Paeds · haematology-oncology-and-transfusion
Aplastic anaemia and bone-marrow failure
Also known as Aplastic anaemia · Severe aplastic anaemia · Bone marrow failure · Acquired aplastic anaemia · Inherited bone marrow failure syndrome · Hypocellular marrow
Fellowship guide to aplastic anaemia and inherited bone marrow failure in children. Covers the Camitta criteria for severe aplastic anaemia of marrow cellularity under 25 percent with two of three peripheral blood thresholds of neutrophils under 0.5 times ten to the nine per litre, platelets under 20 times ten to the nine per litre, and reticulocytes under 60 times ten to the nine per litre, the immune-mediated pathophysiology of cytotoxic T cell destruction of the haematopoietic stem cell, the inherited syndromes of Fanconi anaemia diagnosed by the diepoxybutane chromosomal breakage test, Diamond-Blackfan anaemia, dyskeratosis congenita, and Shwachman-Diamond syndrome, and the two definitive treatments of allogeneic haematopoietic stem cell transplant for the child with a matched sibling donor and immunosuppressive therapy of horse antithymocyte globulin at 40 mg per kg per day for four days with ciclosporin at 5 mg per kg per day and eltrombopag per the RACE trial for the child without one.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A marrow that falls silent, that empties itself of the cells it is meant to make, produces one of the most striking pictures in paediatric haematology. Aplastic anaemia is the syndrome in which the haematopoietic stem cell, the seed of every red cell, white cell, and platelet, is destroyed or fails to function, leaving a marrow that is fatty and empty and a blood count that is low in all three lineages at once. The child arrives pale, bruised, and bleeding, and the blood film shows pancytopenia, the simultaneous fall of the haemoglobin, the neutrophil count, and the platelet count. The bone marrow biopsy, the test that settles the direction, shows a hypocellular marrow with fat where blood-making tissue should be. [2]
The term bone marrow failure is the umbrella under which aplastic anaemia sits, and it carries the central distinction the fellow must hold. The failure is either acquired, in which a previously healthy marrow is injured, most often by an aberrant immune attack on the stem cell, or it is inherited, in which a genetic defect of the stem cell or its DNA repair sets the marrow up to fail over the years of childhood. Acquired aplastic anaemia is the more common, and it is largely an immune disease that answers to immunosuppression. The inherited syndromes, led by Fanconi anaemia, are rarer, they carry extra physical signs, and they change both the treatment and the counselling. [9]
Three ideas make this topic central to the fellowship exam. The first is the Camitta criteria, the numerical thresholds that define severe disease and that decide who is treated aggressively and who is watched. The second is the treatment fork, between the haematopoietic stem cell transplant offered to the child with a matched sibling donor and the immunosuppressive therapy offered to the child without one. The third is the inherited syndrome, especially Fanconi anaemia, whose diagnosis by the chromosomal breakage test and whose cancer risk shape the whole life of the family. The 2024 British Society for Haematology guideline of Kulasekararaj and colleagues is the current standard reference. [8]
Classification
Aplastic anaemia is classified first by severity, because severity decides the treatment, and the severity grading rests on the Camitta criteria of 1976. Severe aplastic anaemia is defined by a bone marrow cellularity under 25 percent, or a cellularity between 25 and 50 percent with under 30 percent residual haematopoietic cells, together with at least two of three peripheral blood thresholds. The three thresholds are a neutrophil count under 0.5 times ten to the nine per litre, a platelet count under 20 times ten to the nine per litre, and a corrected reticulocyte count under 60 times ten to the nine per litre. Very severe aplastic anaemia is reserved for the child whose neutrophil count falls under 0.2 times ten to the nine per litre, the group at highest risk of overwhelming sepsis. [1]
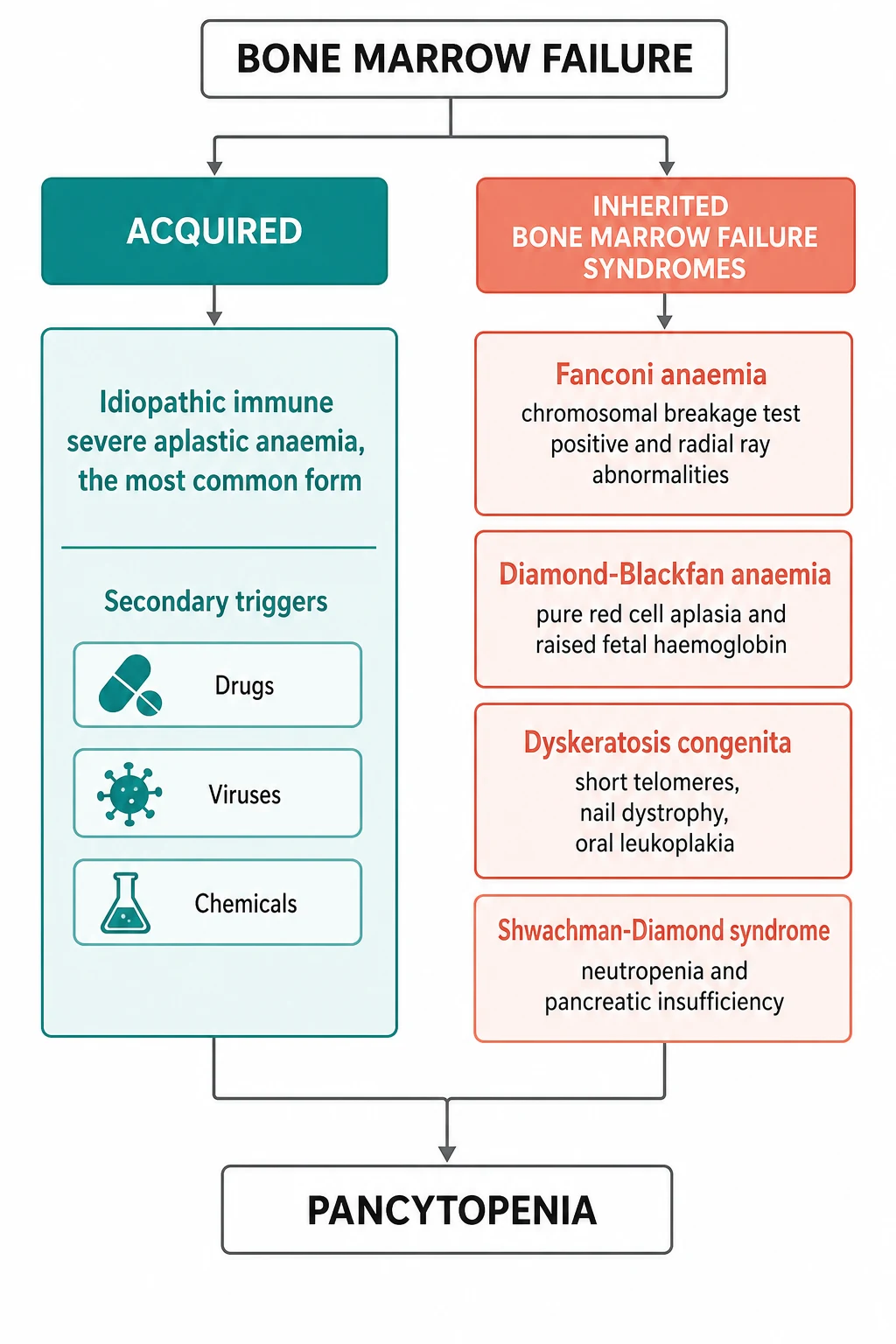
The second axis of classification runs alongside severity and asks whether the disease is acquired or inherited, because the answer changes the prognosis, the treatment, and the counselling. Acquired aplastic anaemia is the more common form in the developed world, and in most children no cause is found, so it is called idiopathic. A minority of cases follow a recognised trigger, the chief of which are the drug exposures, the viral infections, and the chemical or toxin exposures. The inherited bone marrow failure syndromes are the rarer but the more consequential group, because they carry physical signs, cancer risk, and implications for the whole family. Fanconi anaemia is the prototype, and it is joined by Diamond-Blackfan anaemia, the telomere biology disorders once called dyskeratosis congenita, and Shwachman-Diamond syndrome. [9]
A third, functional distinction matters at the bedside and it is the one the fellow must not miss. Aplastic anaemia is a failure of production, which means the marrow is empty and the blood film shows low counts with normal-looking cells. This stands against the destructional causes of pancytopenia, such as leukaemia that fills the marrow, or hypersplenism that traps the cells, or the infections that consume them. The empty marrow of aplastic anaemia is the finding that sends the fellow down the pathway this topic describes, and the marrow full of blasts is the finding that sends the child down a different one. [8]
Epidemiology & Risk Factors
Acquired aplastic anaemia is a rare disease, with an incidence in the Western world of about two new cases per million people each year, and it is two to three times more common in East Asia. The epidemiological study of Young and Kaufman established the age distribution, which is bimodal, with one peak in the teenage years and the early twenties and a second in older adults. The paediatric and adolescent peak is the one the fellow meets, and it reflects the immune origin of the disease, which favours the young immune system that is most prone to the autoreactive T cell response that drives the marrow failure. [2]
The risk factors for acquired aplastic anaemia are the triggers that, in a susceptible child, set off the immune attack on the stem cell. The drug exposures are the best known, and they include the chloramphenicol that is now rare in paediatric practice, the non-steroidal anti-inflammatory drugs, the antiepileptics, and the chemotherapy agents. The viral triggers include the seronegative hepatitis that precedes a minority of cases, the Epstein-Barr virus, the cytomegalovirus, and the parvovirus B19 that more often causes a pure red cell aplasia. The toxin and chemical exposures, including the benzene and the pesticides, are the occupational and environmental triggers that matter in some regions. Most cases, however, have no identifiable cause, which is why the term idiopathic dominates. [2]
The risk factors for the inherited syndromes are genetic, and they are the mutations the child is born with. Fanconi anaemia is autosomal recessive, caused by mutations in any of the over twenty FANC genes that govern the repair of DNA cross-links, and it is the most common of the inherited marrow failure syndromes. Diamond-Blackfan anaemia is caused by mutations in the ribosomal protein genes, most often RPS19, and is largely autosomal dominant. The telomere biology disorders are caused by mutations in the telomerase genes TERT and TERC or the shelterin genes, and they may be autosomal dominant or recessive. A family history of early marrow failure, of early greying, or of pulmonary fibrosis points to a telomere disorder and changes the counselling for the relatives. [9]
Pathophysiology
The marrow of the child with acquired aplastic anaemia is empty, and the question is what emptied it. The answer, worked out over decades and built on the observation that the disease answers to immunosuppression, is that the haematopoietic stem cell is destroyed by an aberrant immune attack. A population of oligoclonal cytotoxic T cells expands, and these T cells release cytokines, chiefly interferon-gamma and tumour necrosis factor-alpha, that turn on the death machinery of the stem cell. The stem cell carries the Fas receptor, and when the cytokines bind, the Fas pathway fires and the cell undergoes programmed death. The marrow is left fatty and empty, and the blood counts fall in all three lineages. [2]
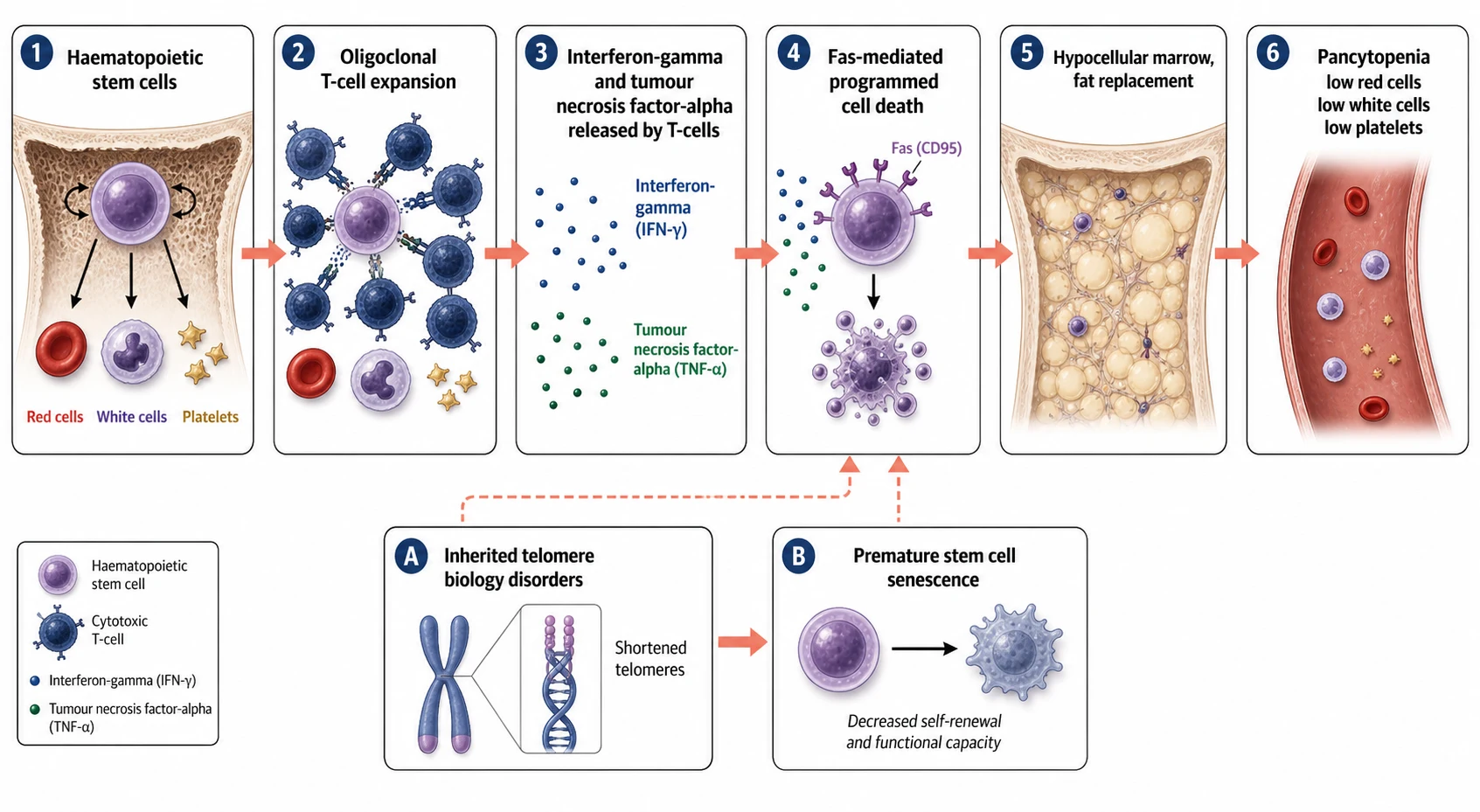
The immune attack is the reason the disease answers to antithymocyte globulin and ciclosporin, the two drugs that form the backbone of immunosuppressive therapy. Antithymocyte globulin is a polyclonal antibody preparation that depletes the T cells, and ciclosporin blocks the calcineurin pathway that drives the T cell activation. The two together suppress the autoreactive clone and allow the surviving stem cells to recover, which is why the response to immunosuppression is itself the strongest evidence that the disease is immune-mediated. The cytokine interferon-gamma is also the reason that eltrombopag, the thrombopoietin receptor agonist, is added, because it directly stimulates the surviving stem cells to divide and make platelets and, in time, the other lineages. [4]
The inherited marrow failure syndromes fail by a different mechanism, and the fellow must hold the two apart. In Fanconi anaemia, the defective DNA cross-link repair means that the stem cell accumulates chromosomal damage every time it divides, so it senesces and dies and the marrow empties over the years of childhood. The diepoxybutane test, which adds a cross-linking agent to the child's cells in the laboratory, shows the chromosomes breaking and forming the radial figures that are the hallmark of the disease. In the telomere biology disorders, the stem cell runs out of telomere, the cap that protects the chromosome end, because the telomerase that should renew it is defective, so the cell stops dividing after too few divisions. Both pathways end in the same hypocellular marrow, but the route there is genetic rather than immune. [9]
Clinical Presentation
The child with aplastic anaemia presents with the consequences of the falling counts, and the presentation reflects which lineage falls first and fastest. The most common first symptom is bleeding, because the platelet count falls early and steeply, and the child presents with bruising that is out of proportion to the trauma, with petechiae on the lower limbs, with epistaxis, with gum bleeding, or with menorrhagia in the adolescent girl. The pallor and the tiredness of the anaemia follow, and the parents describe a child who has slowed down, who is short of breath on exercise, and who looks washed out. The fever and the mucosal ulcers of the neutropenia bring the child in last and most urgently, because the neutropenic child can be septicaemic within hours. [2]
The inherited syndromes declare themselves with the physical signs that the fellow elicits at the bedside, and these signs are the reason the examination of the pancytopenic child is so important. The child with Fanconi anaemia is short, with café-au-lait spots and hypopigmented areas, with radial ray abnormalities of the absent, hypoplastic, or triphalangeal thumbs and the absent or hypoplastic radii, with renal anomalies, and with microcephaly and the characteristic facies. The child with Diamond-Blackfan anaemia is short, with the craniofacial anomalies of the cleft or high palate and the hypertelorism, and occasionally with the thumb anomalies of the triphalangeal thumb. The telomere biology disorders show the nail dystrophy, the oral leukoplakia, and the abnormal reticular skin pigmentation of the neck and chest. [9]
The tempo of the presentation differs between the acquired and the inherited disease, and the fellow uses it as a clue. The acquired disease comes on over weeks to a few months, in a previously well child, and the counts fall together. The inherited syndrome comes on more slowly, often over years, and it may be preceded by a long history of short stature, of congenital anomalies, or of a single cytopenia such as the macrocytosis that precedes the full marrow failure in Fanconi anaemia and the telomere disorders. A raised fetal haemoglobin and a raised mean corpuscular volume in a child with a falling count are the quiet signs of an inherited syndrome, and they should prompt the genetic testing before any transplant is planned. [11]
Differential Diagnosis
The pancytopenic child with a hypocellular marrow has aplastic anaemia, but the fellow must separate it from the other causes of pancytopenia, and the marrow biopsy is the test that sets the direction. A marrow full of blasts is acute leukaemia, and it is the diagnosis the fellow most fears and most needs to exclude. A marrow infiltrated by the Langerhans cells, by the storage cells of a metabolic disease, or by the fibrous tissue of myelofibrosis is a marrow infiltration syndrome. A marrow that looks empty but is in fact trapped in the spleen is hypersplenism, and the spleen is enlarged on examination. The empty marrow of aplastic anaemia is distinct from all of these, and it is the finding that sends the child down the pathway of this topic. [8]
Acquired aplastic anaemia
hypocellular marrow, immune
- Pancytopenia with a hypocellular marrow under 25 percent
- No congenital anomalies, previously well child
- Answers to antithymocyte globulin and ciclosporin
- Negative diepoxybutane chromosomal breakage test
Fanconi anaemia
inherited, DNA repair defect
- Autosomal recessive, FANC gene mutations
- Radial ray anomalies, cafe-au-lait spots, short stature
- Positive diepoxybutane test with radial chromosomal figures
- High cancer risk of MDS, AML, and head and neck carcinoma
Diamond-Blackfan anaemia
ribosomal protein mutation
- Pure red cell aplasia in infancy, raised fetal haemoglobin
- Craniofacial anomalies, triphalangeal thumb
- RPS19 and other ribosomal protein gene mutations
- Corticosteroid responsive in many, transfusion dependent in others
Telomere biology disorders
short telomeres, dyskeratosis
- Nail dystrophy, oral leukoplakia, reticular pigmentation
- Short telomeres on the flow cytometry assay
- TERT, TERC, and shelterin gene mutations
- Family history of early greying, pulmonary fibrosis, or marrow failure
The single most important distinction within the hypocellular marrow group is between acquired aplastic anaemia and Fanconi anaemia, because the treatment differs. A child with Fanconi anaemia is transplanted with a reduced-intensity, fludarabine-based conditioning regimen, because the DNA repair defect makes the child exquisitely sensitive to the chemotherapy and radiation of a standard transplant regimen. A child with acquired aplastic anaemia is transplanted with a cyclophosphamide-based regimen. The diepoxybutane test must therefore be done and be negative before any transplant conditioning begins, because the consequences of giving a standard regimen to a child with Fanconi anaemia can be fatal. [9]
The other diagnosis the fellow must not miss is the hypocellular myelodysplastic syndrome, which can mimic aplastic anaemia early in its course. The clues to a myelodysplastic syndrome are the dysplastic cells on the blood film, the clonal cytogenetic abnormality on the marrow, and the presence of blasts even in small numbers. Flow cytometry and the cytogenetics on the marrow biopsy help separate the two, because aplastic anaemia has a normal karyotype and no clonal population, and the myelodysplastic syndrome does not. The distinction matters because the treatment and the prognosis diverge, and the fellow who treats a myelodysplastic syndrome with immunosuppression loses time. [8]
Clinical & Bedside Assessment
The assessment of the pancytopenic child begins at the bedside with a search for the severity, the cause, and the inherited syndrome. The history asks about the onset and the tempo of the bleeding, the pallor, and the infections, and about the drug, the viral, and the chemical exposures that might have triggered an acquired disease. A family history of marrow failure, of congenital anomalies, of early greying, or of pulmonary fibrosis points to an inherited syndrome, and a consanguineous family raises the chance of an autosomal recessive Fanconi anaemia. The growth chart is reviewed, because the short stature of an inherited syndrome often predates the marrow failure. [9]
The examination looks for the signs of the falling counts and for the stigmata of the inherited syndromes. The skin is inspected for the bruising, the petechiae, and the pallor of the acquired disease, and for the café-au-lait spots, the hypopigmentation, and the reticular pigmentation of Fanconi anaemia and the telomere disorders. The hands are examined for the radial ray anomalies, the triphalangeal thumb, and the nail dystrophy. The mouth is inspected for the oral leukoplakia and the high or cleft palate, the abdomen for the spleen and the liver, and the head for the microcephaly and the craniofacial anomalies. A thorough dysmorphology examination of every pancytopenic child is the act that finds the inherited syndrome. [9]
The structured assessment of the pancytopenic child
Take the history of onset, drug and viral exposures, and the family history of marrow failure, early greying, or pulmonary fibrosis
Plot the growth and review the congenital anomalies, because short stature predates the marrow failure in the inherited syndromes
Examine the skin for bruising, petechiae, café-au-lait spots, and pigmentation, and the hands for radial ray and nail anomalies
Order the full blood count, the film, the reticulocyte count, and the blood group, and send the viral and the autoimmune screens
Book the bone marrow aspirate and trephine with cytogenetics, and the diepoxybutane chromosomal breakage test
Start the supportive care of transfusion, antibiotics, and bleeding precautions while the results return
The severity is graded from the blood count, and the grading is the act that decides the treatment. The fellow calculates the absolute neutrophil count, reads the platelet count, and checks the corrected reticulocyte count, and the child who meets two of the three Camitta thresholds with a hypocellular marrow has severe disease. The child who meets none of the thresholds has non-severe disease and is often managed supportively and observed. The child whose neutrophil count is under 0.2 times ten to the nine per litre has very severe disease, and this is the group that carries the highest risk of sepsis and that is treated most urgently. [1]
Investigations
The diagnosis of aplastic anaemia rests on the bone marrow biopsy, and it is the test the fellow must both request and interpret. The aspirate shows a hypocellular marrow with fat spaces and scattered lymphocytes and plasma cells, and the trephine biopsy confirms the low cellularity, which is measured against the age of the child and is reported as a percentage. A cellularity under 25 percent, or between 25 and 50 percent with under 30 percent residual haematopoietic cells, meets the marrow criterion of severe aplastic anaemia. The cytogenetics on the marrow are normal, and a clonal abnormality redirects the diagnosis toward a myelodysplastic syndrome. [8]
The peripheral blood confirms the pancytopenia and excludes the other causes. The full blood count shows the low haemoglobin, the low neutrophil count, and the low platelet count, and the blood film shows the absence of the blasts and the dysplasia that would point to leukaemia or myelodysplasia. The reticulocyte count is inappropriately low for the degree of anaemia, which is the hallmark of a production failure. The mean corpuscular volume may be raised, a clue to an inherited syndrome or to the recovering marrow, and the fetal haemoglobin may be raised in Diamond-Blackfan anaemia and in the telomere disorders. [11]
The diepoxybutane chromosomal breakage test is the test that confirms or excludes Fanconi anaemia, and it is done on a sample of the child's peripheral blood lymphocytes in a specialist cytogenetics laboratory. The test adds the cross-linking agent to the cells and counts the chromosomal breaks and the radial figures, which are markedly increased in Fanconi anaemia and normal in the acquired disease. The test is available in the major paediatric centres in Australia, Aotearoa New Zealand, the United Kingdom, the United States, and Canada, and the fellow should know the local referral pathway, because a positive result changes the transplant conditioning regimen.
[9]The molecular testing has a growing place, and the next-generation sequencing panel for the inherited bone marrow failure genes is now part of the workup of every child with a hypocellular marrow. The panel sequences the FANC genes, the ribosomal protein genes, the telomerase and shelterin genes, and the genes of the rarer syndromes, and it confirms the inherited diagnosis when the breakage test or the telomere assay is borderline. The telomere length, measured by flow cytometry on the blood, is the test for the telomere biology disorders, and a length under the first percentile for age in a child with marrow failure is diagnostic. The HLA typing of the child and the siblings is done early, because the result decides between transplant and immunosuppression. [9]
Management — Resuscitation
The child with severe aplastic anaemia is at risk of the three acute complications of the low counts, and the fellow meets each with a clear plan before the definitive treatment begins. The first and the most urgent is the neutropenic fever, which in the child with a neutrophil count under 0.5 times ten to the nine per litre is a medical emergency. The response is a prompt blood culture and a parenteral broad-spectrum antibiotic that covers the gram-negative organisms and the pneumococcus, given within the hour, because the death from neutropenic sepsis is measured in hours. The child is admitted, the fever is investigated, and the antibiotic is continued until the cultures are negative and the count recovers. [8]
The second acute risk is the bleeding of the severe thrombocytopenia. The child with a platelet count under 10 times ten to the nine per litre, or under 20 times ten to the nine per litre with active bleeding or fever, is transfused with platelets to keep the count above the threshold. The red cell transfusion is given for the symptomatic anaemia, keeping the haemoglobin above 70 g per litre, or higher in the child with cardiac symptoms or with active bleeding. The transfusions are leucodepleted and irradiated throughout, and the child who may go on to a transplant is transfused sparingly and from a restricted donor pool to minimise the alloimmunisation that complicates the future cross-matching. [8]
[8]The third acute risk is the iron overload of the repeated transfusion, which builds up over the months of supportive care and which the fellow monitors with the ferritin. The iron is chelated when the ferritin exceeds 1000 micrograms per litre, with deferasirox as the usual first line agent in children. The prophylaxis against the opportunistic infections is added for the child on immunosuppressive therapy, with the antifungal and the antiviral cover for the prolonged neutropenia and the T cell suppression, because the invasive fungal infection is a leading cause of death in the treated child. The resuscitation and the supportive care are not the afterthought of the definitive treatment, they are its foundation. [8]
Management — Definitive & Stepwise
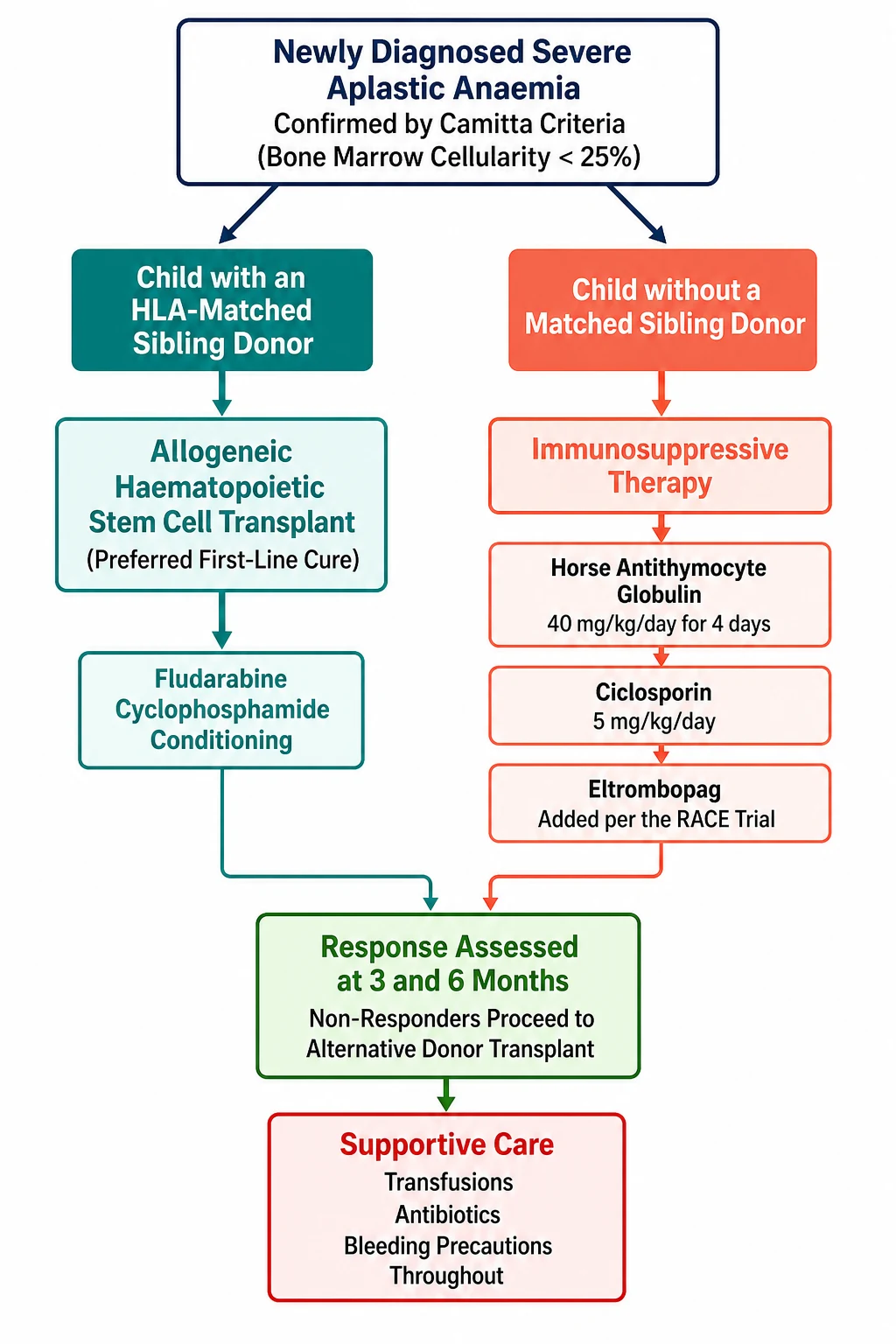
The definitive treatment of severe aplastic anaemia rests on a single decision that is made at diagnosis, and it is the decision that defines the whole topic. The child who has an HLA matched sibling donor is offered an allogeneic haematopoietic stem cell transplant as the first line treatment, because it is the only cure and it gives the best long term survival. The child who does not have a matched sibling donor is offered immunosuppressive therapy, the combination of antithymocyte globulin, ciclosporin, and eltrombopag, which suppresses the immune attack and allows the marrow to recover. The decision is made once, early, and on the basis of the HLA typing of the family. [8]
The haematopoietic stem cell transplant is the treatment of choice for the child with a matched sibling donor, and the survival in the modern era exceeds 90 percent in children. The conditioning regimen is based on cyclophosphamide, often with antithymocyte globulin added, and the stem cell source is the bone marrow rather than the peripheral blood, because the bone marrow source gives the lower rate of the chronic graft-versus-host disease that blights the long term survivor. The transplant is performed at a specialist centre, the donor is confirmed by the full HLA typing, and the family is counselled on the risks of the graft failure, the graft-versus-host disease, and the late effects. [1]
Haematopoietic stem cell transplant conditioning for acquired severe aplastic anaemia
Dose
Cyclophosphamide based regimen, typically 50 mg per kg per day for four days, with antithymocyte globulin added for the unrelated donor, using bone marrow as the stem cell source
The immunosuppressive therapy is the treatment for the child without a matched sibling donor, and it rests on the combination established by the German Aplastic Anemia Study Group in 1991. The randomised trial of Frickhofen and colleagues showed that the addition of ciclosporin to the antilymphocyte globulin and methylprednisolone improved the response rate and the survival, and the combination became the standard of care. The regimen is horse antithymocyte globulin at 40 mg per kg per day for four days, given by intravenous infusion, with ciclosporin at 5 mg per kg per day, adjusted to the trough level and the renal function, continued for at least six months. [3]
Standard immunosuppressive therapy for severe aplastic anaemia
Dose
Horse antithymocyte globulin 40 mg per kg per day intravenously for four days, with ciclosporin 5 mg per kg per day orally in two divided doses adjusted to a trough of 200 to 400 nanograms per millilitre, and eltrombopag added from day 14
The choice of the antithymocyte globulin matters, and the randomised trial of Scheinberg and colleagues in 2011 settled it. The trial compared the horse antithymocyte globulin with the rabbit antithymocyte globulin in the front line treatment of acquired aplastic anaemia, and the horse preparation was superior, with a response rate of 68 percent at six months against 37 percent for the rabbit. The horse preparation is therefore the first line choice, and the rabbit is reserved for the child who reacts to the horse, never substituted on the grounds of tolerability alone. The fellow who remembers this trial and the 68 percent response rate carries the key immunosuppressive fact. [4]
[4] [8]Eltrombopag is the modern addition to the immunosuppressive regimen, and it has changed the response rates. The trial of Olnes and colleagues in 2012 showed that eltrombopag, a thrombopoietin receptor agonist, improved the blood counts in the child with refractory aplastic anaemia, and the follow up work confirmed that it restored the trilineage haematopoiesis, not only the platelets. The RACE trial of Peffault de Latour and colleagues, published in the New England Journal of Medicine in 2022, then showed that adding eltrombopag to the front line immunosuppression improved the complete response rate at three months, and the eltrombopag is now a standard part of the front line regimen. The fellow monitors the child on eltrombopag for the clonal evolution, the emergence of a myelodysplastic clone, with the marrow cytogenetics at the regular intervals. [6][7]
The child who fails the first immunosuppressive course, or who relapses after a response, is offered the second line treatment, and the options are the alternative donor transplant or the second course of immunosuppression. The alternative donor transplant, once a poor option, has improved dramatically with the fludarabine based conditioning and the post-transplant cyclophosphamide, and the retrospective study of Bacigalupo and colleagues from the European Society for Blood and Marrow Transplantation showed the improved survival with the fludarabine, cyclophosphamide, and antithymocyte globulin regimen for the alternative donor. The matched unrelated donor, the haploidentical donor, and the umbilical cord blood are all now viable sources, and the choice rests on the HLA match, the cell dose, and the urgency. [12]
Eltrombopag in severe aplastic anaemia
Dose
Added to the front line immunosuppression from day 14, at a starting dose based on weight, 150 mg once daily for the child over six years, reduced in the East Asian population and in the hepatic impairment, for six months
The high dose cyclophosphamide without a transplant was once proposed as an alternative, and the randomised trial of Tisdale and colleagues in 2000 is the answer the fellow gives when asked about it. The trial compared the high dose cyclophosphamide with the antithymocyte globulin and ciclosporin, and it showed no benefit and a higher toxicity, with more fungal infections and a slower count recovery. The high dose cyclophosphamide is therefore not the standard of care, and the trial is the evidence the fellow cites to explain why. The cyclophosphamide remains the backbone of the transplant conditioning, but it is not a substitute for the immunosuppression in the non-transplanted child. [5]
Specific Subtypes & Scenarios
Fanconi anaemia is the inherited syndrome the fellow must know in detail, because it changes every part of the management. The diagnosis is made by the diepoxybutane chromosomal breakage test, which shows the increased breaks and the radial figures, and it is confirmed by the sequencing of the FANC genes. The child is short, with the café-au-lait spots and the radial ray anomalies, and the marrow failure usually declares itself in the first decade. The treatment of the marrow failure is the haematopoietic stem cell transplant, with a reduced intensity fludarabine based regimen that spares the child the toxicity of the standard cyclophosphamide. The transplant cures the marrow failure but it does not correct the DNA repair defect, so the child remains at the high risk of the solid tumours, chiefly the head and neck squamous cell carcinoma, for life. [9]
The cancer risk in Fanconi anaemia is the counselling that shapes the long term follow up, and the study of Alter and colleagues in 2003 quantified it. The cumulative incidence of the haematological malignancy, the myelodysplastic syndrome and the leukaemia, reaches about a third by the age of 40, and the incidence of the solid tumour, especially the squamous cell carcinoma of the head and neck and the gynaecological tract, rises steeply after the transplant and with the age. The child is therefore followed with the annual marrow cytogenetics and the regular screening for the solid tumours, and the avoidance of the tobacco and the alcohol and the human papillomavirus vaccination are the preventive measures the fellow reinforces at every visit. [10]
Diamond-Blackfan anaemia is the pure red cell aplasia of infancy, and the international consensus statement of Wlodarski and colleagues in 2024 is the current reference. The child presents in the first year of life with a severe macrocytic anaemia, a reticulocytopenia, and a normal neutrophil and platelet count, and the marrow shows the absence of the red cell precursors with the normal white cell and platelet production. The fetal haemoglobin is raised and the adenosine deaminase is elevated, and the ribosomal protein gene mutation, most often in the RPS19 gene, confirms the diagnosis. The treatment is the corticosteroid, to which about half the children respond, and the transfusion for the rest, with the iron chelation, and the transplant for the transfusion dependent child with a matched donor. [11]
The first years of a child with an inherited marrow failure syndrome
The telomere biology disorders, once grouped under dyskeratosis congenita, are the inherited syndromes of the short telomere, and they present with the classic triad of the nail dystrophy, the oral leukoplakia, and the abnormal skin pigmentation, together with the marrow failure, the pulmonary fibrosis, and the hepatic disease. The diagnosis rests on the very short telomere length on the flow cytometry assay and the mutation in the TERT, the TERC, or the shelterin genes. The transplant is offered for the marrow failure, but the child is also monitored for the pulmonary and the hepatic complications, and the family is screened because the autosomal dominant forms are common and the relatives may carry the mutation with a milder or a later presentation. [9]
Complications & Pitfalls
The first pitfall is the failure to exclude an inherited syndrome before the transplant, and it is the error with the gravest consequences. A child with Fanconi anaemia who is given the standard cyclophosphamide conditioning for an acquired aplastic anaemia suffers a severe and potentially fatal toxicity, because the DNA repair defect makes the child exquisitely sensitive to the chemotherapy and the radiation. The diepoxybutane chromosomal breakage test must therefore be done and be negative before any transplant conditioning begins, and the fellow who plans a transplant for a pancytopenic child holds the conditioning until the result returns. The test takes weeks, and the wait is the price of the safety. [9]
The second pitfall is the use of the rabbit antithymocyte globulin in place of the horse preparation. The rabbit preparation is more readily available and it is sometimes preferred for its tolerability, but the trial of Scheinberg and colleagues showed that it is inferior, with a response rate of 37 percent against 68 percent for the horse. The fellow who uses the rabbit preparation because it is easier to give loses the response rate and exposes the child to the second line treatment sooner. The horse preparation is the first line choice, and the switch to the rabbit is made only for the child who cannot tolerate the horse, never as the default. [4]
[9]The third pitfall is the serum sickness of the antithymocyte globulin, which appears in the second week of the treatment with the fever, the rash, and the arthralgia, and which the fellow must recognise rather than mistake for sepsis. The serum sickness is a type three hypersensitivity reaction to the foreign protein, and it is treated with the corticosteroid and the symptomatic relief, and the antithymocyte globulin is continued or tapered according to the severity. The child on the antithymocyte globulin is also at the risk of the infusion reaction, the anaphylaxis, and the hypotension, and the premedication with the antihistamine and the corticosteroid and the close monitoring during the infusion are the standard precautions. [3]
The fourth pitfall is the clonal evolution of the eltrombopag, which is the modern concern that accompanies the modern drug. The eltrombopag stimulates the surviving stem cells to divide, and a small fraction of the treated children develop a clonal cytogenetic abnormality, the loss of a chromosome arm or the emergence of a monosomy 7, that heralds the myelodysplastic transformation. The marrow cytogenetics are therefore checked at the regular intervals during and after the eltrombopag, and the appearance of a clone redirects the treatment toward the transplant. The fellow who uses the eltrombopag without the cytogenetic surveillance runs the risk of missing the evolution. [6][7]
Prognosis & Disposition
The outlook for the child with severe aplastic anaemia has improved dramatically with the modern treatment, and the survival now exceeds 80 to 90 percent in the specialist centres. The child who has a matched sibling donor transplant has the best outlook, with a survival above 90 percent, because the transplant cures the disease and the matched donor minimises the graft-versus-host disease. The child who receives the immunosuppressive therapy has a response rate of 60 to 80 percent, and the responders live with a recovered marrow, though they carry the lifelong risk of the relapse, the clonal evolution, and the late myelodysplasia. [8]
The disposition of the child is shared between the primary centre and the specialist paediatric haematology and transplant service, and the fellow coordinates the two. The primary team holds the supportive care, the transfusion, and the neutropenic fever management, while the specialist service runs the transplant or the immunosuppression, the monitoring, and the long term follow up. The child is reviewed frequently during the treatment, with the blood counts, the ciclosporin trough, and the marrow cytogenetics, and the transition to the adult haematology service is planned in good time for the teenager who carries the late risks. [8]
The long term outlook for the inherited syndromes is shaped by the cancer risk, and this is the counselling the fellow revisits at every visit. The child with Fanconi anaemia, even after a successful transplant, carries the high risk of the head and neck squamous cell carcinoma, and the regular screening, the human papillomavirus vaccination, and the avoidance of the tobacco and the alcohol are the measures that reduce it. The child with a telomere biology disorder carries the risk of the pulmonary fibrosis and the hepatic disease, and the lung function and the liver tests are monitored alongside the blood counts. The fellow who treats the marrow failure and forgets the cancer risk treats only half the disease. [10]
Special Populations
The child from a consanguineous family carries a higher chance of an autosomal recessive inherited syndrome, and the fellow who works in a region with a mixed population plans for it. Fanconi anaemia is autosomal recessive, and the carrier rate is higher in the consanguineous and the founder populations, so the child with a pancytopenia and the stigmata of the syndrome is tested without delay. The family is counselled on the one in four risk in each pregnancy, the siblings are offered the testing, and the prenatal and the preimplantation genetic diagnosis are discussed for the future pregnancies. [9]
MARROW
The adolescent who is transitioning to the adult service carries the late risks that the fellow addresses before the handover. The young person on the ciclosporin is counselled on the renal toxicity and the blood pressure, the young woman on the eltrombopag is counselled on the hepatic toxicity, and the young person with Fanconi anaemia is counselled on the reproductive and the cancer risks. The transition is planned over a year or more, the young person is introduced to the adult team, the records and the care plans are transferred, and the aim is a young person who arrives in the adult service connected to a team rather than lost to the follow up. [10]
The child in a rural or remote setting, far from the specialist centre, is managed by the local team in partnership with the centre through the telehealth and the shared protocols. The local team holds the supportive care, the transfusion, and the neutropenic fever plan, and the child is transferred to the centre for the transplant or the immunosuppression. The fellow who works in such a setting values the local network and the retrieval service, because the child who is connected to the centre through it does as well as the child who lives next door to it. [8]
Evidence, Guidelines & Regional Differences
The evidence base for the treatment of aplastic anaemia is built on a series of landmark trials and studies, and the fellow names them by the author and the year. The Camitta criteria of 1976 defined the severe disease and the thresholds that still guide the treatment. The German Aplastic Anemia Study Group trial of Frickhofen in 1991 established the addition of ciclosporin to the antilymphocyte globulin. The cyclophosphamide trial of Tisdale in 2000 showed that the high dose cyclophosphamide was not a substitute. The horse versus rabbit trial of Scheinberg in 2011 settled the choice of the antithymocyte globulin. The eltrombopag trial of Olnes in 2012 and the RACE trial of Peffault de Latour in 2022 established the thrombopoietin receptor agonist. [1][3][4][7]
Frickhofen 1991 - ciclosporin added to antilymphocyte globulin
Key finding
The addition of ciclosporin to the antilymphocyte globulin and methylprednisolone improved the response rate and the survival in severe aplastic anaemia, establishing the combination as the standard of care.
Scheinberg 2011 - horse versus rabbit antithymocyte globulin
Key finding
Horse antithymocyte globulin gave a response rate of 68 percent at six months against 37 percent for the rabbit preparation, in the front line treatment of acquired aplastic anaemia.
Peffault de Latour 2022 - the RACE trial of eltrombopag added to immunosuppression
Key finding
The addition of eltrombopag to the horse antithymocyte globulin and ciclosporin improved the complete response rate at three months in the newly diagnosed severe aplastic anaemia.
The guidelines that translate this evidence into practice are set by the national haematology societies, and the fellow names the one being followed. The British Society for Haematology guideline of Kulasekararaj and colleagues, published in 2024, is the current comprehensive reference, and it sets the diagnostic criteria, the severity grading, the treatment algorithm, and the monitoring. The European Society for Blood and Marrow Transplantation sets the transplant practice through its working party, and the local paediatric haematology service sets the referral pathway. The fellow follows the local guideline and explains the choice to the family. [8]
The chief controversy in the modern treatment is the place of the eltrombopag, the timing of the alternative donor transplant, and the management of the clonal evolution. The eltrombopag improves the response rate but it carries the risk of the clonal evolution, and the balance between the early response and the late risk is weighed at each centre. The alternative donor transplant, once reserved for the salvage, is now offered earlier as the outcomes improve, and some centres offer it as the first line for the child with a well matched unrelated donor. The fellow follows the local protocol and explains the rationale to the family, because the evidence is still evolving and the practice varies by region. [6][12]
Exam Pearls
The single most testable fact is the Camitta criteria, and the fellow must know the three thresholds and the marrow cellularity exactly. Severe aplastic anaemia is a marrow cellularity under 25 percent with two of three peripheral blood criteria, the neutrophils under 0.5 times ten to the nine per litre, the platelets under 20 times ten to the nine per litre, and the reticulocytes under 60 times ten to the nine per litre. Very severe disease is the neutrophils under 0.2 times ten to the nine per litre. The fellow who can state these four numbers, the cellularity and the three counts, carries the diagnostic heart of the topic. [1]
The immunosuppressive regimen is the second most testable set, and it is asked as the drugs, the doses, and the evidence. Horse antithymocyte globulin at 40 mg per kg per day for four days, ciclosporin at 5 mg per kg per day, and eltrombopag added per the RACE trial. The horse preparation is preferred over the rabbit, with the 68 percent versus 37 percent response rate of the Scheinberg trial, and the ciclosporin was added by the Frickhofen trial of 1991. The fellow who gives the regimen without the doses, or who confuses the horse and the rabbit, loses the marks. [3][4]
[9]The inherited syndromes round out the high yield set, and the fellow links each to its single defining feature. Fanconi anaemia is the diepoxybutane positive chromosomal breakage and the radial ray anomalies, and it is transplanted with a fludarabine based reduced intensity regimen. Diamond-Blackfan anaemia is the pure red cell aplasia of infancy with the raised fetal haemoglobin and the ribosomal protein mutation. The telomere biology disorders are the very short telomeres with the nail dystrophy and the oral leukoplakia. The fellow who holds these three inherited syndromes, the Camitta criteria, the transplant versus immunosuppression fork, and the horse versus rabbit choice, carries the whole topic. [9][11]
References
- [1]Camitta BM, Thomas ED, Nathan DG, et al Severe aplastic anemia: a prospective study of the effect of early marrow transplantation on acute mortality. Blood, 1976.PMID 779871
- [2]Young NS, Kaufman DW The epidemiology of acquired aplastic anemia. Haematologica, 2008.PMID 18379007
- [3]Frickhofen N, Kaltwasser JP, Schrezenmeier H, et al Treatment of aplastic anemia with antilymphocyte globulin and methylprednisolone with or without cyclosporine. The German Aplastic Anemia Study Group. N Engl J Med, 1991.PMID 2017225
- [4]Scheinberg P, Nunez O, Weinstein B, et al Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med, 2011.PMID 21812672
- [5]Tisdale JF, Dunn DE, Geller N, et al High-dose cyclophosphamide in severe aplastic anaemia: a randomised trial. Lancet, 2000.PMID 11075769
- [6]Olnes MJ, Scheinberg P, Calvo KR, et al Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med, 2012.PMID 22762314
- [7]Peffault de Latour R, Kulasekararaj A, Iacobelli S, et al Eltrombopag Added to Immunosuppression in Severe Aplastic Anemia. N Engl J Med, 2022.PMID 34986284
- [8]Kulasekararaj A, Cavenagh J, Dokal I, et al Guidelines for the diagnosis and management of adult aplastic anaemia: A British Society for Haematology Guideline. Br J Haematol, 2024.PMID 38247114
- [9]Auerbach AD Fanconi anemia and its diagnosis. Mutat Res, 2009.PMID 19622403
- [10]Alter BP, Greene MH, Velazquez I, et al Cancer in Fanconi anemia. Blood, 2003.PMID 12584146
- [11]Wlodarski MW, Vlachos A, Farrar JE, et al Diagnosis, treatment, and surveillance of Diamond-Blackfan anaemia syndrome: international consensus statement. Lancet Haematol, 2024.PMID 38697731
- [12]Bacigalupo A, Socie G, Lanino E, et al Fludarabine, cyclophosphamide, antithymocyte globulin, with or without low dose total body irradiation, for alternative donor transplants, in acquired severe aplastic anemia: a retrospective study from the EBMT-SAA Working Party. Haematologica, 2010.PMID 20494932