Paeds · genetics-dysmorphology-and-metabolism
Williams syndrome
Also known as Williams-Beuren syndrome · Williams-Beuren syndrome (WBS) · 7q11.23 microdeletion syndrome · Idiopathic infantile hypercalcaemia with supravalvular aortic stenosis (historical) · WBS
A fellowship approach to Williams syndrome: recognise the multisystem pattern of a 7q11.23 microdeletion (distinctive facies, supravalvular aortic stenosis and elastin arteriopathy, infantile hypercalcaemia, the hypersocial personality and Williams cognitive profile), confirm the deletion with chromosomal microarray, stage the cardiovascular disease as the leading cause of mortality, and run an age-based multidisciplinary surveillance plan anchored to the AAP 2020 health-supervision clinical report.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
The fellowship mark goes to the candidate who thinks in three layers at once. The first is the child's heart: the elastin arteriopathy is the leading cause of morbidity and mortality, and the coronary arteries are its most dangerous and most easily missed expression. The second is the gene dose: a microdeletion of about 1.5 to 1.8 megabases at 7q11.23 removes one copy of elastin and a cluster of neurodevelopmental genes, and the phenotype is the sum of those haploinsufficiencies. The third is the family and the function: an affected parent changes the recurrence risk from background to one in two, and a sociable, verbal child still needs real support because the speech conceals a genuine intellectual and daily-living disability. [2] [5]
Overview & Definition
Williams syndrome, also called Williams-Beuren syndrome, is a multisystem contiguous-gene microdeletion syndrome caused by the hemizygous deletion of approximately 1.5 to 1.8 megabases at chromosome 7q11.23, a region containing 26 to 28 genes. The deletion produces a recognisable constellation of distinctive facies, a cardiovascular arteriopathy dominated by supravalvular aortic stenosis, infantile hypercalcaemia, intellectual disability with a characteristic cognitive profile, and a hypersocial personality. It affects roughly one in 7,500 to one in 10,000 live births, with no racial or sex predilection. [1] [2]
Clinically, Williams syndrome is the paradigm of how the loss of a handful of dosage-sensitive genes produces a coherent multisystem phenotype. Haploinsufficiency of the elastin gene (ELN) accounts for the cardiovascular and connective-tissue features — the narrowed, thickened arteries, the herniae, the joint laxity, the soft skin. Loss of the neighbouring GTF2I, GTF2IRD1, and LIMK1 genes accounts for the intellectual disability, the craniofacial gestalt, the hypersocial personality, and the visuospatial construction deficit. The syndrome is therefore best taught and best examined as the sum of its haploinsufficiencies, not as a single disease. [2] [5]
Historically, the syndrome was assembled from two separate descriptions: in 1961, the New Zealand cardiologist J. C. P. Williams reported supravalvular aortic stenosis with a distinctive facies and intellectual disability, and in the same year the German physicians A. J. Beuren and colleagues described a similar constellation. The historical name idiopathic infantile hypercalcaemia with supravalvular aortic stenosis captures the two features that first brought the syndrome to clinical attention, and the obsolete lay term elfin facies is now avoided because it is considered pejorative; use distinctive or characteristic facies. [8]
Classification
The classification that matters most is molecular, because the size and gene content of the 7q11.23 deletion set the cardiovascular and cognitive phenotype and the counselling. The typical deletion is 1.5 to 1.8 megabases flanked by low-copy repeats that mediate its formation by unequal crossing-over, and it removes the full set of candidate genes including ELN, GTF2I, GTF2IRD1, and LIMK1. [2] [5]
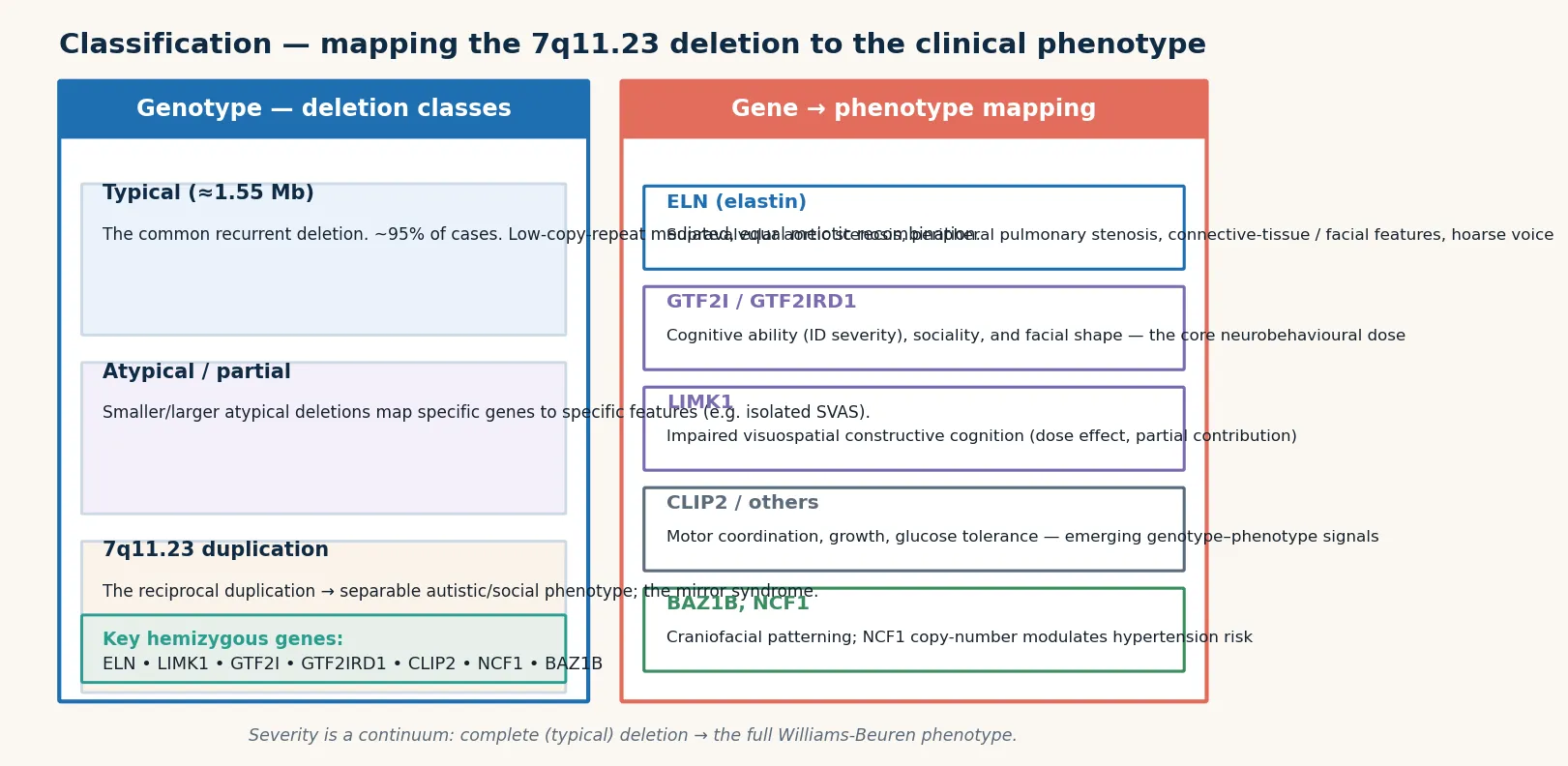
A typical deletion (about 95 percent of cases) removes the common 1.5 to 1.8 megabase block and produces the full Williams-Beuren phenotype. An atypical larger deletion extends beyond the common block and is associated with a more severe phenotype, including more profound intellectual disability, seizures, and autistic features. An atypical smaller deletion confined to a subset of genes produces a partial phenotype — for example, a deletion sparing GTF2I may preserve cognitive function while still conferring the cardiovascular burden. The crucial distinction for counselling is isolated familial supravalvular aortic stenosis, caused by an ELN point mutation or a deletion restricted to the ELN region: the affected person has the narrowed arteriopathy but normal facies and normal cognition, so confusing it with Williams syndrome misleads the family about developmental outcome. [5] [2]
Epidemiology & Risk Factors
Williams syndrome is one of the more common microdeletion syndromes, with a birth prevalence of approximately one in 7,500 to one in 10,000 live births. There is no clear racial, ethnic, or socioeconomic predilection, and males and females are affected equally, so the diagnosis rests on pattern recognition and testing rather than on pre-test probability. [2] [8]
The majority of cases are sporadic — the deletion arises as a new (de novo) event in roughly 75 percent of affected individuals. The mechanism is low-copy-repeat-mediated unequal meiotic recombination at the 7q11.23 region: the flanking segmental duplications misalign during meiosis, and the crossover deletes the intervening block. In some families a parental balanced inversion of 7q11.23 predisposes to the deletion in offspring, which is one reason a careful family history matters even when the index case appears sporadic. [2]
The risk factor that changes counselling is an affected parent. Because the deletion behaves as an autosomal dominant trait, a parent who carries the 7q11.23 deletion has a 50 percent chance of transmitting it to each child. For this reason, once a child is diagnosed, the parents should be assessed clinically and, where indicated, offered testing, and the family should receive formal genetic counselling about recurrence in future pregnancies, including prenatal and preimplantation options. [1]
Pathophysiology
The molecular story is one of haploinsufficiency: the loss of one functional copy of a gene reduces its product below the threshold needed for normal development. The 7q11.23 region contains 26 to 28 genes, and the phenotype is the integrated effect of losing several of them. The best characterised is elastin (ELN), whose loss explains the entire cardiovascular and connective-tissue phenotype. [5] [2]
Elastin is the core protein of the elastic lamellae of the arterial media. With only one functional copy, the developing artery lays down fewer, thinner elastic lamellae, and the vessel wall compensates with excessive smooth-muscle cell proliferation and a thickened, disordered media. The result is a diffuse, stenotic arteriopathy — narrowed at branch points and at the supravalvular aorta — rather than the aneurysmal dilation seen in disorders of matrix integrity such as Marfan syndrome. This is why the hallmark lesion is supravalvular aortic stenosis, why the peripheral pulmonary arteries are narrowed, and why the renal and coronary arteries can be involved, producing hypertension, renovascular disease, and ischaemic sudden death. [5] [3]
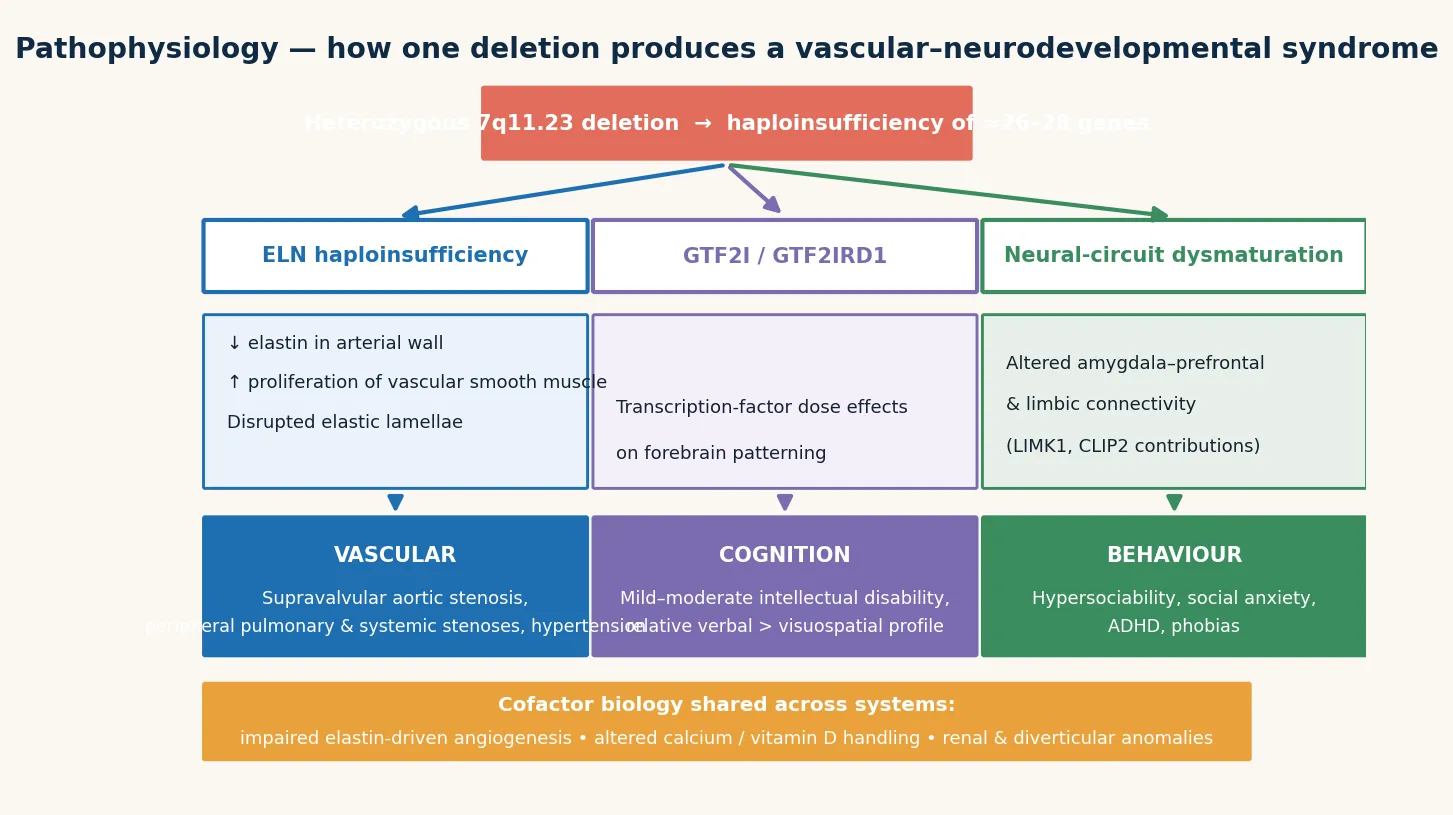
The neurocognitive phenotype is driven by the neighbouring genes. GTF2I and GTF2IRD1 are transcription factors whose haploinsufficiency contributes to the intellectual disability, the characteristic craniofacial gestalt, and the hypersocial, over-friendly personality that is so distinctive. LIMK1, a kinase regulating the actin cytoskeleton in neurons, contributes to the visuospatial construction deficit that defines the Williams syndrome cognitive profile — the striking dissociation between relatively preserved verbal ability and markedly impaired drawing, construction, and spatial reasoning. Other candidate genes, including BAZ1B (cardiac and growth), CLIP2, and FKBP6, add to the integrated phenotype. [2]
The infantile hypercalcaemia is less well explained mechanistically but is clinically important. It reflects abnormal vitamin D and calcium metabolism and is usually transient, peaking in infancy and often resolving, but it can occasionally be severe enough to cause dehydration, irritability, feeding difficulty, failure to thrive, and nephrocalcinosis. The metabolic disturbance is the rationale for screening calcium in infancy and restricting calcium and vitamin D intake when it is elevated. [6] [1]
Clinical Presentation
The presentation is age-dependent, and the diagnosis is often made in infancy when the distinctive facies is recognised alongside a cardiac murmur and failure to thrive with irritability or colic. The craniofacial gestalt includes bitemporal narrowing, periorbital fullness with puffiness around the eyes, a stellate or lacy iris pattern (especially in blue-eyed infants), a short upturned nose with a flat nasal bridge, a long philtrum, a wide mouth with full lips and full cheeks, and a small chin. The obsolete term elfin facies is avoided; the features are described as distinctive or characteristic. [1] [8]
The cardiovascular disease is the central clinical concern. Supravalvular aortic stenosis (SVAS) is present in approximately 60 to 75 percent and is the hallmark lesion — a narrowing of the ascending aorta just above the valve that produces a loud, harsh ejection systolic murmur radiating to the neck and sometimes a carotid bruit. Peripheral pulmonary artery stenosis is also common, particularly in infancy, and may produce a continuous murmur over the lung fields. Critically, the coronary arteries may be narrowed by the same arteriopathy, and coronary ostial or mid-vessel stenosis is the substrate for sudden cardiac death, particularly under anaesthesia, with exertion, or during catheter manipulation. Systemic hypertension is common and reflects involvement of the aorta and renal arteries. [3] [4]
The neurodevelopmental phenotype is unmistakable but deceptive. Children have mild to moderate intellectual disability (typical IQ around 50 to 60) with a characteristic cognitive profile: relatively strong verbal and auditory-rote memory skills, good face recognition and musical affinity, but a marked deficit in visuospatial construction — the child who can talk fluently but cannot assemble a simple puzzle or draw a coherent figure. The hypersocial personality is the most celebrated feature: these children are over-friendly, socially fearless, and unusually empathic, greeting strangers with disarming warmth, but this coexists with anxiety, specific phobias, attention-deficit hyperactivity disorder, and difficulty interpreting social intent despite the surface sociability. Hyperacusis (sensitivity to loud sound) is common and distressing. [1] [2]
The multisystem features complete the picture: infantile colic, feeding difficulty, gastro-oesophageal reflux and chronic constipation; hypothyroidism and early puberty; short stature (best tracked on the Williams-specific growth chart); joint laxity in early childhood giving way to contractures, scoliosis, and kyphosis later; renal anomalies including bladder diverticula and enuresis; dental anomalies (microdontia, malocclusion, enamel hypoplasia); strabismus and refractive errors; and chronic otitis media with effusion. Growth is often restricted prenatally as well as postnatally. [1] [8]
Differential Diagnosis
The differential is the differential of a dysmorphic child with a cardiac murmur, developmental delay, and a distinctive facies, and the chromosomal microarray resolves most of it. The single most important distinction is familial isolated supravalvular aortic stenosis, caused by an ELN point mutation or a deletion restricted to the ELN region: the arteriopathy is present but the facies and cognition are normal, so the developmental prognosis and counselling are entirely different. [5]
From the other microdeletion and dysmorphism syndromes — Smith-Magenis, deletion 1p36, and the RASopathies — Williams syndrome is distinguished by the specific combination of supravalvular aortic stenosis, hypercalcaemia, the distinctive facies, and the hypersocial cognitive profile, and confirmed by the chromosomal microarray demonstrating the 7q11.23 deletion. When the clinical picture fits and the microarray is normal, targeted FISH or MLPA for the 7q11.23 region should be considered to detect an atypical deletion that the array may have missed. [1] [2]
Clinical & Bedside Assessment
The bedside assessment is a structured multisystem examination that converts pattern recognition into a staged investigation and referral plan. Begin with growth — measure length, weight, and head circumference and plot on the Williams-specific growth chart, because general charts over-diagnose failure to thrive. Take a careful perinatal and developmental history: prenatal growth restriction, infantile colic and irritability, feeding difficulty, the timing of milestones, and a three-generation family history of cardiac disease, learning difficulty, or a known deletion. [1] [8]
The cardiovascular examination is the highest-yield bedside manoeuvre. Listen for the harsh ejection systolic murmur of supravalvular aortic stenosis radiating to the neck, the continuous murmur of peripheral pulmonary stenosis over the lung fields, and any bruits. Measure blood pressure in all four limbs: the arteriopathy can produce inter-limb gradients and involve the renal arteries, so a four-limb reading screens for aortic and renovascular disease simultaneously. A difference between upper- and lower-limb pressure raises coarctation or aortic involvement, and hypertension mandates renal-artery evaluation. [3] [4]
Examine for the connective-tissue features — joint laxity, herniae, soft skin, rectal prolapse — and the ophthalmic, dental, and auditory findings (strabismus, stellate iris, dental anomalies, otitis media with effusion). Assess the developmental and behavioural profile formally: the cognitive strengths (verbal, musical, face recognition) and the weaknesses (visuospatial construction, numeracy, daily-living skills), the hypersocial manner, and the anxiety, attention, and hyperacusis that shape the support plan. A neurodiversity-affirming, strength-based framing builds rapport and documents the real functional profile. [1] [2]
Investigations
The molecular investigation confirms the deletion, and the cardiovascular and metabolic work-up stages the disease. Chromosomal microarray is the first-line molecular test: it detects the 7q11.23 deletion, defines its size, and screens the rest of the genome for other copy-number variants in a dysmorphic child. Fluorescence in situ hybridisation (FISH) with a probe for the 7q11.23 region was the historical gold-standard confirmatory test and remains useful for family cascade testing and for confirming an atypical deletion, and MLPA is an alternative targeted method. A conventional karyotype will miss the submicroscopic deletion and should not be relied upon. [1] [2]
Why microarray has superseded karyotype and when FISH still matters
The 7q11.23 deletion is submicroscopic — below the resolution of conventional karyotype banding — so a karyotype returns normal and lulls the clinician into a false sense of security. Chromosomal microarray is the first-line test because it detects copy-number loss at this resolution and across the genome. FISH is retained for two specific roles: confirming a microarray finding rapidly for counselling, and testing parents and at-risk relatives in cascade testing, where a targeted probe is cheaper and faster than a whole-genome array. [1] [5]
The cardiovascular work-up is mandatory at diagnosis and serially. Echocardiography defines the supravalvular aortic stenosis (and its Doppler gradient), the peripheral pulmonary artery stenosis, the coronary artery origins, ventricular hypertrophy, and any other lesion. Because the coronary arteries are the most dangerous and the least visible on transthoracic echo, CT coronary angiography or MR angiography is increasingly considered when coronary stenosis, ostial narrowing, or symptoms are suspected, and before any planned anaesthesia or intervention. [3] [4]
The metabolic and endocrine work-up screens for the predictable complications: serum calcium (and ionised calcium where available), vitamin D, renal function and electrolytes, thyroid function, and fasting glucose. Renal and bladder ultrasound looks for structural anomalies, and audiology assesses both hyperacusis and the hearing loss that can accompany it. Baseline ophthalmology, dental, and a developmental or cognitive assessment complete the diagnostic staging. [6] [1]
Management — Resuscitation
Resuscitation in Williams syndrome centres on the two acute threats: critical cardiovascular obstruction or coronary ischaemia, and severe symptomatic hypercalcaemia. A child who presents with chest pain, syncope, exercise intolerance, or collapse is treated as a cardiac emergency: oxygen, cardiac monitoring, an ECG looking for ischaemia, and urgent cardiology referral, with a low threshold for advanced imaging of the coronary arteries and outflow tract. Critically, no sedation, anaesthesia, or procedural stimulation should be undertaken until the coronary and outflow status is defined, because sudden death under anaesthesia is a recognised and feared event. [3] [7]
Severe symptomatic hypercalcaemia in infancy presents with dehydration, vomiting, polyuria, irritability, and failure to thrive. Management is intravenous hydration to promote calciuresis, withdrawal of calcium and vitamin D supplementation, and a low-calcium formula where indicated; in the rare hypercalcaemic crisis, intravenous bisphosphonates and corticosteroids have been used under specialist metabolic guidance. Most hypercalcaemia is milder and resolves with dietary restriction and monitoring, but the severe end is a metabolic emergency. [6] [1]
Symptomatic hypertension from the arteriopathy and renovascular involvement is lowered in a controlled, gradual fashion — aggressive reduction risks ischaemia — and investigated for a renal-artery cause. In all acute presentations, the underlying principle is that Williams syndrome changes the threshold to act and the safety of intervention: assume coronary and outflow disease until proven otherwise, and treat any anaesthetic or procedure as high-risk. [3] [7]
Management — Definitive & Stepwise
Definitive management is a multidisciplinary health-supervision framework anchored to the AAP 2020 clinical report: confirm the molecular diagnosis, stage the cardiovascular disease, screen for and manage hypercalcaemia, and assemble developmental, endocrine, renal, sensory, and dental surveillance that travels with the child for life. No step cures the deletion, but each changes the trajectory — and cardiac surveillance is the step that saves the life. [1]
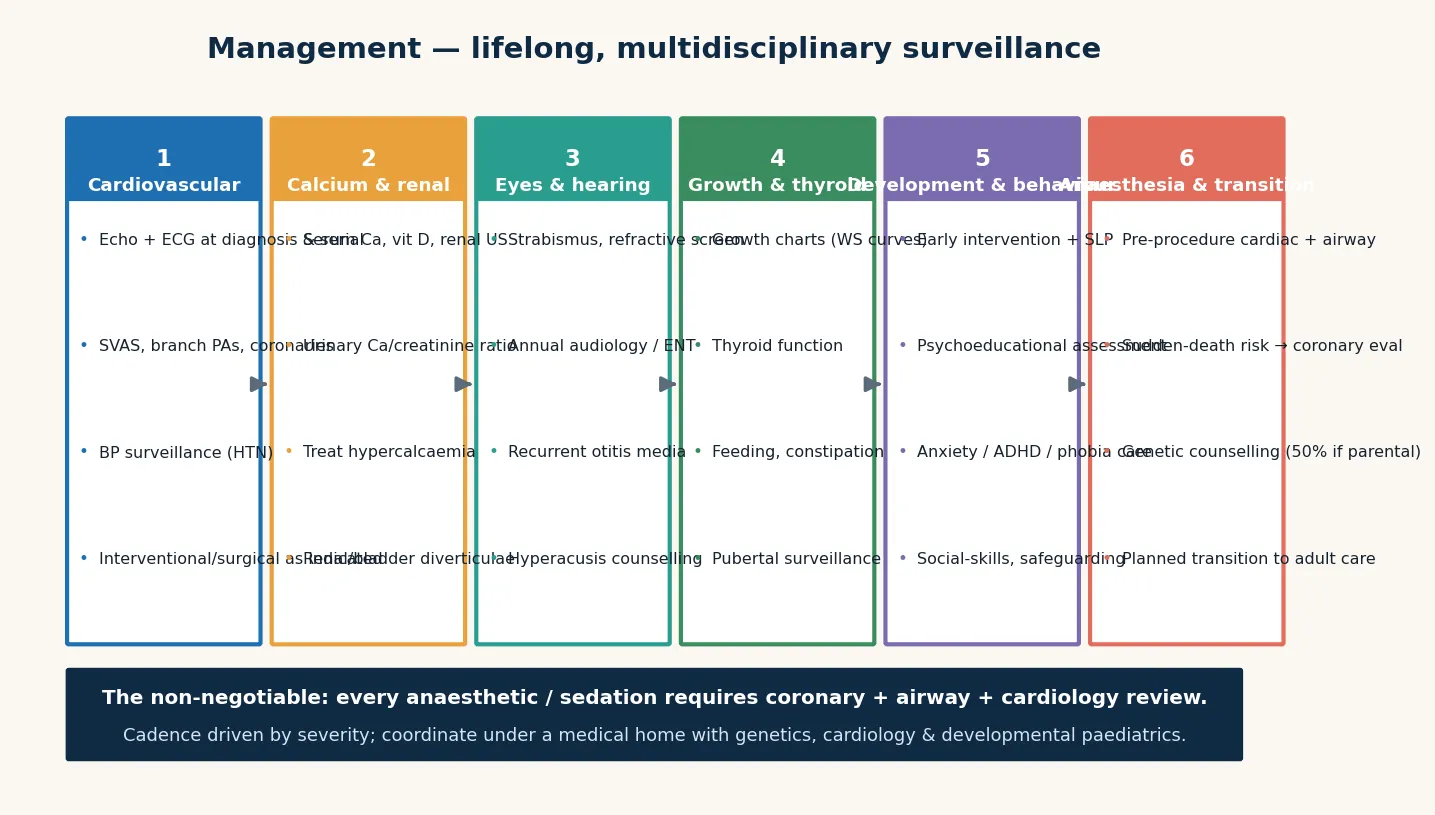
Cardiovascular surveillance is cardiology-led and lifelong. A baseline echocardiogram at diagnosis defines the anatomy, and serial echocardiography tracks the supravalvular aortic stenosis gradient and peripheral pulmonary stenosis. Blood pressure is monitored at every visit and investigated when elevated. Surgical or catheter intervention is considered for progressive or high-gradient supravalvular aortic stenosis (the classic operation is an aortic-widening patch angioplasty, sometimes called the brom three-patch repair), for significant peripheral pulmonary stenosis, and for coronary lesions that threaten ischaemia. Because the arteriopathy is progressive, the heart is never "cleared" — it is reassessed. [3] [4]
Infantile hypercalcaemia is managed with dietary restriction of calcium and vitamin D, avoidance of unnecessary supplementation, and periodic monitoring of serum calcium in the first years; a low-calcium formula is used when levels are elevated. The aim is to prevent nephrocalcinosis, irritability, and failure to thrive without inducing osteopenia from over-restriction. Thyroid replacement treats hypothyroidism, laxatives and fibre manage the chronic constipation, and early puberty is managed endocrinologically when it is distressing or compromises final height. [6] [1]
Developmental and educational support is shaped around the cognitive profile: leverage the verbal, musical, and social strengths while explicitly supporting the visuospatial, numeracy, and daily-living deficits. Early intervention, speech and language therapy (targeting receptive and pragmatic language, not just articulation), occupational therapy for motor and daily-living skills, and an individualised education plan anchor the school years. Anxiety, specific phobias, and ADHD are treated with behavioural strategies first and pharmacotherapy when needed, titrated carefully because medication sensitivity can occur. Hyperacusis is managed with hearing protection and environmental adaptation. [1]
C.A.R.D.S. — the Williams syndrome examination and surveillance set
Specific Subtypes & Scenarios
The infant presenting with failure to thrive, colic, and a murmur is the classic diagnostic scenario. The combination of irritability and feeding difficulty from hypercalcaemia, a distinctive facies, and the murmur of supravalvular aortic stenosis should prompt a calcium level, a four-limb blood pressure, an echocardiogram, and a chromosomal microarray. Recognition in infancy unlocks the entire surveillance pathway at the age when cardiac intervention and calcium management have the most impact. [1] [8]
The child with an atypical 7q11.23 deletion requires phenotype-driven counselling. A larger deletion predicts more severe intellectual disability, seizures, and autistic features, warranting more intensive developmental and neurological support. A smaller deletion sparing GTF2I may preserve cognition while retaining the cardiovascular burden — the child needs full cardiac surveillance but a different developmental prognosis. The microarray deletion size, interpreted with the genetics service, sets the expectation. [2]
The affected parent is the scenario that turns a sporadic case into a familial one. Because the deletion is autosomal dominant, an affected parent has a 50 percent recurrence risk per pregnancy, and the parent themselves needs the full cardiac, calcium, and developmental surveillance. When a deletion is identified in a child, assess both parents clinically and offer testing where there is any suspicion, and arrange formal genetic counselling including prenatal and preimplantation options for future pregnancies. [1]
The child with sudden cardiac symptoms or a family history of sudden death is the highest-stakes scenario. Coronary artery stenosis can present as exertional chest pain, syncope, or collapse, and it is the substrate for anaesthetic and sudden death. Advanced coronary imaging (CT or MR angiography) is considered when symptoms or high-risk anatomy are present, and the family is counselled about exercise restriction and the absolute necessity of pre-procedural cardiac assessment. [3] [4]
Complications & Pitfalls
The complications divide into the cardiovascular events that kill, the metabolic and multisystem problems that accumulate, and the cognitive traps that cost marks. The cardiovascular complications are dominant: supravalvular aortic stenosis can progress to left-ventricular outflow obstruction and heart failure; coronary artery stenosis causes ischaemia and sudden death, especially under anaesthesia and with exertion; and systemic and renovascular hypertension tracks the arteriopathy into adult life. The cardiac burden is the leading cause of the reduced life expectancy associated with the syndrome. [3] [4]
The chief cognitive trap is misreading the loquacity. The articulate, over-friendly manner of a child with Williams syndrome regularly convinces clinicians, teachers, and families that cognitive ability is higher than it is, and the result is a mismatch between expectation and support. The second trap is underestimating the coronary artery: a normal transthoracic echo at the valve level does not exclude coronary stenosis, and proceeding to anaesthesia on that basis is the single most dangerous procedural error. The third is missing the autosomal dominant inheritance when a parent is subtly affected, leaving the family without recurrence counselling. [2] [7]
Prognosis & Disposition
Prognosis is determined chiefly by the severity of the cardiovascular disease, the degree of intellectual disability, and the quality and timing of multidisciplinary support. Life expectancy is reduced compared with the general population, and the dominant contributor is cardiovascular disease and sudden cardiac death — which is precisely why cardiac surveillance is the centre of management. With structured cardiac, metabolic, and developmental care, many individuals achieve a good quality of life, supported employment, and meaningful social participation. [1] [3]
Intellectual outcome follows the characteristic profile: most individuals function in the mild to moderate intellectual disability range, with relative verbal and social strengths that can be leveraged in education and employment, provided the visuospatial, numeracy, and daily-living deficits are explicitly supported. Anxiety and phobias persist into adult life and shape the mental-health need. Hypertension and glucose intolerance become increasingly important in adulthood and require adult-care surveillance. [2]
Disposition is shared, lifelong, medical-home care: the general paediatrician or GP owns coordination and preventive care, cardiology owns the cardiac surveillance and the anaesthetic clearance, clinical genetics owns the counselling and cascade testing, and allied health and education own the developmental and behavioural support. Every transition — into school, into adolescence, and into adult services — is a point at which cardiac surveillance and developmental support can be lost, so the written, reconciled care plan must travel with the child. [1]
Special Populations
The same deletion behaves differently across populations because access, recognition, and service models are unevenly distributed. In remote and Indigenous communities, later presentation, limited cardiology and genetics access, and lower cascade-testing rates mean that the coronary and outflow disease may go unrecognised until a crisis, so culturally safe counselling and outreach or telehealth cardiology are essential. In migrant, refugee, and asylum-seeking families, language barriers, incomplete family histories, and fragmented prior records complicate the pedigree and the counselling, and a professional interpreter must be used at every key consultation. [1]
In families managing complex cardiac and developmental disability, fragmentation of care is the chief threat; a written, shared care plan reconciled at every visit is the intervention that prevents the loss of cardiac surveillance between services. In adolescents transitioning to adult care, the move is a high-risk point for loss of cardiology follow-up, endocrine surveillance, and mental-health support, so the transition plan must explicitly hand over the cardiac record and the surveillance schedule. The affected adult faces progressive hypertension, glucose intolerance, and anxiety, and requires adult cardiology and metabolic care that understands the arteriopathy. [4]
Throughout, the care should be neurodiversity-affirming and strength-based: the characteristic social warmth, verbal ability, and musical affinity of people with Williams syndrome are genuine strengths to be built upon, and the support plan should frame difference as difference, not deficit, while honestly addressing the real intellectual, cardiac, and metabolic needs. [1] [2]
Evidence, Guidelines & Regional Differences
The evidence base rests on consensus clinical guidelines, comprehensive reviews, and the cardiovascular and metabolic literature. The AAP 2020 health-supervision clinical report (Morris and colleagues, PMID 31964759) supersedes the original 2001 AAP guideline (PMID 11331709) and sets the current structure of surveillance, cardiac staging, calcium screening, and family counselling that most national programmes adopt. The Nature Reviews Disease Primers primer (Kozel and colleagues, PMID 34140529) is the definitive modern overview of the genetics, mechanism, and multisystem phenotype. [1] [2] [8]
The cardiovascular literature defines the surveillance and intervention framework. The Collins reviews (PMID 30045083, PMID 39291481) articulate the burden of supravalvular aortic stenosis, peripheral pulmonary stenosis, coronary disease, and hypertension, and the principles of cardiology-led lifelong surveillance and surgical intervention. The Merla elastin arteriopathy review (PMID 23250899) explains the ELN haploinsufficiency mechanism and the distinction from isolated familial supravalvular aortic stenosis. The Sindhar hypercalcaemia study (PMID 27574996) characterises the prevalence and course of hypercalcaemia, and the Twite anaesthesia review (PMID 30811742) defines the perioperative risk and the mandatory precautions. [3] [5] [6] [7]
In Australia and New Zealand, chromosomal microarray is publicly funded when clinical criteria are met, and clinical genetics services deliver counselling and cascade testing with access in metropolitan, regional, and (by outreach or telehealth) remote settings. Paediatric cardiology surveillance is coordinated through tertiary children's cardiac services, and the disability support frameworks (the NDIS in Australia, and the equivalent in New Zealand) fund the allied health and educational supports that anchor long-term care. Anaesthetic care for children with Williams syndrome is centralised at centres with dedicated paediatric cardiac anaesthesia because of the sudden-death risk. Always confirm the current local eligibility criteria for genetic testing, cardiac surveillance intervals, and disability funding, as these change. [1] [7]
Exam Pearls
A fellowship candidate answering on Williams syndrome should land five anchor points and avoid three classic traps. The anchors are the molecular definition (7q11.23 microdeletion, contiguous-gene haploinsufficiency), the elastin arteriopathy (supravalvular aortic stenosis, coronary stenosis, hypertension, and the sudden-death risk under anaesthesia), the cognitive profile (verbal and social strength with visuospatial deficit and real intellectual disability), the infantile hypercalcaemia, and the autosomal dominant inheritance with a 50 percent recurrence risk when a parent is affected. The traps are misreading the loquacity, underestimating the coronary artery, and missing the inheritance. [1] [3]
References
- [1]Morris CA, Braddock SR, Van Bavel-Hintzen PH, et al. Health Care Supervision for Children With Williams Syndrome. Pediatrics, 2020.PMID 31964759
- [2]Kozel BA, Barak B, Kim J, et al. Williams syndrome. Nat Rev Dis Primers, 2021.PMID 34140529
- [3]Collins RT 2nd. Cardiovascular disease in Williams syndrome. Curr Opin Pediatr, 2018.PMID 30045083
- [4]Collins RT 2nd, Beesley A, Crum K, et al. Clinical Care for Cardiovascular Disease in Patients With Williams-Beuren Syndrome. J Am Heart Assoc, 2024.PMID 39291481
- [5]Merla G, Brunetti-Pierri N, Piccolo P, Micale L, de Vries BB. Supravalvular aortic stenosis: elastin arteriopathy. Circ Cardiovasc Genet, 2012.PMID 23250899
- [6]Sindhar S, Lugo M, Xin MingShu, et al. Hypercalcemia in Patients with Williams-Beuren Syndrome. J Pediatr, 2016.PMID 27574996
- [7]Twite MD, Friesen RH. Williams syndrome. Paediatr Anaesth, 2019.PMID 30811742
- [8]Committee on Genetics. American Academy of Pediatrics: Health care supervision for children with Williams syndrome. Pediatrics, 2001.PMID 11331709