Paeds · genetics-dysmorphology-and-metabolism
Tuberous sclerosis complex
Also known as Tuberous sclerosis · TSC · Bourneville disease · Epiloia · Phakomatosis · mTOR-opathy
A fellowship approach to tuberous sclerosis complex: recognise the infant with hypomelanotic macules and infantile spasms or the fetus with a cardiac rhabdomyoma as having TSC, confirm with the 2012 international consensus criteria and TSC1/TSC2 testing, explain the hamartin-tuberin-Rheb-mTORC1 mechanism, build organ-by-organ surveillance, and offer everolimus for growing subependymal giant cell astrocytoma and refractory epilepsy.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The mark-winning candidate keeps three layers in view at once. The first is the child in front of you: their seizures, their skin, their development, and the hamartomas that are and are not there across every organ. The second is the molecular mechanism: a lost tumour-suppressor brake that fails to restrain mTORC1-driven cell growth, and which a drug can restore downstream. The third is the family: an autosomal-dominant condition with 50 per cent transmission means every first-degree relative deserves assessment and counselling. [1] [2]
Overview & Definition
Tuberous sclerosis complex is an autosomal-dominant disorder caused by loss-of-function variants in either of two genes: TSC1 on chromosome 9q34, which encodes hamartin, and TSC2 on chromosome 16p13.3, which encodes tuberin. These two proteins join to form a complex that restrains cell growth by braking the mTORC1 signalling pathway. When the complex is lost, cells in the skin, brain, kidney, heart and lung proliferate without control and form the hamartomas and benign tumours that define the condition. [1]
The name "tuberous sclerosis" comes from the cortical tubers, the potato-like brain malformations first described by Bourneville in 1880, but the disease is far more than a brain disorder. It is a tumour-predisposition and malformation syndrome that declares itself differently at every age: a fetal cardiac rhabdomyoma, an infant with spasms, a preschool child with autism, and an adult woman with cystic lung disease can all be the same condition. [2]
TSC is one of the phakomatoses, the neurocutaneous syndromes shared with neurofibromatosis, Sturge-Weber syndrome and von Hippel-Lindau disease, but it is the only one driven by the mTOR pathway. Recognising the cluster of seizures, skin lesions and multi-organ hamartomas, and confirming it with the consensus criteria and molecular testing, sets the surveillance schedule and opens the door to targeted therapy. [1] [2]
Classification
The classification that governs practice is the 2012 International Tuberous Sclerosis Complex Consensus diagnostic criteria, which replaced the older clinical "criterion of certainty" and brought molecular testing to the centre. Under these criteria, a diagnosis is definite when a patient has two major features, or one major plus two minor features, and is also definite when a pathogenic TSC1 or TSC2 variant is identified, even without clinical features. This means a positive genetic test alone closes the diagnosis, which has transformed prenatal and pre-symptomatic detection. [3]
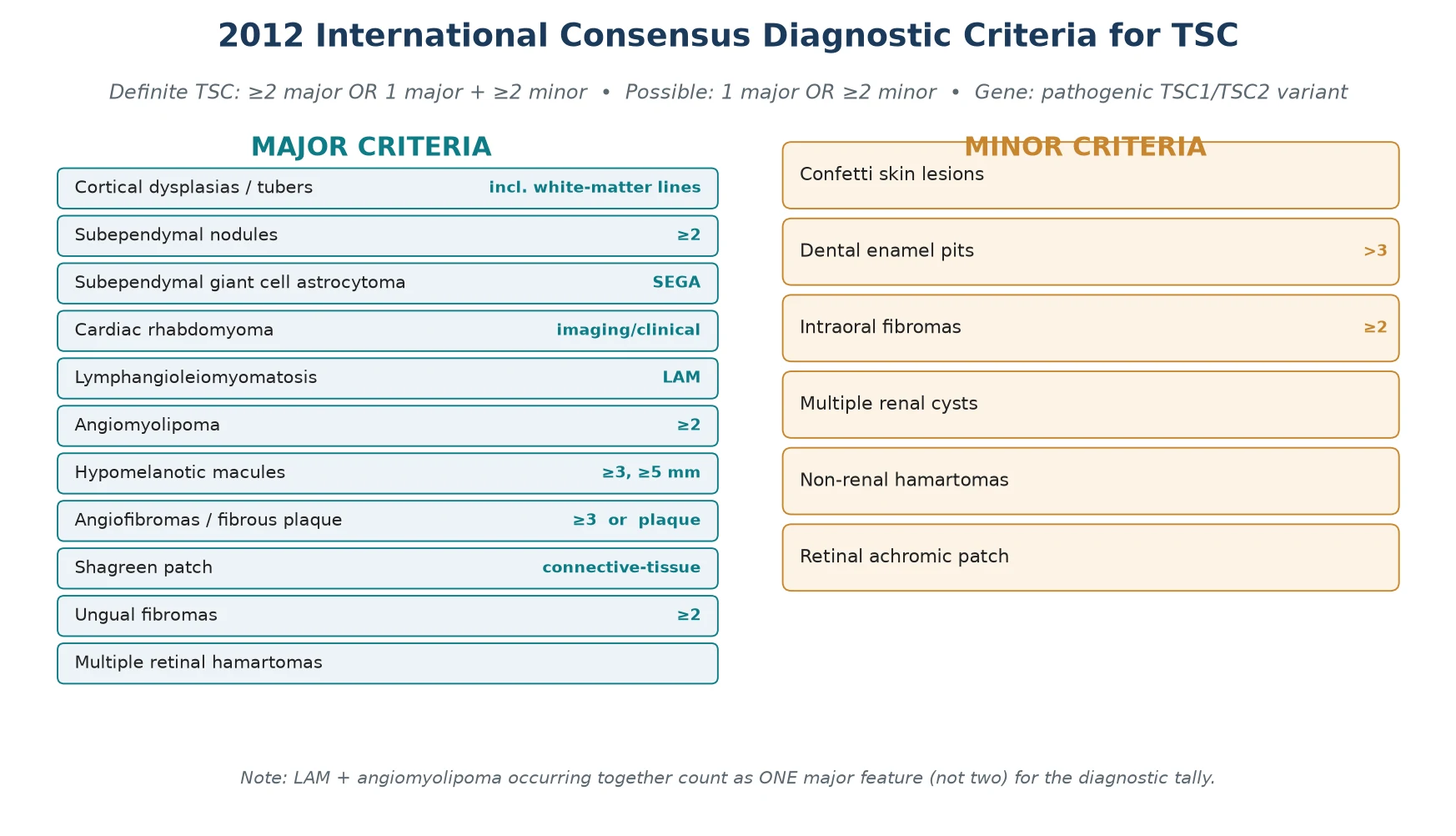
One subtlety that examiners reward is the combined-feature rule. When lymphangioleiomyomatosis and renal angiomyolipomas occur together, they count as a single major feature rather than two, so they do not by themselves confirm a definite diagnosis. The minor features — confetti skin lesions, dental enamel pits, intraoral fibromas, multiple renal cysts, non-renal hamartomas and retinal achromic patches — rarely make the diagnosis alone but tip the balance in an equivocal case. [3]
Major features of TSC \u2014 HAM SURE
Epidemiology & Risk Factors
TSC affects roughly one in 6,000 to one in 10,000 newborns, with no major ethnic predilection and an equal sex distribution. It is the second commonest of the phakomatoses after neurofibromatosis type 1, and it is one of the commonest genetic causes of epilepsy and of autism spectrum disorder. About two-thirds of cases arise from a de novo variant, which means a negative family history never excludes the diagnosis. [1] [2]
The strongest risk factor is a confirmed affected first-degree relative. TSC is autosomal dominant with effectively complete penetrance but highly variable expressivity, so an affected parent has a 50 per cent chance of passing the variant to each child, yet severity within a family can differ enormously — a mildly affected parent may carry a variant that produces severe disease in their child. Because penetrance is complete, an apparently unaffected parent of a sporadic case should be examined for subtle features and offered testing to detect mosaicism. [1] [9]
A clinically important genetic correlation is the TSC2-PKD1 contiguous gene syndrome. A large deletion on chromosome 16 that removes both TSC2 and the adjacent polycystic kidney disease gene PKD1 produces a severe infantile phenotype with early-onset, rapidly progressive polycystic kidney disease, and it is one reason to request deletion analysis rather than sequencing alone. Variants in TSC2 are roughly five times commoner than TSC1 variants and tend to produce a more severe phenotype, with more tubers, earlier seizures and a higher rate of intellectual disability. [1] [9]
Pathophysiology
The whole of TSC begins with the loss of a brake on cell growth. Hamartin and tuberin join to form a complex that acts as a GTPase-activating protein (GAP) for Rheb, a small G-protein. By converting active Rheb-GTP to inactive Rheb-GDP, the complex switches off mTORC1, the master regulator of cell growth, protein synthesis and proliferation. When either hamartin or tuberin is absent, Rheb stays in its active GTP-bound form, mTORC1 runs unchecked, and cells grow and proliferate without restraint. [1]
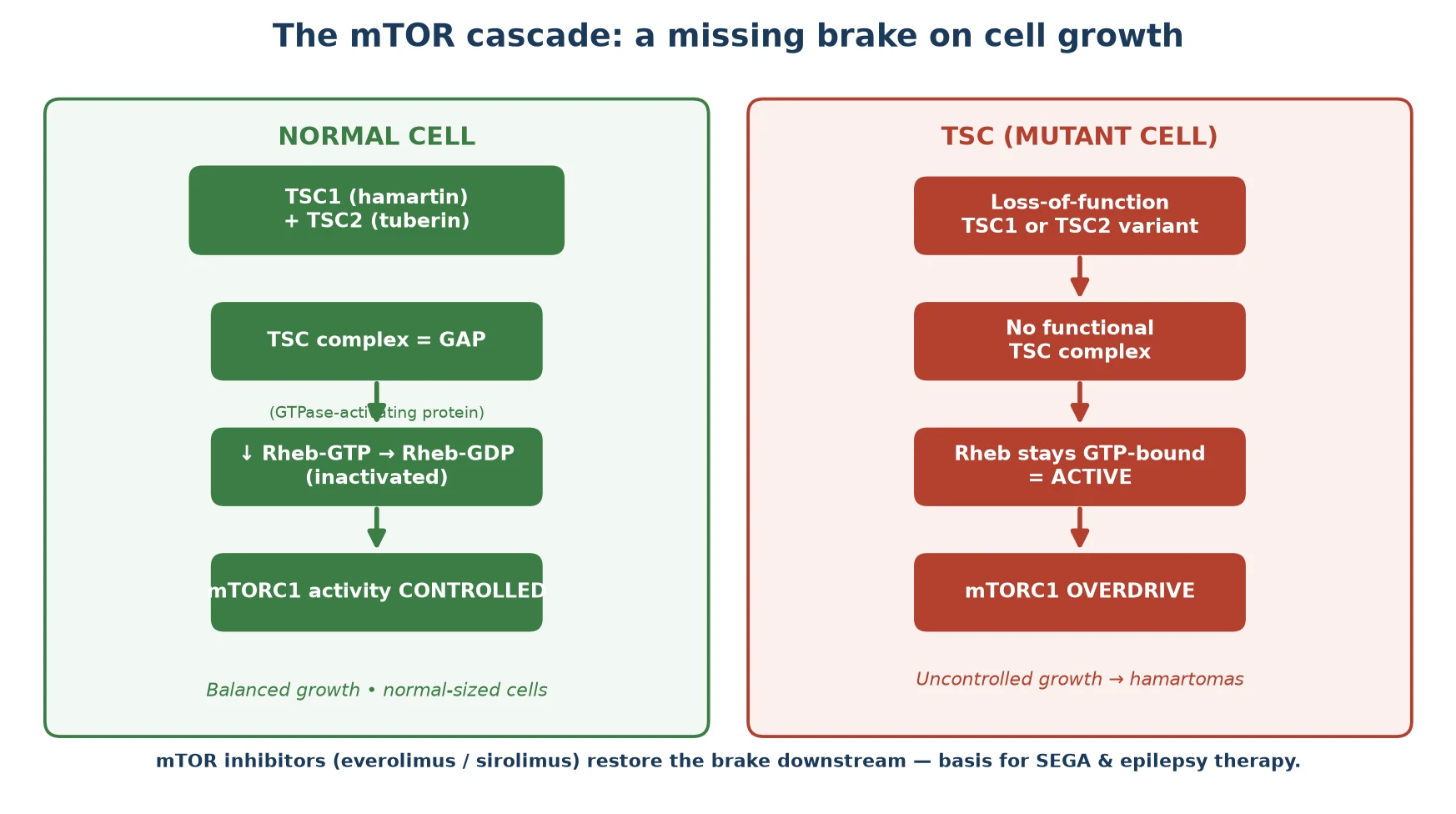
The lesions of TSC appear where this brake matters most. In the brain, abnormal neuronal migration and proliferation produce the cortical tubers that drive epilepsy, the subependymal nodules, and occasionally a subependymal giant cell astrocytoma. In the skin, mTORC1 overdrive in melanocytes and fibroblasts generates hypomelanotic macules, angiofibromas and the shagreen patch. In the kidney, smooth-muscle and fat-lineage cells form angiomyolipomas, and in the heart and lung they form rhabdomyomas and the smooth-muscle proliferation of lymphangioleiomyomatosis. [2] [1]
The mechanism is also the key to therapy, and this is the link examiners test. Because the disease removes an upstream brake, a drug that inhibits mTORC1 directly can restore control downstream, wherever the variant sits. This is why the mTOR inhibitors everolimus and sirolimus shrink subependymal giant cell astrocytomas, reduce renal angiomyolipoma volume, and improve seizure control — they cut in below the broken brake. [5] [6]
[1]Clinical Presentation
TSC presents in a recognisable age-dependent sequence that mirrors the emergence of its features. In the fetus or neonate, a cardiac rhabdomyoma is often the first clue, detected on antenatal ultrasound or presenting with a murmur, arrhythmia or cardiac compromise. In infancy, the dominant presentation is the onset of epilepsy, most characteristically infantile spasms with hypsarrhythmia on EEG (West syndrome), often alongside the first hypomelanotic macules. [2]
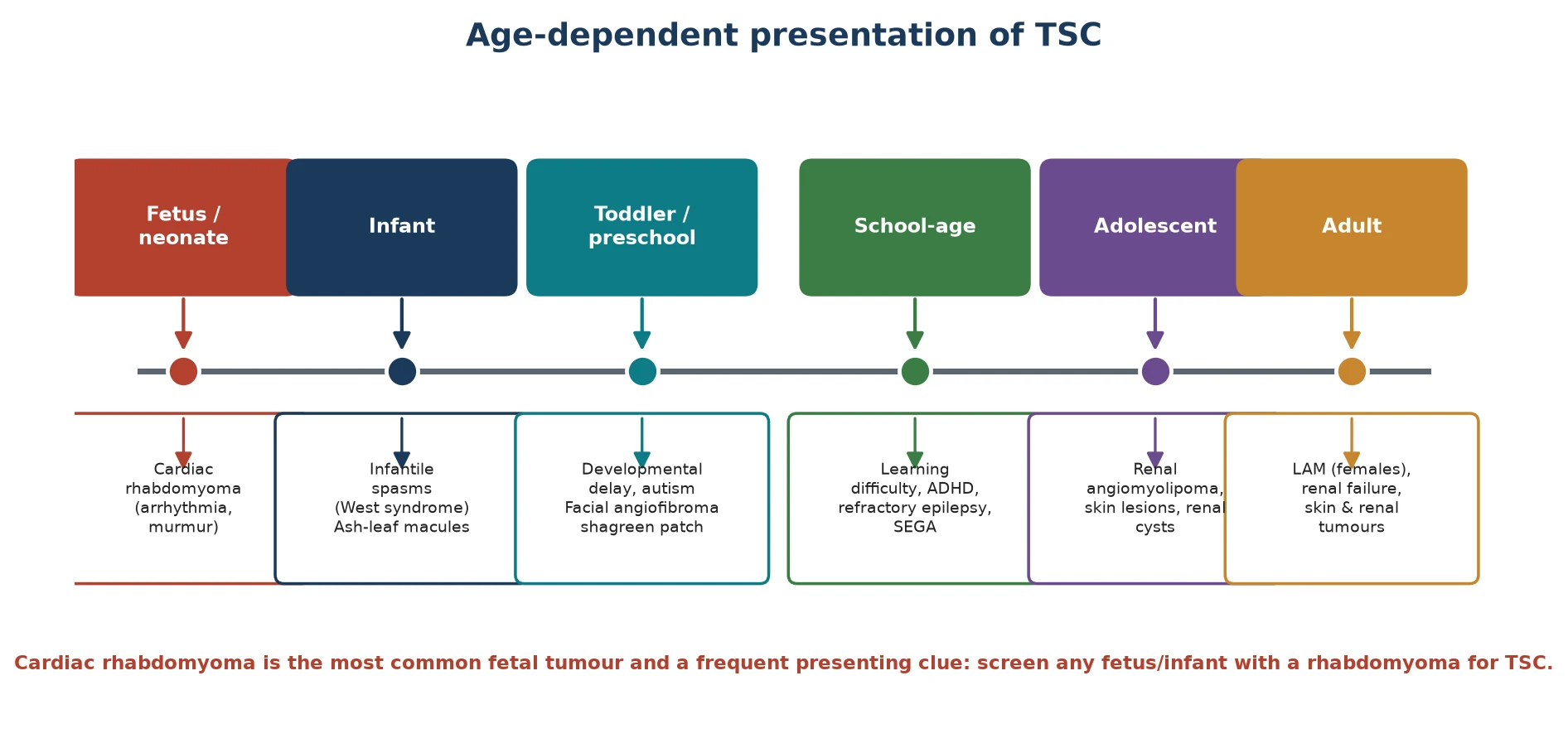
Through the preschool and school-age years, the developmental and behavioural features move to the foreground. Learning difficulty, attention-deficit/hyperactivity disorder and autism spectrum disorder are common and frequently dominate quality of life, while the skin lesions — facial angiofibromas in the butterfly distribution, the shagreen patch over the lower back, and ungual fibromas — become more prominent. A subependymal giant cell astrocytoma may declare itself in this window with growth near the foramen of Monro and the threat of obstructive hydrocephalus. [2] [8]
In adolescence and adulthood the renal and pulmonary complications come to the fore. Renal angiomyolipomas enlarge and may bleed, renal cysts accumulate, and in females lymphangioleiomyomatosis produces cystic lung destruction with dyspnoea and pneumothorax. Epilepsy, when it persists, is often refractory, and the cumulative burden of seizures, surgery and renal disease shapes long-term outcome. Recognising this sequence lets you anticipate the next complication before it arrives. [1] [4]
Differential Diagnosis
The first diagnostic fork is the child with epilepsy and skin lesions, and TSC is the leading diagnosis but not the only one. Neurofibromatosis type 1 brings cafe-au-lait macules, freckling and neurofibromas with optic glioma, driven by neurofibromin and the RAS pathway rather than mTOR, and its seizures are far less prominent. Sturge-Weber syndrome produces a port-wine stain in the trigeminal distribution with leptomeningeal angiomatosis and contralateral seizures, caused by a somatic GNAQ variant. [2]
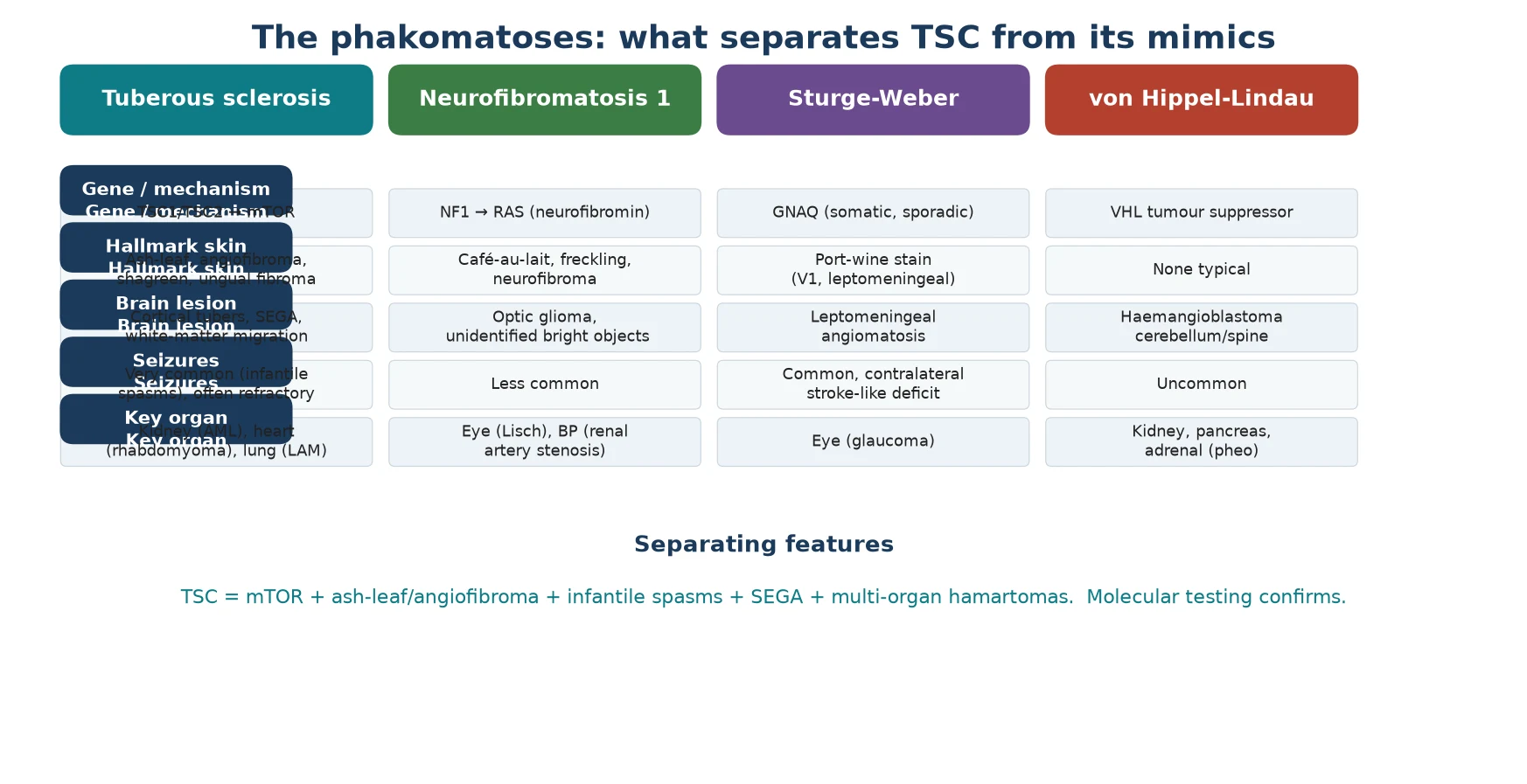
The second fork is the cause of infantile spasms itself. Alongside TSC, the differential includes hypoxic-ischaemic encephalopathy, congenital brain malformations, metabolic disorders such as pyridoxine dependency, and chromosomal abnormalities. Distinguishing TSC matters because it changes the first-line drug (vigabatrin), sets a surveillance programme, and predicts a higher rate of developmental and autistic comorbidity than other aetiologies. [2] [1]
The third fork is the cause of multiple hypomelanotic macules. TSC is the most important diagnosis, but piebaldism, vitiligo, post-inflammatory hypopigmentation and linear hypomelanosis of Ito all produce depigmented patches. Wood's lamp examination accentuates the ash-leaf macules of TSC, and the presence of additional features — angiofibromas, a shagreen patch, seizures or a family history — points toward the diagnosis. Molecular testing resolves equivocal cases. [3]
Clinical & Bedside Assessment
The bedside assessment of a child with suspected TSC is a structured skin, neurological, cardiac, developmental and family examination that converts findings into a diagnostic and surveillance plan. Examine the whole skin under a Wood's lamp, because hypomelanotic macules are more visible under ultraviolet light and may be the only cutaneous clue in a dark-skinned infant. Inspect the face for angiofibromas, the lower back for the shagreen patch, the nails for ungual fibromas, and the teeth for enamel pits. [3] [4]
The neurological and developmental assessment is where outcome is shaped. Screen for seizures and document their semiology, assess tone and motor development, and complete a structured developmental and behavioural assessment looking for learning difficulty, attention-deficit/hyperactivity disorder and autism spectrum features, because these neurocognitive comorbidities are common and dominate quality of life. A full developmental baseline at diagnosis lets you track change over time. [8] [2]
Cardiac and abdominal examination complete the bedside set. Auscultate for the murmur or signs of outflow obstruction of a rhabdomyoma in the neonate, and palpate for a ballotable or enlarged kidney from angiomyolipoma or polycystic disease. Take a three-generation pedigree that explicitly asks about seizures, skin lesions, kidney problems and learning difficulty, and examine both parents, because subtle manifestations in a parent confirm familial disease and reveal an affected family. [1] [4]
Investigations
The diagnosis of TSC is clinical in most patients, made by applying the 2012 consensus criteria. Molecular testing with TSC1 and TSC2 sequencing plus deletion analysis confirms the diagnosis when the phenotype is incomplete, in a fetus with a rhabdomyoma, in a relative of a confirmed case, and whenever the result will guide reproductive counselling. Identifying the familial variant is the gateway to prenatal and preimplantation testing, so it should be requested early rather than held in reserve. [3] [1]
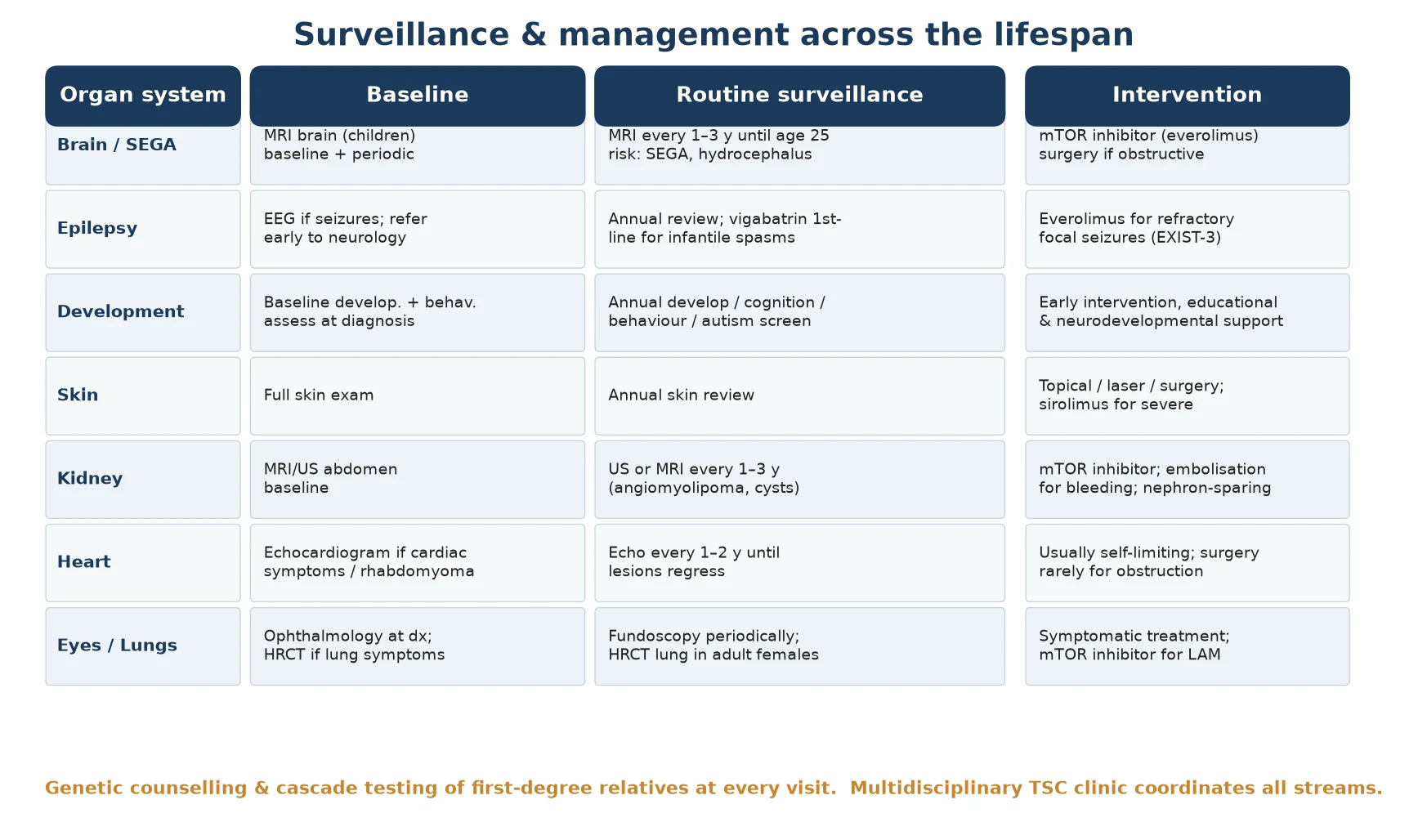
The baseline imaging set maps the multisystem burden at diagnosis. An MRI of the brain defines the cortical tubers, subependymal nodules and any subependymal giant cell astrocytoma. An echocardiogram assesses a cardiac rhabdomyoma and its haemodynamic significance. An abdominal MRI or ultrasound characterises renal angiomyolipomas and cysts, and an electroencephalogram is performed whenever seizures are suspected, looking in particular for the hypsarrhythmia of infantile spasms. [4] [2]
Surveillance investigations follow the consensus schedule and are symptom- and age-driven. Brain MRI is repeated every one to three years through childhood and adolescence to detect a growing subependymal giant cell astrocytoma, renal imaging is repeated at the same interval to monitor angiomyolipoma size, and high-resolution chest CT is offered to adult females to screen for lymphangioleiomyomatosis. During everolimus therapy, blood glucose, lipids, full blood count and renal function are monitored, because the drug affects each of these. [4] [5]
Management — Resuscitation
Acute resuscitation in TSC is uncommon, but four scenarios demand urgent action. The first is infantile spasms or status epilepticus, which requires prompt treatment with vigabatrin first-line for TSC-associated infantile spasms, because delay worsens developmental outcome. The second is obstructive hydrocephalus from a growing subependymal giant cell astrocytoma, presenting with headache, vomiting, visual change or deteriorating consciousness, which needs urgent imaging, neurosurgical decompression or shunting, and consideration of an mTOR inhibitor. [2] [4]
The third emergency is cardiac compromise in the neonate, where a large rhabdomyoma causes outflow tract obstruction or arrhythmia and needs cardiology support, anti-arrhythmic or inotropic management, and occasionally surgery. The fourth is a ruptured or bleeding renal angiomyolipoma, presenting with flank pain and haemodynamic instability, which needs fluid resuscitation, blood transfusion, urgent embolisation by interventional radiology, and nephrology or surgical input. Recognising each of these and acting quickly prevents irreversible harm. [1] [4]
Management — Definitive & Stepwise
Definitive management of TSC is a coordinated surveillance programme that a fellowship candidate can recite as a five-step framework: confirm the diagnosis, assemble a multidisciplinary baseline assessment, run scheduled surveillance, treat complications as they arise, and cascade-test the family with genetic counselling. No step cures TSC, but each prevents disability and detects life-threatening change early. The backbone is a multidisciplinary TSC clinic coordinating neurology, nephrology, dermatology, cardiology, genetics and psychology. [4] [1]
The transformative development of the past decade is targeted medical therapy with the mTOR inhibitor everolimus. In the EXIST-1 trial, everolimus reduced the volume of subependymal giant cell astrocytomas in the majority of patients, offering a medical alternative to neurosurgery for growing or symptomatic lesions. In the EXIST-3 trial, adjunctive everolimus reduced seizure frequency in patients with refractory focal-onset epilepsy, with a post-hoc paediatric analysis confirming benefit in children and adolescents. Everolimus is now a first-line systemic option alongside surgery for SEGA, and an adjunct for drug-resistant epilepsy. [5] [6]
Epilepsy management follows a stepwise ladder. Vigabatrin is first-line for TSC-associated infantile spasms, standard anti-seizure medicines are used for other seizure types, and epilepsy surgery is considered early for drug-resistant focal epilepsy because TSC is a surgically amenable aetiology. Everolimus is added as adjunctive therapy when seizures remain refractory. Skin lesions are managed with topical agents, laser, surgical excision or topical sirolimus, and renal angiomyolipoma is managed with an mTOR inhibitor, prophylactic embolisation when large, and nephron-sparing surgery when required. [6] [4]
| Domain | Surveillance | Intervention |
|---|---|---|
| Brain / SEGA | MRI every 1–3 y until age 25 | Everolimus; surgery if obstructive |
| Epilepsy | Annual review; EEG if seizures | Vigabatrin (spasms); ASMs; surgery; everolimus |
| Development | Annual cognition / behaviour / autism | Early intervention; educational support |
| Kidney | US or MRI every 1–3 y | mTOR inhibitor; embolisation; nephron-sparing surgery |
| Skin | Annual skin review | Topical sirolimus; laser; excision |
| Heart / Lung | Echo if cardiac; HRCT in adult females | Supportive; mTOR inhibitor for LAM |
Specific Subtypes & Scenarios
The fetus or neonate with a cardiac rhabdomyoma is the earliest presentation of TSC and the one most often detected incidentally. Cardiac rhabdomyoma is the most common cardiac tumour of infancy, and when one is found — whether on antenatal ultrasound or after a murmur or arrhythmia — the child should be screened for TSC with a skin examination, family history and ultimately molecular testing. Most rhabdomyomas regress spontaneously over the first years of life, so the management is surveillance rather than surgery unless there is haemodynamic compromise. [1] [4]
The infant with TSC-associated infantile spasms is the scenario where early treatment most changes outcome. Vigabatrin is the first-line drug, started promptly once the diagnosis of spasms is confirmed, with serial EEG to assess response. Developmental surveillance begins immediately, and there is active interest in pre-emptive vigabatrin for infants with TSC and epileptiform EEG changes before clinical spasms emerge. The caveat is vigabatrin's risk of irreversible visual-field loss, which warrants ophthalmic surveillance, though the seizure-control benefit in TSC usually justifies use. [2] [7]
The child with a growing subependymal giant cell astrocytoma is managed jointly by neurology, neurosurgery and oncology. Serial MRI defines growth and any encroachment on the foramen of Monro, and everolimus is the first-line systemic therapy for a growing lesion, reserving surgery for obstructive hydrocephalus or non-response. Because everolimus shrinks rather than cures, therapy is typically continued long-term, with monitoring of its metabolic and haematological effects. [5] [10]
The adolescent or adult female at risk of lymphangioleiomyomatosis needs targeted lung surveillance. High-resolution chest CT detects the cystic changes of lymphangioleiomyomatosis, which progresses insidiously to dyspnoea, pneumothorax and respiratory failure, and an mTOR inhibitor slows decline in symptomatic disease. Counselling on the risks of oestrogen-containing therapies and pregnancy is part of the transition to adult care. [1] [4]
Complications & Pitfalls
The complications of TSC divide into the clinical comorbidities that accumulate over a lifetime and the cognitive traps that cost marks. The clinical comorbidities are refractory epilepsy, intellectual disability and autism spectrum disorder, obstructive hydrocephalus from a subependymal giant cell astrocytoma, renal haemorrhage or failure from angiomyolipoma, cardiac outflow obstruction, and respiratory failure from lymphangioleiomyomatosis. Each is treatable in its own right, and proactive surveillance prevents the secondary disability that neglect produces. [1] [8]
The most modifiable determinant of outcome is seizure control. Early-onset, refractory epilepsy and infantile spasms predict poorer cognition and a higher rate of autism, which is why prompt vigabatrin for spasms and early consideration of epilepsy surgery matter so much. The neurocognitive comorbidities — learning difficulty, attention-deficit/hyperactivity disorder and autism — respond to educational and behavioural support more than to any medical therapy, and early intervention in the preschool years measurably improves function and self-esteem. [8] [2]
The treatment itself carries complications that a candidate must name. Everolimus causes stomatitis, increased susceptibility to infection, hyperglycaemia, hyperlipidaemia and cytopenias, and it requires regular monitoring of glucose, lipids, full blood count and renal function. Vigabatrin carries a risk of irreversible bilateral visual-field constriction that requires ophthalmic surveillance. Failing to mention these adverse effects is a classic examination error, because safe long-term therapy depends on anticipating and monitoring them. [5] [6]
Prognosis & Disposition
Life expectancy in TSC is near-normal in well-surveilled patients but is reduced when epilepsy, renal disease, subependymal giant cell astrocytoma complications or lymphangioleiomyomatosis are severe or untreated. The largest influence on quality of life is not the tumours but the neurocognitive and behavioural comorbidity, which is why developmental surveillance and early intervention deserve as much weight as imaging. Most children with good surveillance and management live into adulthood with a meaningful quality of life. [1] [8]
Neurodevelopmental outcome is shaped by seizure control and tuber burden. Children whose epilepsy is controlled early, and whose infantile spasms are treated promptly, tend to have better cognition and a lower rate of autism than those with refractory seizures. This is the reason vigabatrin-first therapy and early epilepsy surgery are emphasised — they are neuroprotective, not merely anti-convulsant. [2] [7]
For every patient, disposition is to a specialist multidisciplinary TSC clinic that can coordinate neurology, nephrology, dermatology, cardiology, genetics, ophthalmology and psychology. Transition to adult care is a structured process that should begin in early adolescence, because the fragmented handover of complex chronic disease is a well-recognised cause of harm, and the renal and pulmonary complications of adulthood need a prepared adult team ready to receive the patient. [4] [1]
Special Populations
The same TSC diagnosis behaves differently across populations because access and recognition are unevenly distributed. In remote and Indigenous communities, later presentation, limited genetic-service access and lower rates of cascade testing mean that affected children and their relatives are diagnosed late, tumours are larger at detection, and culturally safe genetic counselling is essential to rebuild trust and engagement. In migrant, refugee and asylum-seeking families, language barriers, incomplete family histories and interrupted care make the multigenerational assessment that TSC demands especially hard. [1]
In Australia and New Zealand, specialist TSC clinics and clinical genetics services are concentrated in major centres, and children in rural and remote areas rely on visiting outreach or telehealth for surveillance. For Aboriginal and Torres Strait Islander and Maori whanau, engaging an Indigenous health worker and offering culturally appropriate genetic counselling improves uptake of cascade testing and long-term engagement. Families should be connected to the Tuberous Sclerosis Associations of Australia and New Zealand for peer and practical support, and access to everolimus is coordinated through specialist drug programmes.
Children with intellectual disability and autism make up a large share of the TSC population, because these neurocognitive features are core to the condition, and their management needs an education plan, behavioural support and a strengths-based, neurodiversity-affirming frame. The adolescent female approaching the age of lymphangioleiomyomatosis risk needs targeted lung surveillance and counselling on oestrogen-containing therapy and pregnancy. Across all groups, a named care coordinator maintains continuity and prevents the surveillance gaps that harm patients with complex chronic disease. [8] [4]
Evidence, Guidelines & Regional Differences
The evidence base rests on three pillars: the consensus diagnostic criteria, the surveillance and management recommendations, and the randomised trials of everolimus. The 2012 International Tuberous Sclerosis Complex Consensus criteria, published by Northrup and Krueger in 2013, are the current reference standard for diagnosis and for organ-by-organ surveillance, and they remain the framework against which care is planned. The Henske 2016 disease primer and the Curatolo 2008 Lancet review set the comprehensive clinical and scientific background. [3] [4]
The therapeutic evidence is dominated by two randomised trials. The EXIST-1 trial showed that everolimus reduces subependymal giant cell astrocytoma volume and made a medical therapy available for a lesion previously managed only by surgery. The EXIST-3 trial showed that adjunctive everolimus reduces seizure frequency in refractory focal-onset epilepsy, with a paediatric post-hoc analysis confirming the benefit in children and adolescents. Together these trials established mTOR inhibition as a cornerstone of modern TSC care. [5] [6]
Regional differences are mainly in service delivery rather than biology. Australia and New Zealand follow RACP and RCPCH-aligned pathways through specialist TSC clinics, North American practice is shaped by the consensus guidelines and specialist centres, and many low- and middle-income settings adapt the same surveillance principles with more limited access to molecular testing and everolimus. An ongoing controversy is the role of routine pre-emptive vigabatrin versus surveillance EEG in infants with TSC, an area where evidence is still maturing. [1] [7]
Exam Pearls
A fellowship candidate answering on tuberous sclerosis complex should land these anchors and avoid these traps, then close with the family. The strongest answer names the gene and pathway, applies the diagnostic criteria precisely, sets out a surveillance plan tailored to the child's age and organ involvement, identifies the red flags for hydrocephalus and bleeding, and frames the whole discussion around autosomal-dominant inheritance and cascade testing. Holding the mTOR cascade in mind ties the skin, brain, kidney, heart and lung features to a single mechanism and to the drug that restores control. [1] [2]
References
- [1]Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers, 2016.PMID 27226234
- [2]Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet, 2008.PMID 18722871
- [3]Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Group. Pediatr Neurol, 2013.PMID 24053982
- [4]Krueger DA, Northrup H; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Group. Pediatr Neurol, 2013.PMID 24053983
- [5]Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, et al. Everolimus for subependymal giant cell astrocytoma in patients with tuberous sclerosis complex: 2-year open-label extension of the randomised EXIST-1 study. Lancet Oncol, 2014.PMID 25456370
- [6]French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis complex (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet, 2016.PMID 27613521
- [7]Curatolo P, Nabbout R, Lagae L, Aronica E, Ferreira JC, Feucht M, et al. Adjunctive everolimus for children and adolescents with treatment-refractory seizures associated with tuberous sclerosis complex: post-hoc analysis of the phase 3 EXIST-3 trial. Lancet Child Adolesc Health, 2018.PMID 30169322
- [8]de Vries P, Humphrey A, McCartney D, Prather P, Bolton P, Hunt A, TSC Behaviour Consensus Panel. Consensus clinical guidelines for the assessment of cognitive and behavioural problems in Tuberous Sclerosis. Eur Child Adolesc Psychiatry, 2005.PMID 15981129
- [9]Overwater IE, Rietman AB, Bindels-de Heus K, Looman CW, Rizopoulos D, Sibindi TM, et al. Genotype and brain pathology phenotype in children with tuberous sclerosis complex. Eur J Hum Genet, 2016.PMID 27406250
- [10]Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med, 2010.PMID 21047224