Paeds · genetics-dysmorphology-and-metabolism
Urea-cycle disorders and hyperammonaemia
Also known as Urea cycle disorders · UCD · Hyperammonaemia of metabolic origin · Ornithine transcarbamylase deficiency · OTC deficiency · Carbamoyl phosphate synthetase 1 deficiency
A fellowship approach to the urea cycle disorders: recognise hyperammonaemia as a time-critical metabolic emergency that mimics sepsis in the neonate and presents with bizarre behaviour or coma in the older child, treat on suspicion with calorie loading, nitrogen scavengers and dialysis before the enzyme diagnosis returns, confirm with plasma amino acids and urinary orotic acid, and lock in long-term protein-restricted medical and — for severe forms — transplant-based management.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who thinks in three layers at once. The first layer is the child in front of you: an encephalopathic neonate or a confused older child, where the immediate question is not "what is the enzyme defect" but "what is the ammonia and how fast is it rising". The second is the biochemistry: a cycle that sits half in the mitochondrion and half in the cytosol, with each block producing a recognisable pattern of accumulating metabolites. The third is the family: an X-linked diagnosis in OTC deficiency obliges carrier testing of the mother and sisters and reproductive counselling, because the recurrence risk and the carrier phenotype change lives. [1] [3]
Overview & Definition
The urea cycle disorders are a group of inherited metabolic diseases caused by deficiency of one of the six enzymes or two transporters that convert ammonia, the toxic product of nitrogen metabolism, into urea for renal excretion. The cycle is split between the mitochondrion — where N-acetylglutamate synthase (NAGS), carbamoyl phosphate synthetase 1 (CPS1) and ornithine transcarbamylase (OTC) act — and the cytosol, where argininosuccinate synthetase (ASS1, deficient in citrullinaemia type I), argininosuccinate lyase (ASL) and arginase (ARG1) complete the pathway. A defect at any step blocks nitrogen disposal, ammonia accumulates, and the clinical consequence is hyperammonaemic encephalopathy. [1] [3]
Clinically, the urea cycle disorders sit within the family of intoxication-type inborn errors of metabolism: conditions in which a blocked catabolic pathway generates a circulating toxin that is itself the disease. This framing matters because it dictates management — the priority is toxin removal, not enzyme replacement, and the intervention that changes outcome is the speed of that removal. Ornithine transcarbamylase deficiency is the most common and the only one inherited in an X-linked pattern; the remainder are autosomal recessive, and citrullinaemia type II is caused by deficiency of the citrin transporter (SLC25A13). [1] [8]
Classification
The classification that matters clinically is biochemical and anatomical, because the position of the block within the cycle determines the accumulating metabolites, the diagnostic pattern, and often the severity. The proximal defects (NAGS, CPS1, OTC) sit in the mitochondrion and tend to produce the most severe neonatal hyperammonaemia, while the distal defects (ASS1, ASL, ARG1) are generally milder and carry additional organ-specific complications such as the hepatopathy and hypertension of ASL deficiency or the spastic diplegia of arginase deficiency. [1] [3]
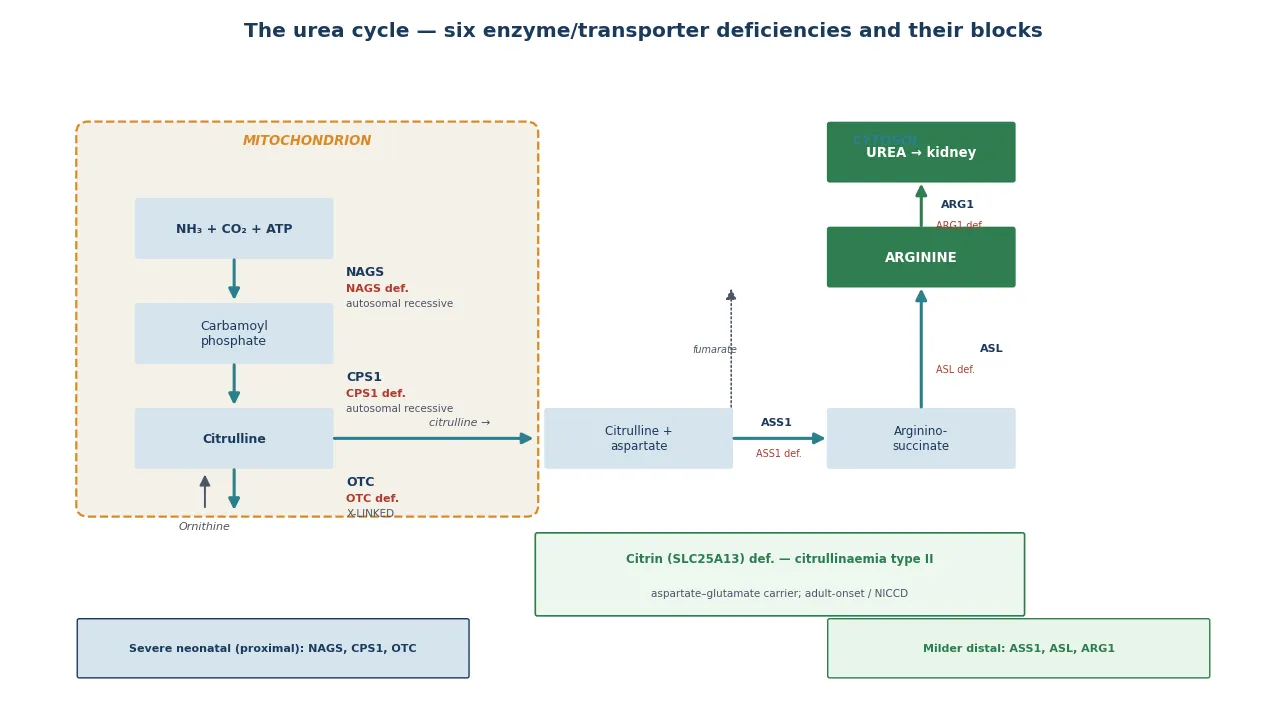
A proximal block at NAGS or CPS1 produces very low citrulline and arginine, because nothing distal is generated; at OTC the block is one step later, so carbamoyl phosphate is shunted into the pyrimidine pathway and excreted as urinary orotic acid — the key discriminator between OTC deficiency (orotic acid high) and CPS1/NAGS deficiency (orotic acid low or absent). A distal block at ASS1 causes citrulline to accumulate (citrullinaemia type I), at ASL argininosuccinic acid accumulates, and at ARG1 arginine accumulates. These patterns convert a single blood test — plasma amino acids — plus urinary orotic acid into a near-diagnostic first-tier screen before molecular confirmation. [1] [2]
Epidemiology & Risk Factors
The combined incidence of the urea cycle disorders is roughly one in 8,000 to 10,000 live births, with ornithine transcarbamylase deficiency the most common single entity at around one in 14,000. Because OTC is X-linked, affected males usually present with severe neonatal hyperammonaemia while heterozygous females show a variable phenotype driven by skewed X-inactivation — some are asymptomatic, others decompensate postpartum or after a protein load, and this female variability is itself a high-yield counselling point. [1] [6]
The Japanese nationwide long-term outcome study by Kido and colleagues is one of the most informative cohort datasets, showing that despite improved emergency management, survival and neurodevelopmental outcome remain strongly predicted by peak ammonia and the time to definitive treatment — children who survive a neonatal crisis with an ammonia above 300 to 500 micromoles per litre frequently carry a significant cognitive burden. This is the epidemiological engine behind the "treat on suspicion" principle: the disease is rare, but the consequence of delay is permanent and large. [6]
The major risk factor for a late-onset or recurrent presentation is a catabolic trigger — intercurrent infection, fasting, surgery, trauma, the postpartum period, valproate therapy, or a high-protein meal. A previously well child or adolescent who becomes acutely confused, ataxic or comatose after any of these should have ammonia measured. A family history of neonatal or unexplained death, maternal hyperemesis or postpartum confusion, or known consanguinity raises the pre-test probability further. [1] [4]
Pathophysiology
The molecular story begins with nitrogen. Dietary and endogenous protein catabolism generates ammonia, which the liver detoxifies through the urea cycle: ammonia and bicarbonate are condensed into carbamoyl phosphate by CPS1 (activated by N-acetylglutamate, itself made by NAGS), carbamoyl phosphate joins ornithine to form citrulline (OTC), and the cycle then runs in the cytosol through ASS1, ASL and ARG1 to regenerate ornithine and release urea. A block at any step halts this disposal, and ammonia accumulates in plasma. [3] [1]
Ammonia crosses the blood–brain barrier freely. In astrocytes it is fixed into glutamine by glutamine synthetase, and it is the accumulation of glutamine — not ammonia directly — that drives the brain injury. Glutamine is osmotically active: it draws water into the astrocyte, the cell swells, mitochondrial function fails, reactive oxygen species accumulate, and the result is astrocyte swelling, cytotoxic and vasogenic cerebral oedema, raised intracranial pressure, and ultimately neuronal death and herniation. The injury is proportional to both the peak ammonia and its duration, which is why speed of reduction is the dominant determinant of outcome. [3]

The genotype–phenotype relationship is best worked out for OTC deficiency. The functional study by Lo and colleagues, which assessed 1,570 individual amino acid substitutions in the OTC protein, demonstrated a wide functional spectrum that predicts residual enzyme activity and, in turn, the likelihood of neonatal versus late-onset presentation — a molecular confirmation that severity is graded by how much enzyme function survives. This residual-activity principle explains why some OTC-deficient males and many heterozygous females present late, and it underpins the modern move toward severity-adjusted treatment, including transplantation. [12]
Clinical Presentation
The presentation is bimodal, and the distinction between neonatal and late-onset disease is the single most useful clinical framework. Neonatal presentation is the classic, dramatic form: a term baby, well at birth, who deteriorates after 24 to 72 hours of protein-containing feeds with poor feeding, lethargy, vomiting, hypothermia, hyperventilation (from ammonia-driven central respiratory stimulation, producing a respiratory alkalosis) and progressing to seizures, coma and apnoea. The hyperventilation is a subtle but high-yield sign that distinguishes a hyperammonaemic baby from one with a primary respiratory or infectious process. [1] [4]
Late-onset presentation is increasingly recognised and is the form most often missed. It occurs in older infants, children and adults with partial enzyme deficiency, and it is typically precipitated by a catabolic stressor — infection, fasting, surgery, the postpartum period, or a high-protein load. The picture is episodic encephalopathy: vomiting, ataxia, confusion, agitation or bizarre behaviour, progressing to seizures and coma, often labelled initially as a gastroenteritis, a toxin, or a psychiatric presentation. A low threshold to measure ammonia in any unexplained encephalopathy is the lesson. [1] [6]
Arginase deficiency is the third presentation, and it is the great cerebral-palsy mimic. Rather than acute hyperammonaemia, it produces a chronic, progressive spastic diplegia with intellectual disability, seizures and growth failure, often with only mildly elevated ammonia. A child labelled "cerebral palsy" who has a progressive course, an abnormal metabolic screen, or a family history of a similar pattern warrants plasma arginine measurement, because arginase deficiency is treatable with protein restriction and nitrogen-scavenging therapy. [8]
Differential Diagnosis
The differential is the differential of hyperammonaemia itself, and the urea cycle disorders are one — important but not the only — cause. The first task is to separate the urea cycle disorders from the other causes of secondary hyperammonaemia, because the management differs. The organic acidaemias (propionic, methylmalonic, isovaleric), the fatty acid oxidation defects, and the mitochondrial hepatopathies all cause hyperammonaemia through secondary inhibition of the cycle, and they are distinguished by metabolic acidosis (with a high anion gap), ketosis, lactic acidaemia, and an abnormal acylcarnitine profile. [1]
Reye syndrome — now rare — and other causes of acute liver failure produce hyperammonaemia from hepatocellular dysfunction, and transient hyperammonaemia of the newborn (THAN), classically in preterm infants with respiratory distress, is a separate, often self-limiting entity that nonetheless requires urgent treatment while the work-up proceeds. Portosystemic shunts, infections with urease-producing organisms, and valproate therapy are acquired causes worth naming. The discriminator is the metabolic profile: a urea cycle disorder typically shows a normal anion gap and normal glucose with a respiratory alkalosis, whereas the organic acidaemias show acidosis and ketosis. [1] [4]
Clinical & Bedside Assessment
The bedside assessment has two speeds: the acute stabilisation, where the question is resuscitation and the ammonia level, and the diagnostic characterisation, where the question is which defect and what it means for the family. In the acute presentation, take a focused history focused on onset relative to feeds, the presence of a catabolic trigger, and the family history, while simultaneously drawing the blood gas, glucose, ammonia (free-flowing, on ice, to the laboratory urgently), and the metabolic first-tier tests. Do not delay resuscitation for a complete history. [1] [4]
In the more stable or recovering child, build a structured picture: a three-generation pedigree asking explicitly about neonatal deaths, unexplained developmental delay or intellectual disability, maternal hyperemesis or postpartum confusion, consanguinity, and reactions to protein or fasting. The examination looks for the stigmata of chronic disease in the late-onset case — growth failure, developmental delay, hepatomegaly — and for the spasticity of arginase deficiency, while the neonate is examined for the signs of encephalopathy and cerebral oedema that drive the urgency. [1] [6]
The bedside assessment converts directly into the investigation plan: plasma amino acids and urinary orotic acid to localise the defect, quantitative amino acids to guide arginine and citrulline supplementation, plasma glutamine as a treatment-monitoring marker, and molecular genetic testing of the candidate gene (or a metabolic gene panel / exome) for confirmation and family counselling. In a neonatal crisis the enzyme diagnosis will not return before treatment decisions, so the acute plan is driven by the ammonia trajectory and the metabolic first-tier pattern. [1] [2]
Investigations
The first-tier investigation of hyperammonaemia is a coordinated metabolic panel interpreted together. Plasma ammonia must be measured from a free-flowing sample (venous or arterial, not heel-prick, to avoid contamination), transported on ice and run urgently, because falsely elevated values from poor handling are a common and dangerous confounder. Plasma amino acids quantify glutamine (the treatment-monitoring marker), citrulline (low in proximal defects, high in citrullinaemia type I), and arginine (high in arginase deficiency). Urinary orotic acid discriminates OTC deficiency (high) from CPS1 and NAGS deficiency (low or absent). [1] [2]
The supporting tests exclude the mimics and define the biochemical context: blood gas and lactate for acid–base and the anion gap, glucose and ketones, liver function and coagulation to separate primary hepatopathy, and plasma acylcarnitines and urinary organic acids to identify the organic acidaemias and fatty-acid oxidation defects that secondarily inhibit the cycle. A normal anion gap with a respiratory alkalosis and raised glutamine points toward a primary urea cycle defect; a high anion-gap metabolic acidosis with ketosis points away from it. [1] [4]
Why a single normal ammonia does not always close the case
Late-onset and partial urea cycle disorders produce intermittent hyperammonaemia, so a single normal ammonia drawn when the child is well and fed does not exclude the diagnosis. In a child with a suggestive history — episodic encephalopathy triggered by illness or protein — repeat ammonia during an acute episode, and supplement it with plasma amino acids and urinary orotic acid at the same moment, because the diagnostic metabolite pattern is only visible when the cycle is under stress. [1] [6]
Molecular confirmation is then undertaken by sequencing the candidate gene suggested by the biochemical pattern, or by a metabolic gene panel or exome when the pattern is ambiguous. Confirming the molecular defect is essential for three reasons: it defines the exact diagnosis, it enables cascade carrier testing and prenatal or preimplantation diagnosis for the family, and — for OTC deficiency specifically — it identifies at-risk female relatives whose own phenotype and reproductive risk must be counselled. Enzyme assay on liver biopsy is now largely reserved for cases where sequencing is uninformative. [1] [12]
Management — Resuscitation
Resuscitation is the section that earns or loses the most marks, because outcome is determined here. The principle is to treat on suspicion before the enzyme diagnosis returns: the moment hyperammonaemia is identified in an encephalopathic child, start the emergency protocol. The first moves are to stop all protein intake, establish intravenous access, and begin aggressive calorie provision — 10 percent glucose with intralipid, with insulin as needed to drive anabolism — because switching the child from catabolism to anabolism is the single most important physiological intervention to reduce endogenous ammonia generation. [1] [4]
Nitrogen-scavenging drugs are then introduced. Sodium benzoate and sodium phenylbutyrate (or sodium phenylacetate) provide alternative pathways for nitrogen excretion: benzoate conjugates glycine to form hippurate, and phenylbutyrate conjugates glutamine to form phenylacetylglutamine, both renally excreted and each removing a mole of nitrogen. Arginine is given intravenously to replenish the cycle downstream — essential in ASS1 and ASL deficiency and useful in the proximal defects — and citrulline is substituted for arginine in the proximal defects. N-carbamylglutamate (carbaglu) is the specific therapy for NAGS deficiency and may augment CPS1 in some organic acidaemias. [1] [9]
The decision to dialyse is made on ammonia thresholds and trajectory. The consensus guidance — reflected in the Raina continuous kidney-replacement-therapy guidelines and the regional protocols — is to proceed to haemofiltration or haemodialysis when ammonia exceeds roughly 500 micromoles per litre in a neonate or a lower threshold (often 200 to 300) with encephalopathy or a rising trend despite medical therapy, and not to wait for neurological deterioration. Continuous kidney replacement therapy is preferred in haemodynamically unstable neonates. The objective is to halve the ammonia within hours, because every hour of cerebral oedema adds to permanent injury. [5] [4]
Management — Definitive & Stepwise
Once the acute crisis is controlled, definitive management is a long-term, multidisciplinary framework built around four pillars: a protein-restricted diet with essential amino acid supplementation, chronic nitrogen-scavenging therapy, prevention of catabolism during intercurrent illness, and — for the most severe forms — liver transplantation. The diet is the cornerstone: natural protein restricted to tolerance, with essential amino acid supplements and adequate non-protein calories to maintain anabolism and prevent catabolism-induced decompensation. [1] [9]

Chronic nitrogen-scavenging therapy uses oral sodium benzoate and sodium phenylbutyrate between episodes, titrated to metabolic control; the randomised trial infrastructure developed by Ah Mew and colleagues established the rigorous, intervention-trial approach needed to evaluate these agents during acute decompensation. Prevention of catabolism is taught to every family as an emergency sick-day plan: at the first sign of illness, stop protein, increase calories from glucose and lipids, and present early for intravenous management. A medic alert and a written emergency letter travel with the child. [7] [1]
Liver transplantation is the definitive therapy for the severe neonatal-onset urea cycle disorders, particularly OTC and CPS1 deficiency, because it corrects the hepatic enzyme defect, restores metabolic stability, and liberates the child from dietary restriction and the constant threat of decompensation. The indications and timing are refined by the severity-adjusted outcome data of Posset and colleagues and the transplant-centre series of García Vega and colleagues, which together show that early transplant in a metabolically stable window substantially improves survival and neurodevelopmental outcome, though it does not reverse established brain injury — reinforcing that transplant is complementary to, not a substitute for, excellent emergency care. [10] [11]
H.A.L.T. the ammonia \u2014 the acute protocol
Specific Subtypes & Scenarios
Ornithine transcarbamylase deficiency is the prototype and the most common urea cycle disorder. Affected males typically present with severe neonatal hyperammonaemia; heterozygous females range from asymptomatic to severely affected depending on X-inactivation, and some present for the first time in the postpartum period with confusion and coma. The biochemical signature is high urinary orotic acid with low citrulline, and the X-linked inheritance makes carrier testing of the mother and sisters a mandatory part of the family work-up. Early liver transplantation is increasingly the standard for severe neonatal OTC disease. [1] [12]
ASL deficiency (argininosuccinic aciduria) deserves special mention because it carries complications beyond hyperammonaemia: hypertension, hepatomegaly and hepatic fibrosis, trichorrhexis nodosa (fragile hair), and developmental disability that may progress despite good metabolic control. The hypertension and hepatopathy are thought to reflect the toxicity of accumulated argininosuccinic acid itself, which is why ASS1 and ASL deficiency receive arginine to push metabolism toward argininosuccinate excretion — a paradox that the fellowship candidate should be able to explain. [1] [6]
Arginase deficiency is the cerebral-palsy mimic and the one urea cycle disorder that does not typically present as acute neonatal hyperammonaemia. Its hallmarks are progressive spastic diplegia, intellectual disability, seizures and growth failure, with a chronically elevated plasma arginine. Early diagnosis and a low-arginine, protein-restricted diet with nitrogen scavengers can slow or halt the neurological progression, which is why every atypical or progressive "cerebral palsy" warrants a plasma amino acid profile. [8]
The NAGS-deficient patient is a scenario with a uniquely satisfying answer, because NAGS deficiency is the one urea cycle disorder with a specific, oral, disease-modifying drug: N-carbamylglutamate (carbaglu), an analogue of N-acetylglutamate that activates CPS1. The work of Singh and colleagues confirms that carbaglu reshapes the nutritional management of confirmed NAGS deficiency, and because NAGS deficiency is clinically indistinguishable from CPS1 deficiency on first-tier testing, a therapeutic trial of carbaglu is a reasonable and often diagnostic early move when a proximal defect with low orotic acid is suspected. [9] [1]
Complications & Pitfalls
The complications divide into the acute neurological injuries, the chronic multisystem burden, and the cognitive traps that cost marks. The acute injuries are cerebral oedema, seizures, stroke-like episodes, coma and death, and the risk of permanent cognitive impairment rises steeply with peak ammonia above 300 to 500 micromoles per litre and with prolonged duration — this is the evidence base from the Japanese nationwide cohort that underpins the urgency of treatment. The chronic burden includes intellectual disability, seizures, behavioural and psychiatric disturbance, growth failure, and the hepatic and renal complications of specific defects and their therapy. [6] [1]
The chief cognitive trap is delay, which takes three forms: failure to measure ammonia in an encephalopathic neonate because sepsis seems more likely; failure to start treatment before the diagnosis is confirmed; and failure to dialyse because the ammonia "might come down with medication". The second trap is misclassification — confusing a urea cycle disorder with an organic acidaemia, which changes the diet and the scavenger choice. The third is missing the family, particularly in OTC deficiency, where an affected mother's carrier status and an at-risk sister's reproductive risk are obligations of the index diagnosis. [1] [3]
Prognosis & Disposition
Prognosis is determined by three factors: the peak ammonia and its duration during the presenting crisis, the specific defect (with the proximal defects carrying the worst acute risk), and the quality and consistency of long-term metabolic management. A child who survives a neonatal crisis with a very high ammonia frequently carries a significant cognitive and neurological burden, whereas late-onset disease caught early and managed well can allow near-normal development — a gradient that makes the speed of the acute response the single most powerful modifier of the lifelong outcome. [6] [1]
Liver transplantation changes the trajectory for severe disease by correcting the metabolic defect and removing the constant threat of decompensation, though it cannot reverse established neurological injury, which is why it is best performed in a metabolically stable window rather than as a rescue. Disposition is shared, lifelong, multidisciplinary care: a specialist metabolic service owns the diet, the scavenger regimen, and the transplantation pathway; the general paediatrician or GP owns coordination, immunisation, and the emergency sick-day plan; and the family owns the day-to-day vigilance that prevents decompensation. Every transition — into school, into adolescence, and into adult metabolic services — is a high-risk point, so the emergency plan and the metabolic record must travel with the child. [10] [11]
Special Populations
The same urea cycle disorder behaves differently across populations because access, recognition, and service models are unevenly distributed. In remote and Indigenous communities, later presentation, distance from a metabolic service, and the need for aeromedical retrieval during a neonatal crisis mean that the window between onset and treatment is longer and outcomes are worse — so a written, location-specific emergency plan and a low threshold to measure ammonia in any encephalopathic neonate are disproportionately important. In migrant, refugee, and asylum-seeking families, consanguinity raises the pre-test probability of autosomal recessive defects, language barriers complicate the emergency teaching of a sick-day plan, and an interpreter must be used at every key consultation. [1]
In adolescents transitioning to adult metabolic care, the move is a high-risk point: dietary adherence often slips, the emergency plan may not transfer, and pregnancy becomes a dual concern of maternal metabolic control and the risk to a heterozygous (for OTC) or affected fetus. Affected girls and women are a special population in their own right — an OTC-heterozygous female may be asymptomatic for years and decompensate in the postpartum period, and pre-pregnancy counselling and a metabolic obstetric plan are essential. In families managing complex chronic metabolic disease, fragmentation of care is the chief threat, and a written, shared, reconciled care plan is the intervention that matters most. [6] [1]
Evidence, Guidelines & Regional Differences
The evidence base rests on three pillars: consensus clinical guidelines, longitudinal cohort outcome data, and emerging molecular and therapeutic evidence. The Häberle first-revision guidelines (2019) are the current international standard for diagnosis and management, building on the original 2012 document, and they set the structure of acute management, long-term dietary and pharmacological therapy, transplantation, and family counselling that most national programmes adopt. The Alfadhel regional protocol and the Raina continuous kidney-replacement-therapy consensus translate these principles into operational detail for emergency and dialysis thresholds. [1] [4] [5]
The longitudinal cohort data rest largely on nationwide registries, of which the Japanese study by Kido and colleagues is among the most informative, demonstrating that survival and neurodevelopmental outcome remain strongly predicted by peak ammonia and time to treatment despite modern care. The molecular and therapeutic evidence is moving fast: the functional OTC variant study by Lo and colleagues grounds severity prediction in protein biochemistry, the severity-adjusted transplant outcome work of Posset and colleagues refines who benefits from liver transplantation and when, and the carbaglu evidence of Singh and colleagues consolidates NAGS deficiency as the one urea cycle disorder with a specific oral disease-modifying drug. [6] [9] [11] [12]
In Australia and New Zealand, neonatal and paediatric hyperammonaemia is managed through the state-based metabolic services (most coordinated through the major children's hospitals in each state and Starship in New Zealand), with newborn screening programmes detecting some but not all urea cycle disorders — the proximal defects are not reliably captured by tandem mass spectrometry on the bloodspot, so a normal newborn screen does not exclude a urea cycle disorder. Aeromedical retrieval to a tertiary metabolic and intensive-care centre is the expected pathway for a neonatal crisis, and continuous kidney replacement therapy is available at the paediatric intensive care units that receive these patients. Genetic counselling, carrier testing, and prenatal or preimplantation genetic diagnosis are coordinated through clinical genetics services, and the NDIS (Australia) and equivalent disability support (New Zealand) fund the allied health, dietetic, and developmental supports that anchor long-term care. Always confirm the current local retrieval pathway and metabolic service contact, as these vary by state.
[1][6]Exam Pearls
A fellowship candidate answering on the urea cycle disorders should land six anchor points and avoid three classic traps. The anchors are the bimodal presentation (neonatal after feeds, late-onset after catabolism), the ammonia–glutamine mechanism of cerebral oedema, the metabolic first-tier panel and how plasma amino acids plus urinary orotic acid localise the defect, the "treat on suspicion" emergency protocol (stop protein, calorie-load, scavenge, arginine, dialyse), the role of liver transplantation for severe disease, and the X-linked counselling implications of OTC deficiency. The traps are delay (waiting for the diagnosis), misclassification (confusing UCD with an organic acidaemia), and missing the family. The candidate who can name arginine as essential in ASS1 and ASL deficiency, arginase deficiency as the cerebral-palsy mimic, and carbaglu as the specific NAGS therapy will stand out. [1] [3]
References
- [1]Häberle J, Burlina A, Chakrapani A, Dixon M, Karnebeek C, Lindner M, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. Eur J Pediatr / J Inherit Metab Dis, 2019.PMID 30982989
- [2]Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis, 2012.PMID 22642880
- [3]Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr, 1996.PMID 8794176
- [4]Alfadhel M, Mutairi FA, Makhseed N, Jasmi FA, Al-Thihli K, Al-Jishi E, et al. Guidelines for acute management of hyperammonemia in the Middle East region. Ther Clin Risk Manag, 2016.PMID 27099506
- [5]Raina R, Bedoyan JK, Lichter-Konecki U, Jouvet P, Picca S, Mew NA, et al. Consensus guidelines for management of hyperammonaemia in paediatric patients receiving continuous kidney replacement therapy. Nat Rev Nephrol, 2020.PMID 32269302
- [6]Kido J, Matsumoto S, Häberle J, Nakajima Y, Wada Y, Mochizuki N, et al. Long-term outcome of urea cycle disorders: Report from a nationwide study in Japan. J Inherit Metab Dis, 2021.PMID 33840128
- [7]Ah Mew N, Cnaan A, McCarter R, Choi H, Simpson PM, Atkinson JM, et al. Conducting an investigator-initiated randomized double-blinded intervention trial in acute decompensation of inborn errors of metabolism: Lessons from the N-Carbamylglutamate Consortium. Transl Sci Rare Dis, 2018.PMID 30613471
- [8]Sin YY, Baron G, Schulze A, Funk CD. Arginase-1 deficiency. J Mol Med (Berl), 2015.PMID 26467175
- [9]Singh RH, Bourdages MH, Kurtz A, MacLoed E, Gropman A, Barth M, et al. The efficacy of Carbamylglutamate impacts the nutritional management of patients with N-Acetylglutamate synthase deficiency. Orphanet J Rare Dis, 2024.PMID 38637895
- [10]García Vega M, Andrade JD, Morais A, Frauca E, Díaz C, Aldamiz-Echevarría L, et al. Urea cycle disorders and indications for liver transplantation. Front Pediatr, 2023.PMID 36937980
- [11]Posset R, Garbade SF, Gleich F, Scharre S, Kolker S, Burgard P, et al. Severity-adjusted evaluation of liver transplantation on health outcomes in urea cycle disorders. Genet Med, 2024.PMID 38054409
- [12]Lo RS, Cromie GA, Tang M, Teng K, D'Souza S, Leung JW, et al. The functional impact of 1,570 individual amino acid substitutions in human OTC. Am J Hum Genet, 2023.PMID 37146589