes, posterior or total vault expansion when raised intracranial pressure recurs, and the developmental, educational, hearing and dental surveillance that the syndromes demand. The broad great toe or the mitten hand is corrected by hand surgery, coordinated with the craniofacial timeline; the strabismus and the corneal exposure are managed by ophthalmology; the middle-ear effusion and the conductive loss are managed by ENT. Growth, development and school progress are tracked at every visit, because a plateau often signals a recurrent pressure or an unrecognised airway problem. [12] [6]
Adolescence brings the midface forward. A Le Fort III or monobloc midface advancement corrects the midface hypoplasia, enlarges the nasopharynx and improves both the airway and the exorbitism, and definitive orthognathic surgery is planned at skeletal maturity. The transition to adult craniofacial and psychological care is structured, with the documented genotype, the surgical history, the airway and pressure status, and the psychosocial needs handed over intact, because the adult with a repaired syndromic craniosynostosis still requires craniofacial, airway and mental-health surveillance. [8] [11]
Specific Subtypes & Scenarios
Apert syndrome is the syndromic synostosis a fellowship candidate must know in depth, because it combines the most severe limb anomaly with a characteristic surgical and developmental pathway. The defining feature is severe symmetric syndactyly of the hands and feet — the mitten hand — alongside a tall or short skull from bicoronal fusion, a cleft palate in many, and a cognitive outcome that is variable and often reduced in proportion to the brain abnormality. The surgical pathway therefore layers hand surgery for syndactyly release onto the cranial vault and midface timeline, and the developmental surveillance is more intensive than in Crouzon. [7] [3]
Crouzon syndrome shares the FGFR2 craniofacial dysostosis — bicoronal fusion, exorbitism, midface hypoplasia and a beaked nose — but spares the hands, and cognition is usually preserved, which is the discriminating comfort in a child who otherwise looks severe. A distinct FGFR3-related form, Crouzonodermoskeletal syndrome, adds acanthosis nigricans and skeletal anomalies, and is separated from classic Crouzon by its gene and its skin. The absence of a limb anomaly does not make Crouzon mild, because the exorbitism and the midface airway are fully present, but the preserved cognition and the normal hands do shape the counselling. [6] [7]
Pfeiffer syndrome separates into the milder type 1 and the severe types 2 and 3, and the separation is operationally critical. Type 1 carries the broad thumb and great toe with a variable synostosis and a relatively good prognosis; types 2 and 3 carry the kleeblattschädel, the choanal atresia, the airway and cerebral compromise, and a markedly poorer prognosis. Modern genotype-specific data show that a child with the W290C FGFR2 variant follows a surgical pathway associated with improved developmental outcomes and reduced mortality, and a therapeutic algorithm based on a modified severity grading scale now guides the timing and extent of intervention. Naming the subtype and, where possible, the variant is what lets the team plan rather than react. [9] [11]
Muenke syndrome, from the recurrent FGFR3 p.Pro250Arg variant, is the commonest single-gene syndromic synostosis and the great masquerader. Its coronal synostosis can look isolated, so the child is labelled nonsyndromic — until the broad great toe, the carpal or tarsal fusion, the sensorineural hearing loss, or a reoperation for recurrent fusion surfaces the gene. The data show that the FGFR3 p.Pro250Arg variant increases the risk of reoperation in what appeared to be nonsyndromic coronal disease, and the safeguard is to test the apparently isolated coronal case for the variant, because a positive result restores the recurrence counselling, the hearing surveillance and the surgical expectation. [12] [1]
Saethre-Chotzen syndrome, from TWIST1 or TCF12, carries a distinctive face — a low frontal hairline, ptosis, small ears with a prominent crus, and brachydactyly — alongside a coronal synostosis that is often asymmetric. Craniofrontonasal syndrome, from EFNB1, is the X-linked paradox: heterozygous girls are more severely affected than hemizygous boys, with a coronal synostosis, a bifid nasal tip, wiry hair and developmental difference, and its X-linked inheritance changes the counselling. Carpenter syndrome, from RAB23 or MEGF8, is the autosomal-recessive exception — polysyndactyly, congenital heart defects and short stature — and its 25 per cent sibling recurrence is the counselling fact that distinguishes it from the dominant family. [4] [2] [5]
Why the W290C variant changed Pfeiffer management
The FGFR2 W290C variant disrupts a specific disulfide bond in the third immunoglobulin-like domain, and the cohort data show that children with this variant follow a surgical pathway — earlier and more complete vault and airway intervention — associated with improved developmental outcomes and reduced mortality compared with historical, anatomy-only planning. The lesson is that genotype-specific surgery is replacing purely anatomical planning: the gene predicts the pressure and the airway risk, and the team intervenes before rather than after the complication. [9] [11]
Complications & Pitfalls
The harm in syndromic craniosynostosis comes from the three complications the surveillance exists to catch — raised intracranial pressure, corneal exposure, and airway obstruction — and from the diagnostic errors that delay the label. Raised intracranial pressure is the cardinal complication, because multi-suture fusion restricts the vault while the brain keeps growing, and unrecognised pressure harms the visual system through papilloedema and optic atrophy and the developing brain through cognitive plateau. The safeguard is active, repeated screening — fundoscopy, developmental tracking, and pressure monitoring when the suspicion is borderline — rather than waiting for the pressure to declare itself through harm. [7] [9]
Corneal exposure is the sight-threatening complication, and it is the one most easily under-weighted because the protruding eye looks dramatic but stable. The exposed cornea dries, abrades, ulcerates and can perforate, and the safeguard is to treat exorbitism as an ophthalmic emergency from the first visit: lubrication, moisture chamber, and tarsorrhaphy or orbital reconstruction when exposure threatens the globe. The pitfall is to treat the head shape first and the eye later, because the cornea will not wait for the craniofacial timetable. [3] [7]
Airway obstruction from midface hypoplasia is a lifelong complication, and it is missed without a sleep study because the family under-reports it and the bedside impression is unreliable. The safeguard is a baseline polysomnography at diagnosis and repeat studies through growth and around surgery, with midface advancement and, in the severe, tracheostomy as the surgical answers. Chiari I herniation and hydrocephalus complicate the severe FGFR2 syndromes disproportionately, and the small foramen magnum that accompanies them changes the neurosurgical plan, because a symptomatic Chiari demands decompression alongside the vault expansion. [5] [7]
The diagnostic pitfalls share a common root: substituting an easier label for the harder one. Mistaking a deformational head shape for a fused suture leads to unnecessary surgery; mistaking Muenke for nonsyndromic coronal disease forfeits the recurrence counselling, the hearing surveillance and the surgical expectation; and treating a cosmetic concern before securing the airway, the eye and the pressure inverts the priorities and endangers the child. Over-interpreting a variant of uncertain significance as a classical syndrome is the opposite error, and the safeguard is always to correlate the gene with the gestalt and to hold both when they disagree. [12] [1]
Prognosis & Disposition
The prognosis spans from a near-normal lifespan and cognition — in Crouzon and in many with Muenke — to a significant neurodevelopmental and surgical burden in severe Apert and in Pfeiffer types 2 and 3, and the determinant is less the label than the quality and timing of the multidisciplinary care. Early diagnosis, genotype confirmation, and consistent craniofacial-team management are the single biggest predictors of outcome, because each preventable complication — the unrecognised pressure, the exposed cornea, the silent apnoea — subtracts from the trajectory the surveillance exists to protect. [7] [9]
Long-term quality-of-life data show that adults with syndromic craniosynostosis, followed through a craniofacial programme, achieve meaningful participation in education, work and relationships, and that the visible difference and the surgical history shape — but do not determine — the life. The fellowship answer frames prognosis in those terms rather than in deficit language alone, naming the psychosocial support, the peer contact and the structured transition as the assets that protect the adult outcome, alongside the surgical and medical surveillance. [8] [11]
The general paediatrician owns the coordination, and the disposition is shared, structured care at a craniofacial centre. Neurosurgery and plastic or craniofacial surgery lead the vault and midface operations, ophthalmology owns the cornea and the pressure signs, ENT and audiology own the airway and the hearing, genetics owns the testing and the counselling, and dental, speech, and psychology contribute across the lifespan. A named coordinator prevents the fragmentation that is the enemy of a checklist-based plan, and referral to a specialist craniofacial centre at diagnosis — rather than after a complication — measurably improves the outcome. [5] [8]
Recurrence-risk counselling closes the prognostic picture. When a parent carries an autosomal-dominant variant, the recurrence risk is 50 per cent in each pregnancy, and gonadal mosaicism accounts for a small additional risk even when parental testing is normal; prenatal testing and preimplantation genetic testing are options the family is offered. For the autosomal-recessive Carpenter syndrome the sibling recurrence is 25 per cent, and for the X-linked craniofrontonasal syndrome the inheritance follows the daughters and sons in the pattern the gene dictates. The counselling addresses not only the next pregnancy but the surveillance of the wider family, because the dominant fraction means a positive test opens a cascade of relatives who may themselves be living undiagnosed. [4] [2]
Special Populations
The syndrome interacts with the child's social, cultural and geographic context, and the same surveillance plan behaves differently across populations — access, adherence and late presentation each shape the outcome. A plan that is clinically correct but unattainable for a family living far from the craniofacial centre is no plan at all, and the fellowship answer recognises that the staged schedule is only as good as the family's ability to engage with it. [8] [1]
Indigenous children in Australia and New Zealand may carry a higher background burden of ear and respiratory disease that the midface airway obstruction of a syndromic synostosis amplifies, and reduced access to a craniofacial centre in remote communities intensifies the need for early referral, outreach and telehealth coordination. The standard indigenous-health emphasis on early audiological and respiratory surveillance is even more urgent in the syndromic child, because the airway and the hearing are already compromised by the syndrome, and telehealth and outreach extend the surveillance net into communities that a clinic-based model would miss. [6] [5]
Migrant, refugee and asylum-seeking families may arrive with incomplete records, an uncertain family history and no prior genetic testing, and the diagnosis may not have been made in the country of origin. A careful reconstruction of the history, confirmation of the syndrome and its inheritance by panel testing and parental testing, and an interpreter-mediated explanation of the staged surgical and genetic plan are the foundations. Vaccination and surveillance status are reconciled, and the written schedule is provided in the family's language, because a plan that cannot be read cannot be followed. [4] [7]
Socioeconomic disadvantage shapes late presentation, the feasibility of repeated travel for staged surgery, and the psychosocial support available, because the limiting step is often attendance and transport rather than the medicine. Structuring the surveillance around coordinated centre visits, linking the family to peer-support and travel-and-accommodation programmes, and using telehealth to reduce travel all improve engagement, and a coordinator who knows the local services is the asset that makes a staged plan deliverable. The school-age child with a visible craniofacial difference and possible learning needs is a population in their own right, because the educational and psychosocial support is what protects the developmental trajectory, and trauma-informed, interpreter-supported communication extends to the surgical-consent conversation itself. [8] [11]
Evidence, Guidelines & Regional Differences
The evidence base rests on the Twigg and Wilkie 2015 review of craniofacial malformations, which frames the FGFR biology and the shared-embryology logic that a fellowship answer is built on, and on the Passos-Bueno and Jabs 1999 review of the clinical spectrum of fibroblast growth factor receptor mutations, which maps the FGFR family across the syndromes. The Cohen 2009 sutural-biology perspective supplies the classification and the biological reasoning, and the Kreiborg and Cohen 2010 ocular data anchor the exorbitism and the corneal exposure that drive the ophthalmic emergency. [1] [6] [5]
The genetic evidence is what distinguishes a mature answer from a checklist. The Paznekas 1998 paper established the genetic heterogeneity of Saethre-Chotzen through TWIST1 and FGFR, and the Sharma 2013 Nature Genetics finding identified TCF12 as a frequent cause of coronal craniosynostosis, adding the digenic layer that a contemporary answer must name. The Fernandes 2016 data on Apert and Crouzon cognitive development and brain abnormalities anchor the neurodevelopmental surveillance and the prognostic framing, and the Thomas 2005 data on the FGFR3 p.Pro250Arg reoperation risk is the evidence that drives the testing of apparently nonsyndromic coronal cases. [4] [2] [7] [12]
The management evidence has moved toward genotype-specific planning. The Wenger 2019 cohort showed that a genotype-specific surgical approach for the Pfeiffer W290C variant improved developmental outcomes and reduced mortality, and the Raposo-Amaral 2020 therapeutic algorithm formalised a severity-graded approach to Pfeiffer management. The Sakamoto 2021 quality-of-life data ground the prognostic counselling in measured adult outcomes rather than impression, and the Mai 2019 national birth-defects estimates frame the epidemiology within the broader congenital-anomaly burden. [9] [11] [8] [10]
Where the evidence is weak, a fellowship answer says so honestly. The optimal timing and technique of vault and midface surgery, the role of endoscopic versus open repair in the syndromic forms, the threshold for overnight intracranial pressure monitoring, and the long-term durability of midface advancement are areas of genuine uncertainty and evolving practice, and regional centre practices differ in the availability of three-dimensional imaging, panel testing and the endoscopic option. Naming the uncertainty is a mark of intellectual honesty that examiners reward, and it guards against presenting a contested technique as settled. [5] [9]
In Australia and New Zealand, syndromic craniosynostosis is managed through specialist craniofacial centres in the major cities, where neurosurgery, plastic and craniofacial surgery, genetics, ophthalmology, ENT and audiology coordinate the staged plan. Access to a centre, to three-dimensional imaging and panel genetic testing, and to the endoscopic option varies by jurisdiction, and the great distances of rural and remote ANZ intensify the need for early referral, outreach and telehealth coordination. Surgical timing and technique are centre-specific and guided by the genotype where one is identified, and the transition to adult craniofacial and psychological care is increasingly formalised. [8] [9]
Exam Pearls
A fellowship candidate answering on syndromic craniosynostosis should land five anchor points and avoid three classic traps. The anchors are the framework examiners listen for; the traps are where easy marks are lost. [1] [5]
Anchor one: recognise the syndromic fingerprint. An abnormal head shape with midface hypoplasia, exorbitism or a limb anomaly is syndromic until proven otherwise, because the same genetic signal fuses the suture and shapes the face and the hand. The head, the face and the hand are one diagnosis. [6]
Anchor two: map every fused suture. Low-dose three-dimensional computed tomography maps every suture and the skull base, because the unrecognised additional fusion is what drives the raised intracranial pressure that the surveillance exists to catch. The obviously fused suture is not the whole story. [5]
Anchor three: confirm the gene. A targeted FGFR and TWIST1 panel names the syndrome, predicts the face, the limb, the inheritance and the surgical risk, and a chromosome microarray excludes the chromosomal mimics. Muenke syndrome, from FGFR3 p.Pro250Arg, is the commonest single-gene form and the one most often missed. [2] [12]
Anchor four: secure airway, eye and intracranial pressure. The airway from midface hypoplasia and choanal atresia, the cornea from exorbitism, and the brain from multi-suture raised pressure are the three things that harm, and each is secured before any cosmetic plan for the head. [3] [7]
Anchor five: coordinate an age-based craniofacial team. The vault is remodelled in infancy, the forehead and orbits in the preschool years, the midface in adolescence, and the airway, eye, pressure and development are surveilled throughout, because the cranial operation is one event in a lifelong plan. [8] [9]
The three traps to avoid are treating the head shape before securing the airway, eye and pressure; missing Muenke by labelling a coronal synostosis nonsyndromic and forfeiting the recurrence counselling; and forgetting that most forms are autosomal dominant, so the parents must be tested. The high-yield numbers a candidate holds are these: roughly one in 2000 to 2500 overall, around 15 to 20 per cent syndromic, a 50 per cent recurrence risk for the dominant genes, and FGFR2 the gene for Crouzon, Apert and Pfeiffer — with the limb as the separator. Avoid the traps and land the anchors, and the rest of the answer falls into place. [1] [4]
References
- [1]Twigg SR, Wilkie AO. New insights into craniofacial malformations. Hum Mol Genet, 2015.PMID 26085576
- [2]Sharma VP, Fenwick AL, Brockop MS, McGowan SJ, Goos JA, Hoogeboom AJ, Brady AF, Jeelani NO, Lynch SA, Mulliken JB, Murray DJ, Phipps JM, Sweeney E, Tomkins SE, Wilson LC, Johnson D, Wall SA, van der Spek PJ, Mathijssen IM, Maxson RE, Twigg SR, Wilkie AO. Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat Genet, 2013.PMID 23354436
- [3]Kreiborg S, Cohen MM Jr. Ocular manifestations of Apert and Crouzon syndromes: qualitative and quantitative findings. J Craniofac Surg, 2010.PMID 20856021
- [4]Paznekas WA, Cunningham ML, Howard TD, Korf BR, Lipson MH, Grix AW, Feingold M, Goldberg R, Borochowitz Z, Aleck K, Mulliken J, Yin M, Jabs EW. Genetic heterogeneity of Saethre-Chotzen syndrome, due to TWIST and FGFR mutations. Am J Hum Genet, 1998.PMID 9585583
- [5]Cohen MM Jr. Perspectives on craniosynostosis: sutural biology, some well-known syndromes, and some unusual syndromes. J Craniofac Surg, 2009.PMID 19293680
- [6]Passos-Bueno MR, Wilcox WR, Jabs EW, Sertié AL, Alonso LG, Kitoh H. Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat, 1999.PMID 10425034
- [7]Fernandes MB, Maximino LP, Perosa GB, Abramides DV, Passos-Bueno MR, Yacubian-Fernandes A. Apert and Crouzon syndromes-Cognitive development, brain abnormalities, and molecular aspects. Am J Med Genet A, 2016.PMID 27028366
- [8]Sakamoto Y, Takenouchi T, Miwa T, Kishi K. Assessment of long-term quality of life in patients with syndromic craniosynostosis. J Plast Reconstr Aesthet Surg, 2021.PMID 33039308
- [9]Wenger TL, Hopper RA, Rosen A, Tully HM, Cunningham ML, Lee A. A genotype-specific surgical approach for patients with Pfeiffer syndrome due to W290C pathogenic variant in FGFR2 is associated with improved developmental outcomes and reduced mortality. Genet Med, 2019.PMID 29915381
- [10]Mai CT, Isenburg JL, Canfield MA, Meyer RE, Correa A, Alverson CJ, Lupo PJ, Riehle-Colarusso T, Cho SJ, Aggarwal D, Kirby RS, National Birth Defects Prevention Network. National population-based estimates for major birth defects, 2010-2014. Birth Defects Res, 2019.PMID 31580536
- [11]Raposo-Amaral CE, Denadai R, Máximo G, Raposo-Amaral CA, Ghizoni E. Pfeiffer Syndrome: A Therapeutic Algorithm Based on a Modified Grading Scale. Plast Reconstr Surg Glob Open, 2020.PMID 32440448
- [12]Thomas GP, Wilkie AO, Richards PG, Wall SA. FGFR3 P250R mutation increases the risk of reoperation in apparent 'nonsyndromic' coronal craniosynostosis. J Craniofac Surg, 2005.PMID 15915095