Paeds · genetics-dysmorphology-and-metabolism
Turner syndrome
Also known as 45,X syndrome · Monosomy X · Ullrich-Turner syndrome · Turner's syndrome · Bonnevie-Ullrich syndrome
A fellowship approach to Turner syndrome: recognise the complete or partial loss of one X chromosome as the most common sex chromosome aneuploidy in females, confirm the diagnosis with a karyotype, and build lifelong multidisciplinary surveillance around short stature, gonadal dysgenesis, cardiovascular risk, and the neurocognitive profile — because early growth-hormone therapy, timed oestrogen replacement, and aortic imaging change both the trajectory and the survival of the girl you are looking after.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who thinks in three layers at once. The first layer is the girl in front of you: her height velocity, her pubertal staging, her cardiovascular anatomy, her renal ultrasound, her hearing, her thyroid function, and her school performance and mood. The second is the chromosomal mechanism: the karyotype class that determines the gonadoblastoma risk, the mosaicism that softens or hardens the phenotype, and the haploinsufficiency of SHOX and the lymphogenic genes that build the body. The third is the lifespan: the transition from paediatric to adult care, the fertility window that closes early, the aorta that dilates silently, and the osteoporosis that is preventable if oestrogen is given on time. [1] [8]
Overview & Definition
Turner syndrome is a chromosomal disorder of phenotypic females caused by the complete or partial absence of one X chromosome, with or without mosaicism. The classic karyotype, 45,X (monosomy X), accounts for roughly half of cases; the remainder are mosaics (45,X/46,XX being the most common) or harbour a structurally abnormal X chromosome — an isochromosome of the long arm, a ring X, or a partial deletion. The clinical consequence is a syndrome of short stature, gonadal dysgenesis, cardiovascular and renal anomalies, a characteristic lymphatic phenotype, and a distinctive but variable neurocognitive profile. [2] [1]
Two biological facts anchor the entire topic and recur in every examination. First, Turner syndrome is far more common at conception than at birth: an estimated 99 percent of 45,X conceptuses are lost spontaneously, and Turner syndrome accounts for approximately 10 percent of all first-trimester miscarriages. The live-birth prevalence is therefore about one in 2,500 females — high enough to be common, low enough that the diagnosis is missed when the phenotype is mild. Second, the phenotype is driven by haploinsufficiency of genes that escape X-inactivation and remain active on the single X chromosome, and the most important of these for stature and skeletal patterning is SHOX (short-stature homeobox-containing gene) in the pseudoautosomal region of Xp. [2] [1]
Classification
The classification that matters clinically is karyotypic, because the karyotype determines the gonadal malignancy risk, the severity of the phenotype, and the reproductive counselling. Five karyotype classes run from the classic monosomy through structural variants, and the boundary between 45,X/46,XY mosaicism and the rest is where gonadoblastoma surveillance changes from watchful waiting to prophylactic surgery. [2] [1]
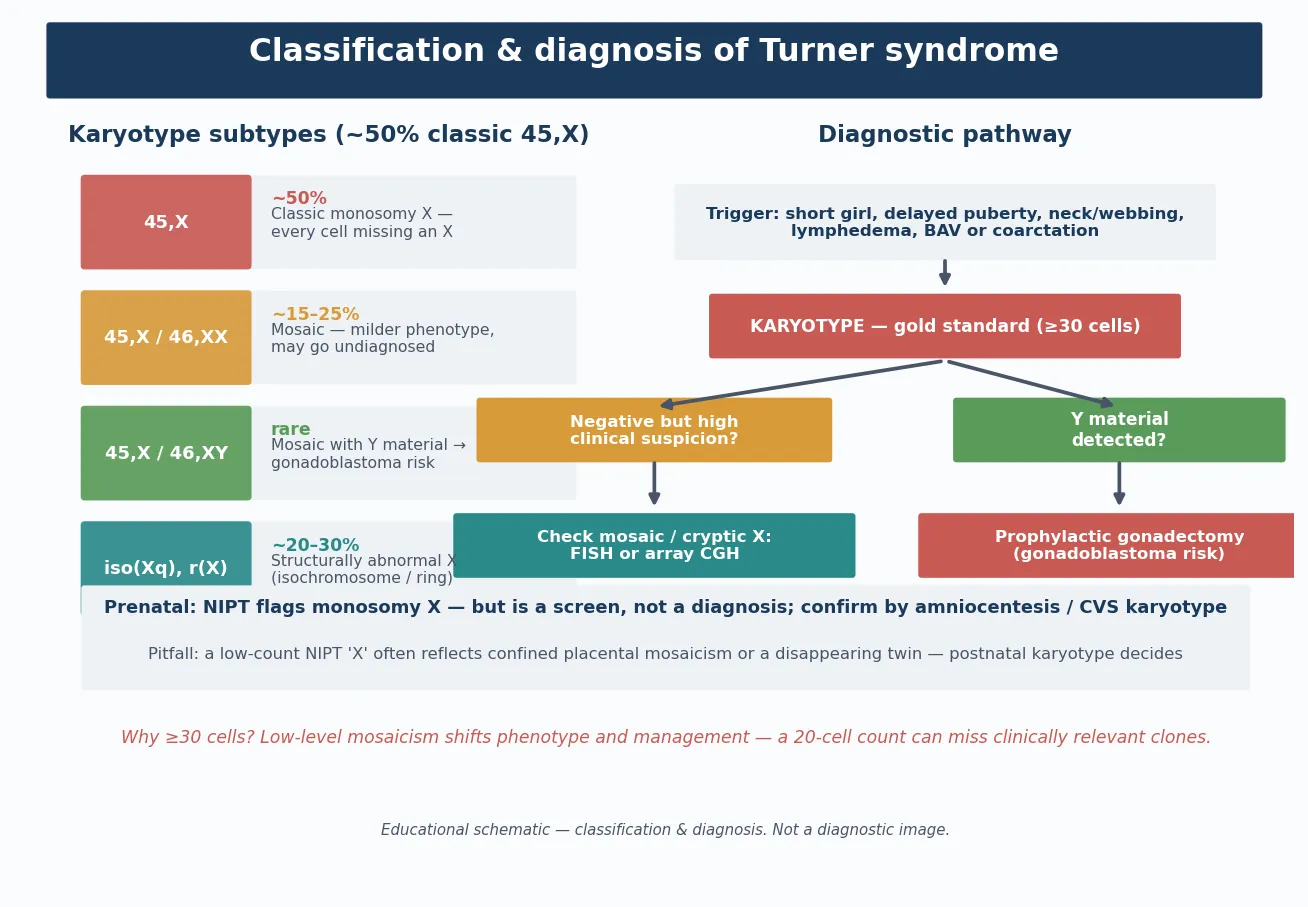
Why the structural variants matter
The structural X abnormalities each carry their own nuance. An isochromosome of the long arm, i(Xq), is functionally a monosomy of the short arm and a trisomy of the long arm; it behaves phenotypically like classic 45,X and is one of the commonest structural variants. A ring X chromosome forms when both ends of the X are lost and the ends join; if the ring is small and lacks the XIST gene, the cell cannot inactivate it, and the resulting overexpression of X-linked genes produces a more severe, sometimes syndromic phenotype. A partial deletion of Xp produces the short stature and skeletal features if SHOX is lost, while a deletion of Xq tends to produce gonadal failure. [2] [1]
Epidemiology & Risk Factors
Turner syndrome is the most common sex chromosome aneuploidy in live-born females, with a prevalence of approximately one in 2,500 female births. It is not inherited in the Mendelian sense and shows no clear racial or ethnic predilection, but it is not rare: at any given time a general paediatrician with a hospital cohort will have several girls under care. The risk is not related to maternal age, which distinguishes it from autosomal trisomies such as Down syndrome — a distinction examiners probe because candidates often conflate the two mechanisms. [2] [1]
The most powerful epidemiological fact is the prenatal lethality of the full 45,X complement. Only about one percent of 45,X conceptuses survive to live birth, which means that most Turner syndrome seen in practice is milder than the textbook image, and the live-born girl frequently has a mosaic or structural karyotype that has rescued some of the phenotype. This survival selection explains why the clinical presentation is so variable and why the diagnosis is still missed in the school-age girl who simply has short stature. [2]
Pathophysiology
The molecular story begins with the genes that escape X-inactivation. In a typical female, one X chromosome is transcriptionally silenced in each cell by XIST-driven lyonisation, but a subset of genes in the pseudoautosomal regions and along the length of the X escape inactivation and remain active on both copies. When one X is wholly or partially lost, these escape genes become haploinsufficient, and it is their reduced dosage that builds the Turner phenotype. [2] [1]
The single most important haploinsufficient gene is SHOX, the short-stature homeobox gene in the pseudoautosomal region of Xp (and Yp). SHOX is expressed in the growth plate and governs chondrocyte proliferation and differentiation, so its loss explains the short stature, the cubitus valgus, the high-arched palate, the short fourth metacarpals, the Madelung deformity, and the characteristic skeletal proportions. Point mutations and deletions of SHOX in otherwise chromosomally normal girls produce Leri-Weill dyschondrosteosis, proving that SHOX haploinsufficiency is the dominant stature gene in Turner syndrome. [2]
The lymphatic phenotype — neck webbing, peripheral lymphoedema, cystic hygroma, low posterior hairline, and the nail and lymphatic anomalies — arises from haploinsufficiency of lymphogenic genes on Xp that impair lymphangiogenesis. The resultant lymph stasis distends the jugular lymph sacs in the fetus (the cystic hygroma that may be detected at the 12-week scan), stretches the neck skin into webbing, and leaves residual peripheral lymphoedema in infancy that usually, but not always, resolves. [2] [6]
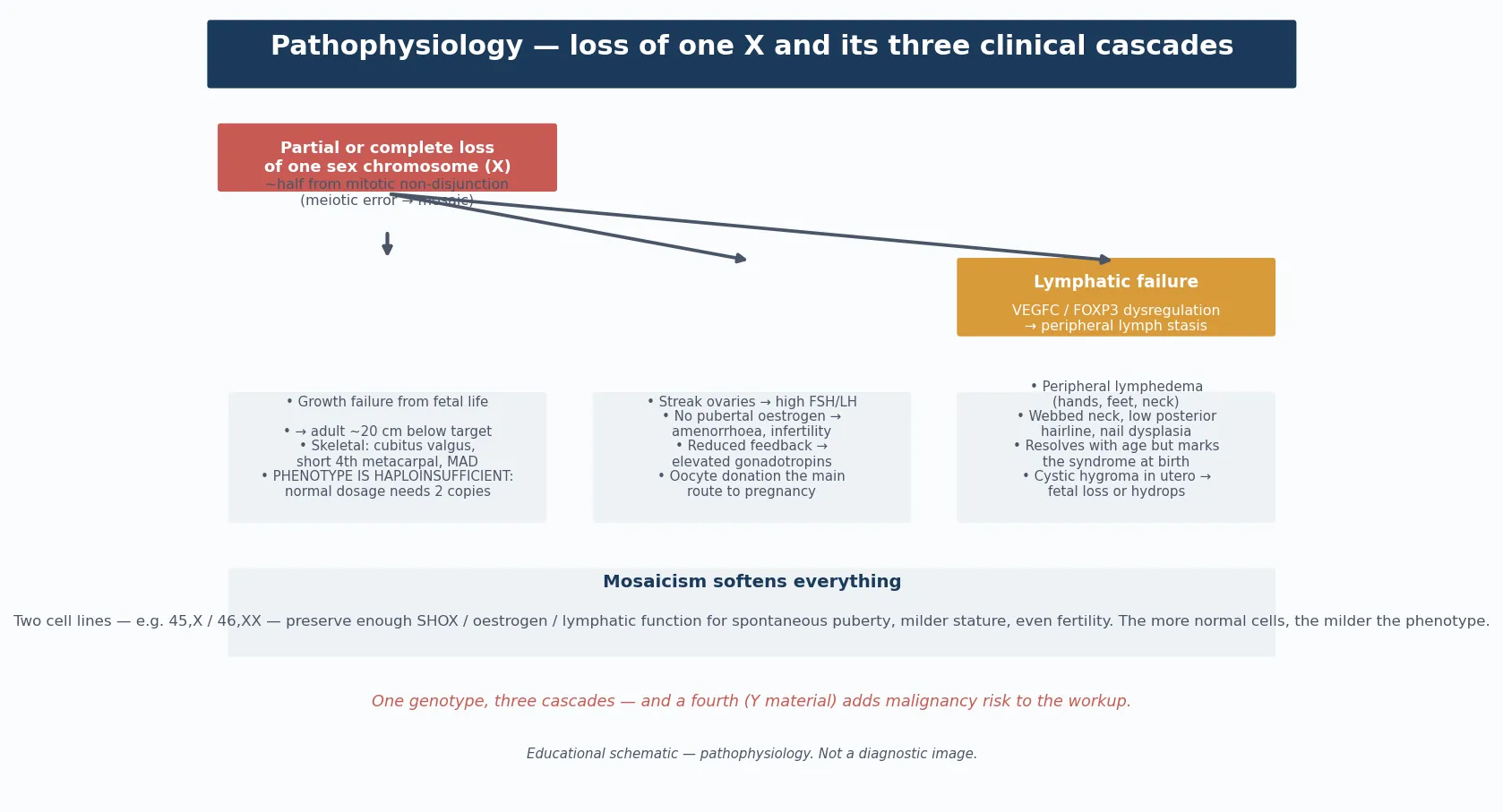
The gonadal dysgenesis arises because the genes governing ovarian differentiation and survival on the second X are absent, so the primordial follicles that are present at mid-gestation undergo accelerated atresia. By birth or early childhood the ovaries are fibrous streaks devoid of follicles, and the resulting oestrogen deficiency produces the failure of puberty, the primary amenorrhoea, the osteoporosis risk, and the infertility. The neurocognitive profile — broadly normal intelligence with relative weakness in visuospatial processing, mathematics, working memory, and social cognition — is attributed to haploinsufficiency of X-linked neural genes and is the basis for the targeted educational support that these girls need. [2] [8]
Clinical Presentation
The presentation is age-dependent, and the same syndrome looks different at every stage of childhood. In fetal life, Turner syndrome presents as increased nuchal translucency, a cystic hygroma, hydrops fetalis, or a cardiac anomaly detected on the 18-to-20-week anatomy scan; coarctation of the aorta is the classic associated finding. Many of these pregnancies miscarry, but those that survive may present in the newborn period. [2] [1]
In the newborn and young infant, the presentation is lymphatic: dorsal hand and foot lymphoedema (puffiness that is often the first sign), a low posterior hairline, neck webbing (pterygium colli), and small, hyperconvex or deep-set nails. A left-sided cardiac lesion such as coarctation of the aorta may bring the infant to medical attention through the duct-dependent or antenatally detected pathway. These infants are often diagnosed early because the phenotype is unmistakable, and the karyotype is sent as part of the dysmorphic work-up. [2]
In childhood, the presentation is growth: short stature, a downward-crossing growth trajectory, and the skeletal stigmata of SHOX haploinsufficiency (cubitus valgus, short fourth metacarpals, high-arched palate, Madelung deformity). The girl is otherwise well, and the absence of a striking dysmorphic gestalt is the reason the diagnosis is often delayed until mid-childhood. Recurrent otitis media with conductive hearing loss is a common and early accompaniment. [2] [1]
In adolescence, the presentation is pubertal: absent or stalled breast development, primary amenorrhoea, and the psychological burden of watching peers mature. Short stature is usually present by this stage but may have been attributed to familial or constitutional factors. The finding of elevated gonadotrophins (hypergonadotrophic hypogonadism) on the pubertal-delay work-up is the biochemical clue that triggers the karyotype. [2] [7]
Differential Diagnosis
The differential depends on the presenting problem, and Turner syndrome sits at one node within each of four differential lists. The first is the differential of short stature in a girl: familial short stature, constitutional delay of growth and puberty, growth hormone deficiency, hypothyroidism, coeliac disease, and chronic systemic disease. Turner syndrome is distinguished by the combination of short stature with skeletal stigmata, delayed puberty, and (eventually) hypergonadotrophic hypogonadism, and the karyotype settles the question. [2] [1]
The second is the differential of delayed puberty and primary amenorrhoea: constitutional delay, hypothalamic and pituitary causes of hypogonadotrophic hypogonadism (functional hypothalamic amenorrhoea, Kallmann syndrome, pituitary tumour), other causes of hypergonadotrophic hypogonadism (premature ovarian insufficiency, galactosaemia, autoimmune oophoritis), and anatomic causes (Mullerian agenesis, androgen insensitivity syndrome). Mullerian agenesis (Mayer-Rokitansky-Kuster-Hauser syndrome) produces primary amenorrhoea with normal stature, normal secondary sexual characteristics, and normal hormone profile — a clean contrast with Turner syndrome. Androgen insensitivity syndrome produces primary amenorrhoea with breast development but absent or sparse pubic hair and a blind-ending vagina, and the karyotype is 46,XY. [2]
The third is the differential of the dysmorphic or syndromic girl with short stature: Noonan syndrome (an autosomal dominant RASopathy that phenotypically mimics Turner syndrome with short stature, neck webbing, and a cardiac lesion — classically pulmonary valve stenosis rather than left-sided — but has a normal 46,XX or 46,XY karyotype and normal ovaries), and the skeletal dysplasias such as Leri-Weill dyschondrosteosis (SHOX mutation without the full Turner phenotype). Noonan syndrome is the single most tested differential because it is the phenocopy that is karyotypically normal. [2]
The fourth is the differential of cystic hygroma and fetal hydrops: Turner syndrome is the leading single cause of a first-trimester cystic hygroma, but the differential includes trisomies 21, 18, and 13, Noonan syndrome and related RASopathies, fetal anaemia, and congenital infection. The cell-free DNA (non-invasive prenatal testing) result that flags a sex chromosome aneuploidy is the modern route to the antenatal diagnosis, but it is a screen and requires confirmatory amniocentesis or postnatal karyotype. [1]
[2]Clinical & Bedside Assessment
The bedside assessment begins with a structured history and examination that anticipates the multisystem involvement, then converts the findings into a karyotype and a surveillance plan. Take a growth history that plots every available measurement on a Turner-specific growth chart, because the trajectory — not the single point — reveals the downward crossing that signals the diagnosis. Ask about pubertal staging and menstrual history in the adolescent, school performance and any learning or visuospatial difficulty, recurrent ear infections and hearing, thyroid symptoms, and the family history of short stature, chromosomal disorders, or early menopause. [1] [2]
The examination is head-to-toe and looks for the stigmata of haploinsufficiency: the facies (low posterior hairline, neck webbing, ptosis, low-set or malformed ears, high-arched palate, micrognathia), the chest (shield chest, widely spaced nipples), the limbs (cubitus valgus, short fourth metacarpals, Madelung deformity, lymphoedema, nail dysplasia), the cardiovascular system (four-limb blood pressures to detect a coarctation gradient, murmur of bicuspid valve or coarctation, radio-femoral delay), the abdomen and renal systems, and the pubertal staging (Tanner stage of breast and pubic hair development, which will be delayed or absent). Measure the height, weight, and head circumference precisely, and calculate the predicted adult height if possible. [2] [1]
Investigations
The diagnostic investigation is the peripheral blood karyotype (standard G-banded chromosome analysis of at least 20 to 30 peripheral lymphocyte metaphases). The karyotype confirms the diagnosis, defines the class (45,X, mosaic, or structural), and — critically — identifies any Y-chromosomal material, which changes the gonadoblastoma surveillance. A rapid fluorescent in-situ hybridisation (FISH) or cell-free DNA screen may have flagged the diagnosis antenatally or in the newborn, but these are screens: the diagnosis is not secure without a confirmatory karyotype, because cell-free DNA can give a false positive from a confined placental mosaicism or a vanished twin. [1] [2]
Once the karyotype is confirmed, the baseline investigations assemble the surveillance map. A cardiac assessment with echocardiography and, in most centres, cardiac magnetic resonance imaging defines the aortic and valvular anatomy — bicuspid aortic valve, coarctation, and aortic dilation — because these are the lesions that shorten life. A renal ultrasound defines the anatomy and screens for the horseshoe kidney, duplicated collecting system, and positional anomalies that occur in roughly a third. Audiology (newborn hearing screen and ongoing assessment) screens for the conductive and sensorineural loss that accumulate. Thyroid function tests (TSH and free T4) and thyroid antibodies screen for the autoimmune hypothyroidism that is markedly increased in Turner syndrome. A bone age and pelvic ultrasound (to assess the uterus and streak ovaries) complete the baseline, alongside a fasting glucose, lipids, and liver function to capture the metabolic risk. [1] [8]
The hormonal work-up in the adolescent or symptomatic girl includes follicle-stimulating hormone (FSH) and luteinising hormone (LH), which are elevated (hypergonadotrophic hypogonadism) in classic gonadal dysgenesis, and oestradiol, which is low. Anti-Mullerian hormone (AMH) is typically undetectable or very low, reflecting the follicle depletion. These are not needed to make the diagnosis if the karyotype is positive, but they define the gonadal axis and the need for oestrogen replacement. [2] [7]
Management — Resuscitation
Resuscitation in Turner syndrome is rarely about an acute collapse at diagnosis, but two scenarios demand urgent action. The first is the duct-dependent coarctation or critical left-heart lesion in the newborn, managed with prostaglandin E1 to maintain ductal patency, prostaglandin-related apnoea precautions, and timely surgical or catheter-based repair. The second, and the one that defines the lifelong surveillance, is aortic dissection, which can occur at any age — even in childhood and young adulthood — and is the leading preventable cause of premature death. A girl with Turner syndrome presenting with acute chest or back pain, a blood pressure differential, or syncope has an aortic dissection until proven otherwise, and the pathway is immediate cross-sectional imaging (CT angiography or MRI), blood pressure control, and cardiothoracic surgical involvement. [1] [6]
Management — Definitive & Stepwise
Definitive management is a lifelong, multi-domain framework that a fellowship candidate can recite and a general paediatrician can coordinate: confirm the karyotype, initiate growth-hormone therapy, induce puberty with timed oestrogen, sustain cardiovascular surveillance, and run the psychosocial and reproductive support. No single intervention cures Turner syndrome, but each changes the trajectory and, in the case of the aortic surveillance, the survival. [1] [3]
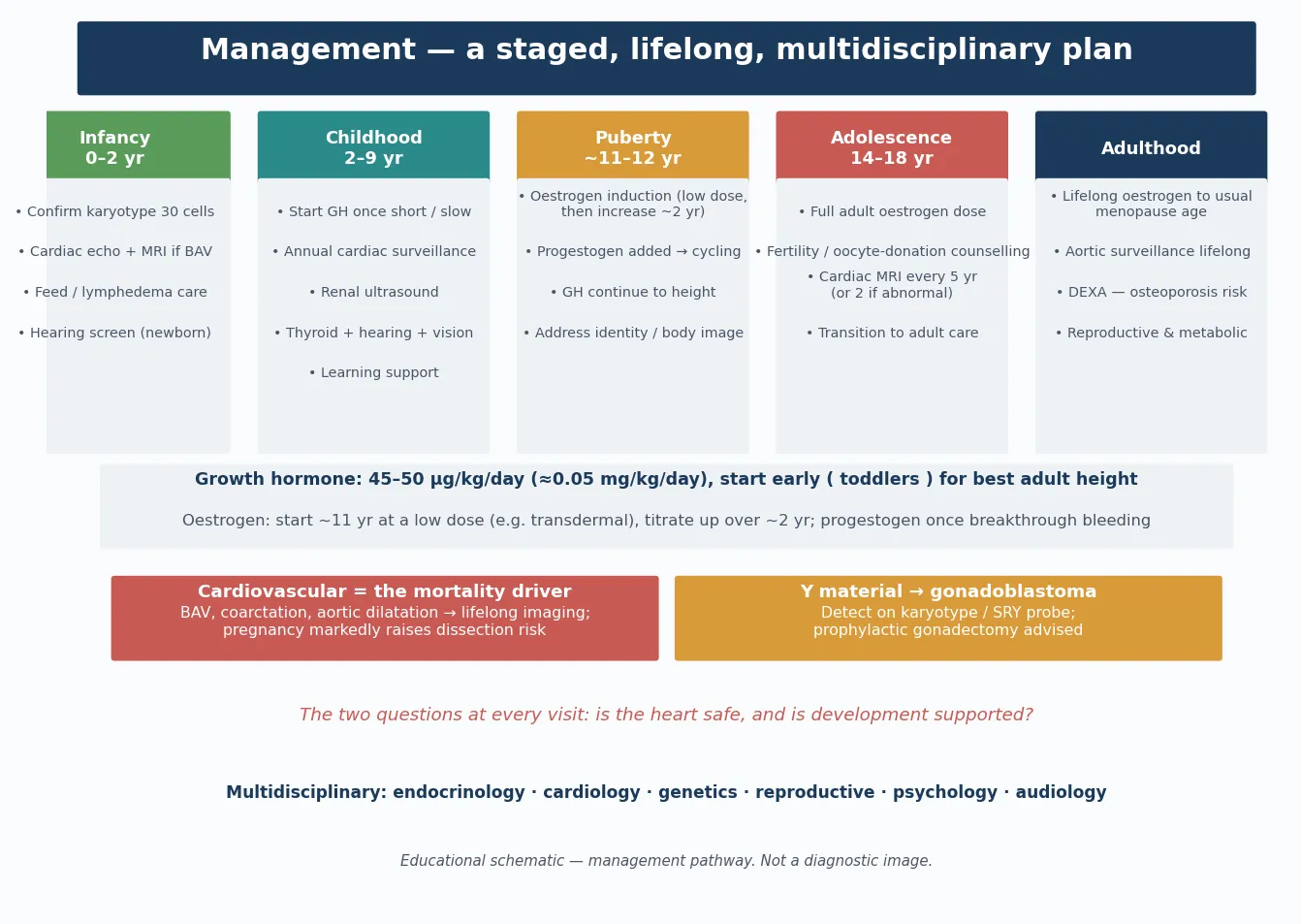
Growth-hormone therapy
Recombinant human growth hormone is the standard of care for short stature in Turner syndrome and is approved for this indication regardless of growth-hormone deficiency status. The Canadian randomised controlled trial demonstrated a gain of approximately 5 to 7 centimetres in final adult height, and therapy is best started early — as soon as the diagnosis is made and growth deviates from the normal trajectory, often by four to six years of age — and continued until near-final height. Adjunctive oxandrolone, a weak anabolic androgen, is added in some protocols after the age of about nine to ten years to further augment height, though it requires monitoring for virilisation and hepatic effects. [3] [5]
Puberty induction and hormone replacement
Oestrogen replacement is essential for the induction of puberty, the development of secondary sexual characteristics, the achievement of adequate uterine size for future fertility options, and the accrual of bone mineral density. The timing is a balance: started too early, it hastens epiphyseal fusion and compromises final height; started too late, it delays puberty beyond the peer group and compromises bone health. The current consensus is to begin low-dose oestrogen at approximately 11 to 12 years of age, titrate gradually over two to three years, and add a progestogen once breakthrough bleeding occurs or after about two years of oestrogen to produce cycling and protect the endometrium. The randomised trial by Cleemann and colleagues demonstrated that higher oestradiol dosing achieves better uterine growth, which matters for future oocyte-donation pregnancy. [1] [7]
Cardiovascular surveillance
The cardiovascular surveillance is the most safety-critical domain. Every girl and woman with Turner syndrome needs a baseline echocardiogram and cardiac MRI to define the aortic and valvular anatomy, and ongoing surveillance imaging — the frequency determined by the aortic size, the growth rate, and the presence of risk factors such as bicuspid aortic valve, coarctation, and hypertension. Prophylactic aortic surgery is recommended at lower aortic dimensions than in the general population, because the Turner aorta is intrinsically abnormal and dissects at smaller sizes. Hypertension must be aggressively treated. Pregnancy — whether spontaneous or assisted — substantially increases the aortic dissection risk and demands dedicated cardio-obstetric input. [1] [6]
Specific Subtypes & Scenarios
The 45,X/46,XY mosaic is the scenario where the karyotype changes the management. The presence of Y-chromosomal material confers a materially elevated risk of gonadoblastoma — a carcinoma in situ of the dysgenetic gonad — because the streak gonad with Y-bearing cells has a high malignant potential. The international guideline recommends prophylactic gonadectomy once the diagnosis is confirmed, and ongoing surveillance for any residual gonadal tissue. This is the single karyotype-specific action point in Turner syndrome, and a fellowship candidate who fails to mention it loses easy marks. [1] [8]
The mosaic with a normal cell line (45,X/46,XX) is generally milder: stature may be less compromised, some spontaneous pubertal development and even menarche may occur, and a minority may achieve spontaneous pregnancy. The surveillance, however, is unchanged — the aortic, thyroid, renal, and audiology risks persist — because the normal cell line does not fully rescue the phenotype. These girls are the ones most likely to be diagnosed late and to be lost to follow-up, which is a recurring system failure. [2] [4]
The adolescent approaching fertility counselling is a scenario where honest, early, and specialist-led conversation changes outcomes. The overwhelming majority of girls with Turner syndrome will be infertile due to streak ovaries, but a small minority — mostly mosaics — retain enough follicles for spontaneous pregnancy, and oocyte donation offers the prospect of pregnancy to the rest. The Hadnott and Bondy cohort from the National Institutes of Health documented that even in Turner syndrome, spontaneous and assisted pregnancies are achievable but carry elevated risks of miscarriage and aortic dissection. The reproductive conversation should begin in early adolescence, with referral to a fertility specialist and, critically, a cardio-obstetric assessment before any pregnancy attempt, because the aortic dissection risk in pregnancy is life-threatening. [4] [1]
Complications & Pitfalls
The complications divide into the clinical morbidities that accumulate over a lifetime and the cognitive traps that cost marks. The clinical morbidities are cardiovascular (bicuspid aortic valve, coarctation, aortic dilation and dissection, hypertension), endocrine (autoimmune hypothyroidism, impaired glucose tolerance and type 2 diabetes, dyslipidaemia, osteoporosis if oestrogen is inadequate), renal (horseshoe kidney, hydronephrosis, recurrent urinary tract infection), audiological (conductive loss from recurrent otitis media, progressive sensorineural loss), gastrointestinal (inflammatory bowel disease, coeliac disease, elevated liver enzymes), and psychosocial (specific learning difficulties, anxiety, depression, social cognition challenges, and the burden of a chronic diagnosis). Each is treatable or preventable in its own right, and proactive surveillance prevents the secondary disability that neglect produces. [1] [8]
The cognitive traps are five, and a fellowship candidate should name them. First, assuming the diagnosis requires a classic phenotype — it does not; short stature or delayed puberty alone warrants a karyotype. Second, forgetting the Y-bearing mosaic and the gonadoblastoma obligation — this is a safety-critical omission. Third, delaying oestrogen to maximise height at the expense of bone health and psychosocial wellbeing — the modern approach starts oestrogen at 11 to 12 years. Fourth, treating the aortic surveillance with generic thresholds — the Turner aorta dissects at smaller dimensions, and the surgery threshold is lower. Fifth, losing the patient at the transition to adult care — the surveillance lapses, the aorta is not imaged, the osteoporosis is not prevented, and the preventable complications occur. [1] [6]
Prognosis & Disposition
Prognosis is determined by the cardiovascular burden, the adequacy of hormone replacement, and — most powerfully — the quality and continuity of the multidisciplinary surveillance. A girl diagnosed early, treated with growth hormone, given timely oestrogen, and enrolled in structured cardiac and metabolic surveillance can expect a near-normal lifespan, though life expectancy remains reduced relative to the general population, driven primarily by cardiovascular disease. The excess mortality is concentrated in young and middle adulthood and is dominated by aortic dissection and ischaemic heart disease, which is precisely why the transition to adult care and the lifelong aortic imaging are the highest-yield interventions. [1] [8]
Intelligence is typically within the normal range, and most girls and women with Turner syndrome live independent and productive lives, with the visuospatial and mathematical weaknesses supported by targeted educational help and the social-cognition challenges supported by psychological input where needed. The psychosocial prognosis is good when the diagnosis is explained openly and developmentally, the family is supported, and the girl is included in her own care planning as she matures. [2] [8]
Special Populations
The same Turner diagnosis behaves differently across populations because access, recognition, and service models are unevenly distributed. In remote and Indigenous communities in Australia and New Zealand, later presentation, limited access to paediatric endocrinology and genetic services, lower rates of cardiac MRI, and the burden of intercurrent disease mean that girls are diagnosed late and surveillance lapses — culturally safe, outreach-supported, telehealth-augmented care is essential, and the aortic surveillance is the non-negotiable element. In migrant, refugee, and asylum-seeking families, language barriers, fragmented histories, and limited continuity of care delay both diagnosis and the hormone replacement that protects bone health, and an interpreter-mediated, written-plan approach prevents loss to follow-up. [1]
In the adolescent and young-adult transition, the risk is the transfer cliff — the loss of the structured paediatric surveillance at the moment the aortic risk is rising and the fertility window is narrowing. A structured transition, with a named adult endocrinologist and cardiologist, a reconciled written summary, and explicit aortic-imaging and bone-density plans, is the intervention that prevents the preventable deaths. In the girl with a co-occurring developmental disability or significant learning profile, the management plan must integrate the educational and psychological support alongside the medical surveillance, and the consent and assent framework must be developmentally appropriate. [8]
Evidence, Guidelines & Regional Differences
The evidence base rests on three pillars: the international clinical practice guidelines, the growth-hormone and oestrogen trial literature, and the cardiovascular risk and pregnancy-outcome cohorts. The 2017 international guideline (Gravholt and colleagues, European Journal of Endocrinology), developed jointly by the European Society of Endocrinology, the Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology, is the current practice standard and sets the structure of karyotyping, growth-hormone and oestrogen therapy, cardiovascular imaging thresholds, and transition that most national programmes adopt. [1]
The growth-hormone evidence rests on the Canadian randomised controlled trial (Stephure and colleagues, 2005), which established the approximately 5 to 7 centimetre adult-height gain, and on the oxandrolone augmentation literature. The oestrogen evidence includes the Cleemann randomised trial of oestradiol dosing for uterine growth, which informed the modern push for adequate uterine priming in preparation for fertility options. The cardiovascular and pregnancy evidence includes the vasculopathy work of Ostberg and Conway, which demonstrated that the Turner aorta is intrinsically abnormal with intimal thickening and dilation independent of flow, and the Hadnott and Bondy pregnancy cohort, which documented the maternal risks. [3] [7] [6] [4]
Regional differences are modest in principle but significant in execution. The ANZ model follows the international guideline and emphasises the multidisciplinary Turner clinic, the cardiac MRI as a baseline, and the structured transition; access is the challenge in rural and remote settings. The UK and European model aligns closely with the international guideline, with national Turner syndrome registers in several countries. The North American model follows the same guideline and emphasises the medical home and the growth-hormone indication. The common thread is that the guideline is broadly shared, but the continuity of surveillance — especially across the transition and into pregnancy — is where systems fail. [1] [8]
Exam Pearls
A fellowship candidate answering on Turner syndrome should land five anchor points and avoid three classic traps. The anchors are the karyotypic definition and the five classes, the SHOX-driven pathophysiology, the growth-hormone and timed-oestrogen management framework, the cardiovascular surveillance with the lower dissection threshold, and the Y-mosaic gonadoblastoma rule. The traps are waiting for a classic phenotype before karyotyping, delaying oestrogen at the expense of bone and psychosocial health, and losing the patient at transition. The communication skill that distinguishes the strong candidate is the honest, developmentally framed explanation of infertility and aortic risk that preserves the adolescent's autonomy and future. [1] [2]
References
- [1]Gravholt CH, Andersen NH, Conway GS, Dekkers OM, Geffner ME, Klein KO, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol, 2017.PMID 28705803
- [2]Sybert VP, McCauley E. Turner's syndrome. N Engl J Med, 2004.PMID 15371580
- [3]Stephure DK, Canadian Growth Hormone Advisory Committee. Impact of growth hormone supplementation on adult height in Turner syndrome: results of the Canadian randomized controlled trial. J Clin Endocrinol Metab, 2005.PMID 15784709
- [4]Hadnott TN, Gould HN, Gharib AM, Bondy CA. Outcomes of spontaneous and assisted pregnancies in Turner syndrome: the U.S. National Institutes of Health experience. Fertil Steril, 2011.PMID 21496813
- [5]Sheanon NM, Backeljauw PF. Effect of oxandrolone therapy on adult height in Turner syndrome patients treated with growth hormone. Int J Pediatr Endocrinol, 2015.PMID 26322078
- [6]Ostberg JE, Donald AE, Halcox JP, Storry C, McCarthy C, Conway GS. Vasculopathy in Turner syndrome: arterial dilatation and intimal thickening without endothelial dysfunction. J Clin Endocrinol Metab, 2005.PMID 15985480
- [7]Cleemann L, Holm K, Fallentin E, Moller N, Heickendorff L, Skouby SO, et al. Effect of Dosage of 17beta-Estradiol on Uterine Growth in Turner Syndrome-A Randomized Controlled Trial. J Clin Endocrinol Metab, 2020.PMID 31613320
- [8]Trolle C, Mortensen KH, Hjerrild BE, Cleemann L, Gravholt CH. Clinical care of adult Turner syndrome--new aspects. Pediatr Endocrinol Rev, 2012.PMID 22946288