Paeds · haematology-oncology-and-transfusion
Bleeding child: diagnostic approach
Also known as Bleeding child · Bruising child workup · Coagulation screen interpretation in children · Bleeding assessment tool · Platelet versus coagulation bleeding
Fellowship guide to the systematic diagnostic approach to the child who presents with bleeding or bruising. Covers recognising normal childhood bruising, quantifying the bleeding history with a bleeding assessment tool, and splitting the presentation into platelet-type (primary hemostasis) and coagulation-type (secondary hemostasis) bleeding, then resolving it with the full blood count, blood film, prothrombin time, activated partial thromboplastin time and targeted factor and Von Willebrand disease assays.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A child bleeds abnormally when the bleeding is out of proportion to injury, recurs without obvious trauma, sits at an unusual site, or follows a recognisable inherited pattern. The diagnostic task is not to list every cause at once, but to move through three ordered questions. Is the bleeding actually abnormal, or is it normal childhood bruising? Does the pattern point to a platelet problem or a clotting-factor problem? And which specific disorder is responsible? Answering these in sequence turns a scattergun panel of tests into a focused, safe workup. [1]
The first question is the hardest and the most often skipped. Normal children bruise their shins, have occasional nosebleeds, and cut themselves. Abnormal bleeding is defined by its site, severity, duration, and disproportion to injury, and it is best quantified with a validated bleeding assessment tool rather than a clinician's impression. The second question is settled by the pattern of bleeding and a small set of first-line tests. A child with petechiae, nosebleeds, and heavy periods has a primary-hemostasis (platelet-type) problem; a boy with a swollen knee and a deep muscle haematoma has a secondary-hemostasis (coagulation-type) problem. [1]
The third question is resolved by targeted confirmatory assays guided by the screening tests. A prolonged activated partial thromboplastin time with deep bleeding demands factor VIII and IX assays, while a normal screen with convincing mucocutaneous bleeding demands Von Willebrand disease testing and a platelet function workup. Holding these three questions in order is what separates a confident diagnostician from one who orders everything and waits. [2]
Classification
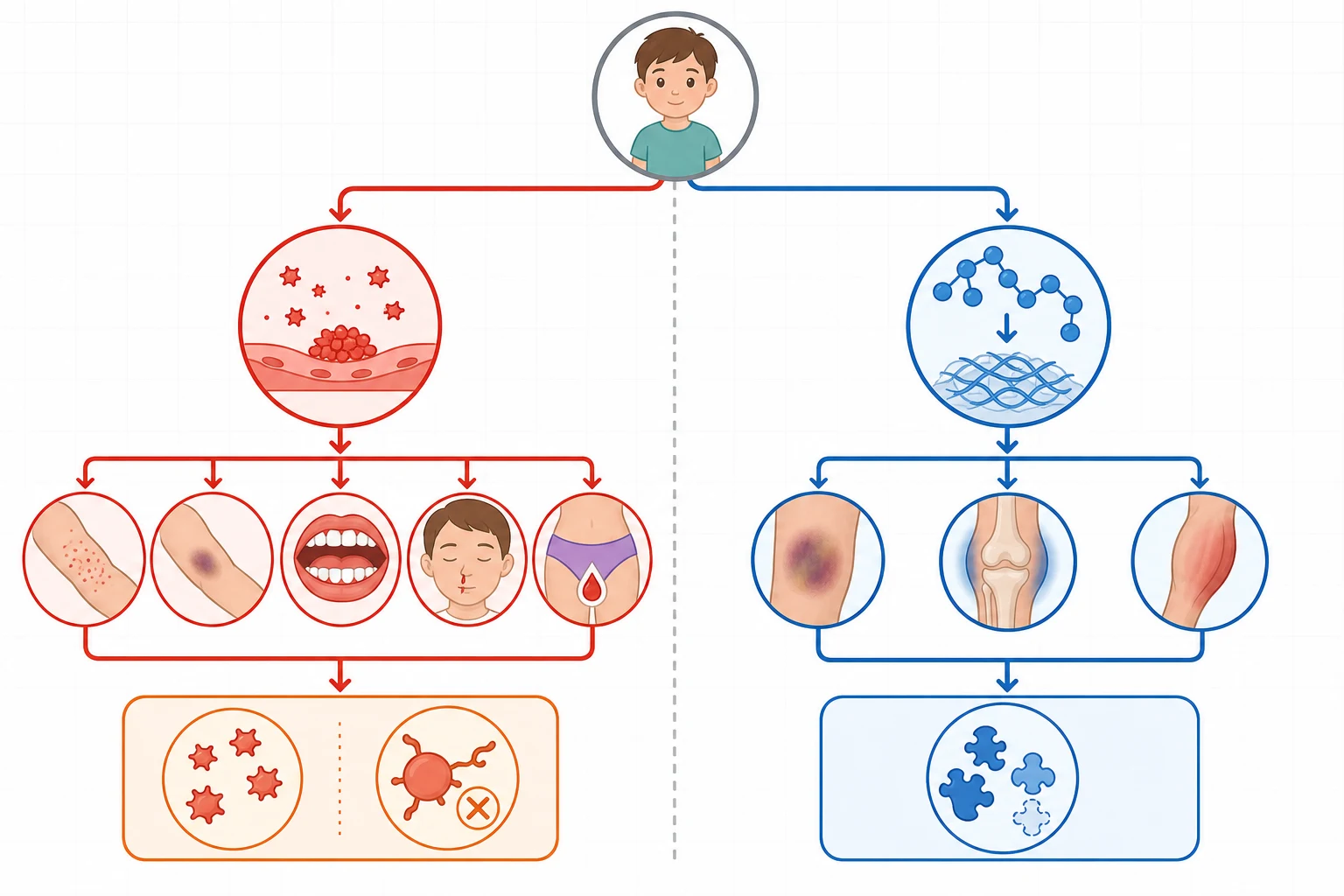
The single most useful branch point is the pattern of bleeding, because the haemostatic system fails in two distinct ways that produce two distinct clinical pictures. Splitting the presentation into primary-hemostasis and secondary-hemostasis bleeding immediately halves the differential and tells the clinician whether to chase a platelet problem or a clotting-factor problem. This division is the backbone of the diagnostic algorithm and should be taught to every trainee before any laboratory interpretation. [1]
Primary-hemostasis bleeding is immediate, mucocutaneous, and superficial. The child has petechiae, easy bruising, prolonged epistaxis, gum bleeding, heavy menstrual bleeding, and prolonged oozing from small cuts, because the platelet plug that should form within seconds at a wound is missing or faulty. Secondary-hemostasis bleeding is delayed by hours and deep. The child has haemarthroses, intramuscular haematomas, and delayed rebleeding after surgery or injury, because the fibrin mesh that should reinforce the platelet plug is missing. [2]
[1]A third category straddles the two. Mixed-pattern bleeding, where a child has both petechiae and deep bleeds, or thrombocytopenia with a prolonged clotting time, points to a combined or consumptive disorder: disseminated intravascular coagulation, liver disease, or Von Willebrand disease. Von Willebrand disease is the classic straddler, because Von Willebrand factor mediates platelet adhesion, giving mucocutaneous bleeding, and also carries and stabilises factor VIII, so a low level can prolong the activated partial thromboplastin time and mimic a coagulation defect. [5]
Epidemiology & Risk Factors
Most children who present with bruising or bleeding do not have a bleeding disorder. Epistaxis and easy bruising are so common in active preschool and school-age children that the diagnostic challenge is separating the normal from the pathological. A bleeding assessment tool exists precisely because clinical impression alone over-diagnoses and under-diagnoses in equal measure. [6]
When a real disorder is present, Von Willebrand disease is the most common inherited bleeding disorder, with a prevalence of symptomatic disease around 1 in 1000, and it is the leading inherited cause of adolescent menorrhagia and recurrent epistaxis. Haemophilia A affects about 1 in 5000 male births and haemophilia B about 1 in 30000; both are X-linked recessive and present in boys. Immune thrombocytopenia is the commonest cause of acute isolated thrombocytopenia in an otherwise well child aged two to five years, classically one to two weeks after a viral illness, with an incidence of 1 to 6 per 100000 children per year. [3]
Risk factors that raise the prior probability of a real disorder include a family history of bleeding or a known bleeding disorder, consanguinity, male sex with a maternal family history of bleeding (X-linked haemophilia), onset of bleeding in infancy, bleeding that is disproportionate to injury, and exposure to medications that impair platelet function or clotting, such as non-steroidal anti-inflammatory drugs and anticoagulants. Vitamin K deficiency bleeding remains relevant in exclusively breastfed infants who did not receive vitamin K prophylaxis, and in children with malabsorption or cholestasis. [2]
Pathophysiology
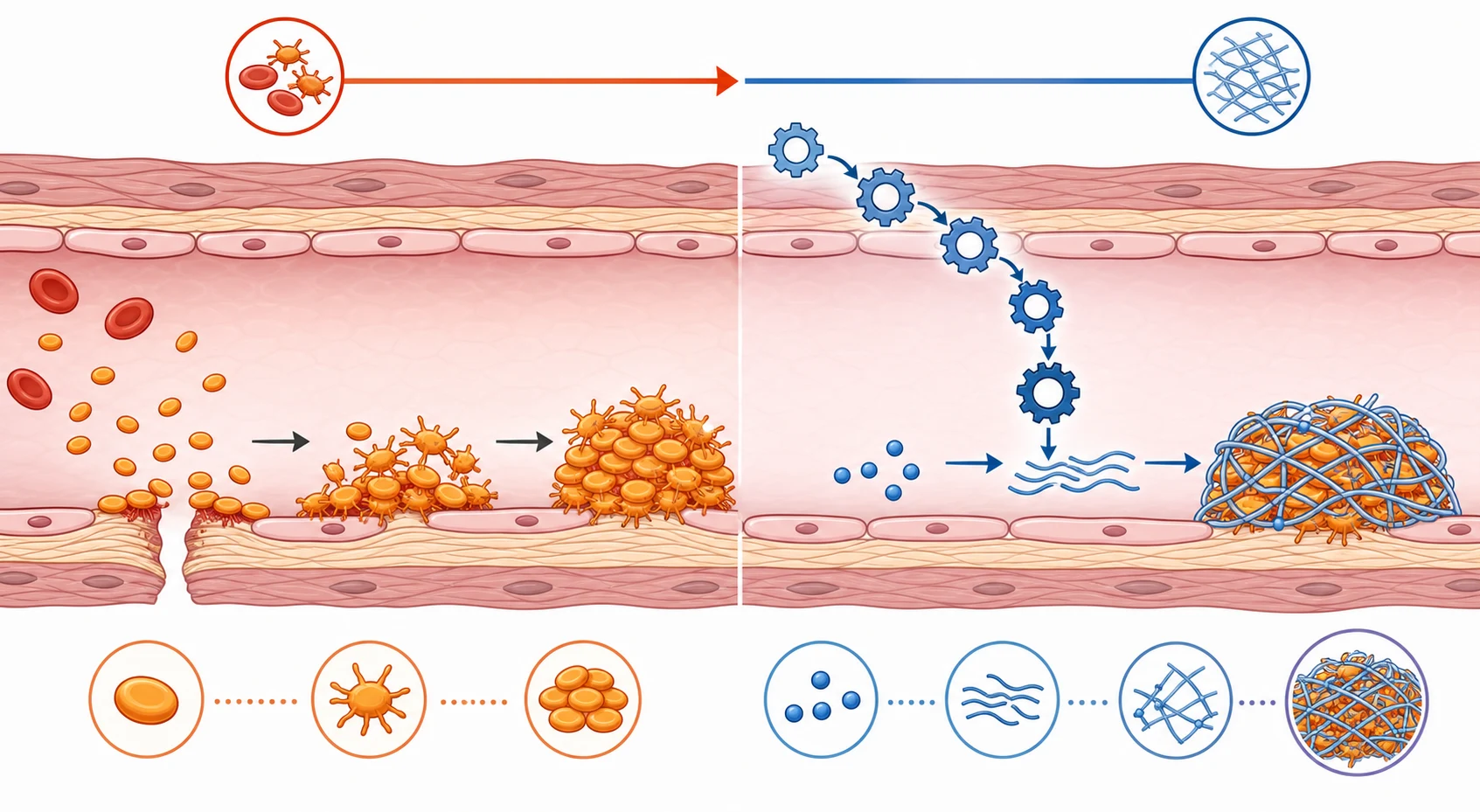
Normal hemostasis is two sequential phases, and the clinical patterns map directly onto them. In primary hemostasis, injured endothelium exposes subendothelial collagen and Von Willebrand factor. Circulating platelets adhere through their glycoprotein Ib-IX-V receptor (a Von Willebrand factor dependent step), then activate and aggregate through glycoprotein IIb-IIIa using fibrinogen as the bridge. The result is a primary platelet plug that forms within seconds. A failure anywhere in this sequence, or simply too few platelets, produces immediate mucocutaneous bleeding and a petechial pattern. [1]
In secondary hemostasis, the coagulation cascade generates thrombin, which converts soluble fibrinogen into an insoluble fibrin mesh that reinforces and stabilises the platelet plug. The tissue-factor (extrinsic) pathway is initiated by factor VII and is tested by the prothrombin time, while the contact (intrinsic) pathway is tested by the activated partial thromboplastin time. A failure here lets the primary plug form but fail to hold, so bleeding is delayed by hours and tends to be deep, into joints and muscles. [2]
Von Willebrand factor straddles both phases, and this is the key to understanding why Von Willebrand disease is so protean. Because it mediates platelet adhesion, its deficiency gives mucocutaneous bleeding like a platelet disorder. Because it carries and protects factor VIII, its deficiency can lower factor VIII and prolong the activated partial thromboplastin time, mimicking a mild coagulation defect. A child with Von Willebrand disease may therefore present with nosebleeds and a borderline prolonged activated partial thromboplastin time, and the diagnosis is missed unless specific Von Willebrand factor assays are sent. [5]
Why each phase fails in a distinct way
Primary hemostasis fails: too few platelets (thrombocytopenia) or faulty adhesion and aggregation (Von Willebrand disease, platelet function defects)
Result: immediate mucocutaneous bleeding with petechiae, because capillary leaks are not plugged in seconds
Secondary hemostasis fails: a clotting factor is missing (haemophilia, vitamin K deficiency, liver disease)
Result: delayed deep bleeding into joints and muscles, because the fibrin reinforcement never forms
Combined failure: consumption or underproduction of platelets and factors together (DIC, liver disease, severe Von Willebrand disease)
The haemostatic system matures through childhood, and this single fact causes more misreading of coagulation screens than any other. Neonates and young infants have physiologically lower vitamin-K-dependent factors and contact-pathway factors than adults, so their prothrombin time and activated partial thromboplastin time are naturally prolonged. Interpreting an infant's results against adult reference ranges labels a healthy baby as coagulopathic. Every coagulation value in a child must be read against age-specific paediatric reference ranges. [9]
Clinical Presentation
The pattern of bleeding is the single most diagnostic feature, and it must be elicited and quantified before any laboratory test. A structured history, ideally formalised with a bleeding assessment tool, is worth more than a battery of early tests. The history should ask systematically about each bleeding site and record the frequency, duration, severity, and precipitant of each symptom. [6]
Platelet-type bleeding is immediate, mucocutaneous, and superficial. The child describes petechiae, which are non-blanching pinpoint red lesions on the skin and mucosa; easy bruising, especially over pressure areas; recurrent epistaxis lasting more than ten minutes despite pressure; gum bleeding with tooth brushing; heavy menstrual bleeding with clots, flooding, and iron deficiency; and prolonged oozing from small cuts or venepuncture sites. Coagulation-type bleeding is delayed and deep. The boy with haemophilia describes painful swollen joints, classically the knees, elbows, and ankles; intramuscular haematomas, especially in the calf, psoas, and forearm; and delayed rebleeding after circumcision, dental extraction, or injury. [2]
The tempo of onset is itself diagnostic. Sudden onset of bruising and petechiae over hours to days in a previously well child points to new-onset immune thrombocytopenia, especially with a recent viral illness. Lifelong recurrent bleeding since infancy or early childhood, or a positive family history, points to an inherited disorder. Bleeding from birth, particularly cephalohaematoma or bleeding after circumcision in a boy, raises haemophilia or vitamin K deficiency bleeding. A single dramatic bleed after minor trauma raises both a severe factor deficiency and, in an infant, non-accidental injury. [1]
A few presentations flag a dangerous cause. A severe headache, vomiting, or altered consciousness in a thrombocytopenic child raises intracranial haemorrhage, which is rare but catastrophic in immune thrombocytopenia. Bruising with prolonged fever, pallor, and systemic illness raises leukaemia, marrow failure, disseminated intravascular coagulation, or sepsis. A painful swollen joint in a boy is a haemarthrosis until proven otherwise. Patterned or symmetric bruises, or bruises away from bony prominences, raise non-accidental injury. [4]
Differential Diagnosis
The differential is anchored on two things together: the bleeding pattern, and the first-line laboratory results. Start by confirming the bleeding is truly abnormal, because several non-disorders mimic it. Normal active children bruise; non-steroidal anti-inflammatory drugs prolong minor bleeding; and a vigorous cough or vomiting bout can cause petechiae around the face and eyes that are harmless. Once the bleeding is judged abnormal, the pattern and the platelet count divide the differential. [1]
A low platelet count with a petechial pattern brings a short, ordered list. Immune thrombocytopenia is the most likely cause in a well child with isolated thrombocytopenia and a recent viral illness. Leukaemia and marrow failure are the dangerous mimics: an ill child, additional cytopenias, blast cells on the film, hepatosplenomegaly, or lymphadenopathy all demand a bone marrow aspirate before any steroid is given. Disseminated intravascular coagulation, haemolytic uraemic syndrome, hypersplenism, and neonatal alloimmune or autoimmune thrombocytopenia complete the thrombocytopenic list. [3]
A normal platelet count with a mucocutaneous pattern redirects the workup to platelet function and Von Willebrand factor. Von Willebrand disease heads the list and is the commonest inherited cause. Inherited platelet function disorders, such as Glanzmann thrombasthenia (a glycoprotein IIb-IIIa defect) and Bernard-Soulier syndrome (a glycoprotein Ib-IX-V defect with giant platelets), are rarer but distinctive. Drug-induced platelet dysfunction from non-steroidal anti-inflammatory drugs is common and reversible. [1]
[2]A prolonged activated partial thromboplastin time with a deep-bleeding pattern demands haemophilia A and B first. A lupus anticoagulant can also prolong the activated partial thromboplastin time, but it is associated with thrombosis rather than bleeding, and a mixing study sorts the two. A prolonged prothrombin time points to vitamin K deficiency, liver disease, disseminated intravascular coagulation, or the rarer factor VII deficiency. Finally, non-accidental injury is the critical non-haematological differential in any child with unexplained bruising, and it must be considered and excluded in every case, especially in a non-mobile infant. [1]
Clinical & Bedside Assessment
The bedside assessment does two jobs at once: it gauges how sick the child is, and it harvests the historical clues that direct the laboratory workup. Begin with a paediatric assessment of haemodynamic stability and the paediatric assessment triangle. Tachycardia, prolonged capillary refill, pallor, and altered consciousness quantify the cardiovascular strain, and their severity decides whether the child can wait for results or needs resuscitation first. [1]
The history is the highest-yield instrument in the whole evaluation. Ask systematically about each bleeding site. For epistaxis, record the frequency, the duration, the side, and the response to pressure. For bruising, record the sites, the size, and the precipitant. For heavy menstrual bleeding, record the cycle length, the pad or tampon count, the presence of clots and flooding, and any iron deficiency. Ask about bleeding after circumcision, dental extraction, the umbilical stump, and venepuncture. Ask about joint and muscle swelling, and about blood in the stool or urine. Quantify the whole history with a structured bleeding assessment tool, because a formal score separates abnormal bleeding from normal childhood bruising far better than impression. [6]
Examination searches for specific signs. Petechiae are best seen on the lower limbs and conjunctivae. The distribution of bruises matters: bruises over the shins and bony prominences of a mobile child are common, but bruises on the ears, neck, buttocks, trunk, or soft tissue, and patterned or symmetric bruises, raise concern. Hepatosplenomegaly and lymphadenopathy suggest leukaemia or hypersplenism rather than a simple bleeding disorder. A warm, swollen, tender joint, or a joint with chronic deformity, suggests recurrent haemarthrosis from haemophilia. Signs of chronic liver disease point to a synthetic coagulopathy. [4]
Two bedside cautions are worth stating. First, avoid intramuscular injections and invasive procedures in any child with a suspected coagulopathy until the workup is complete, because a haematoma can be limb-threatening in haemophilia. Second, a non-mobile infant with bruising must trigger a safeguarding assessment alongside the bleeding-disorder workup, because non-accidental injury and a bleeding disorder can coexist, and missing either is catastrophic. [1]
Investigations
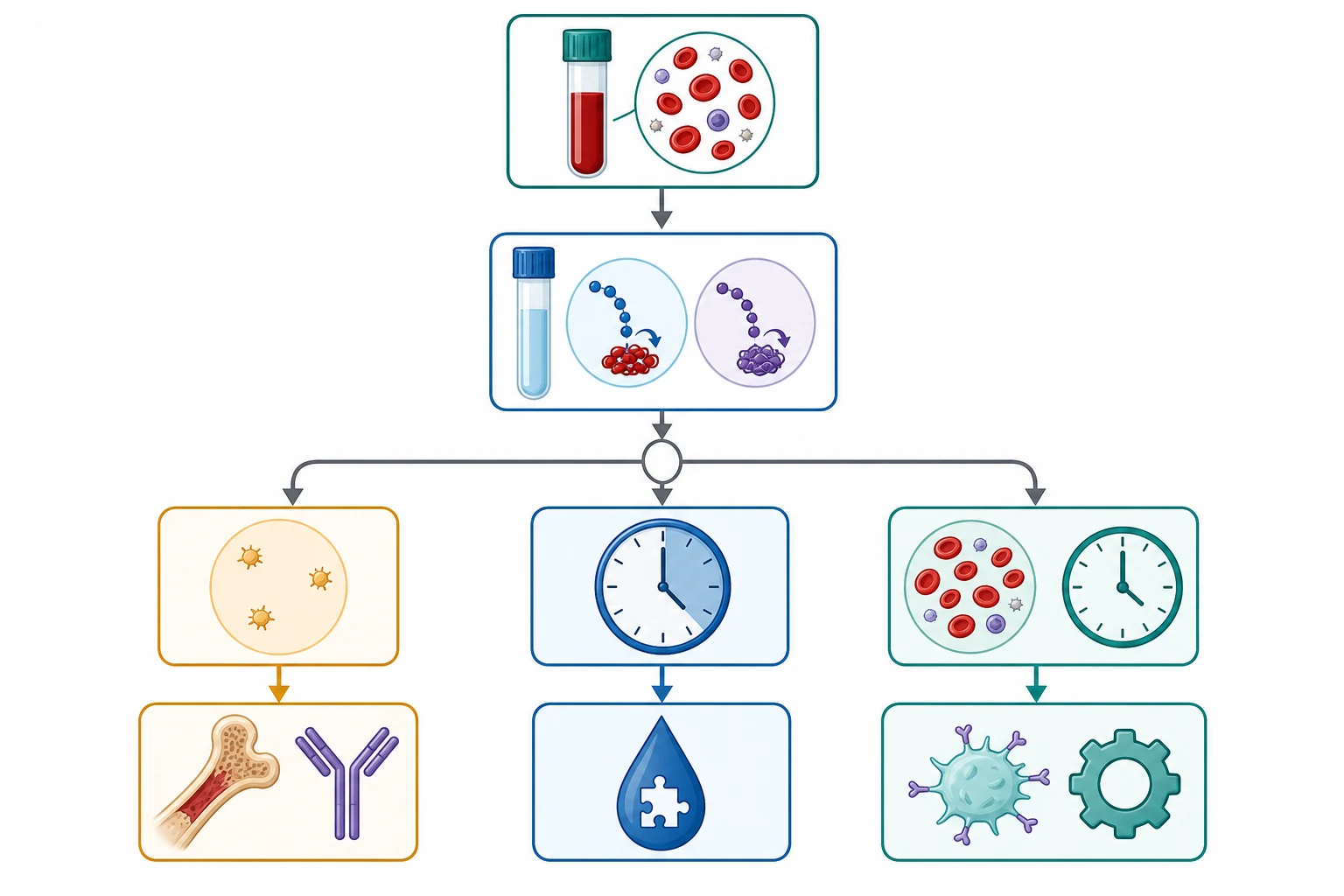
The investigation strategy runs in three tiers: confirm and localise the defect with screening tests, then resolve the specific cause with targeted assays. The first tier is the same for every child and should be sent at the first venepuncture, before any transfusion, so that no sample is wasted. [1]
Tier one is the full blood count and blood film, the prothrombin time and international normalised ratio, the activated partial thromboplastin time, the fibrinogen, and a blood group. The full blood count quantifies the platelet number and excludes anaemia, and the blood film is critical because it confirms the count, shows the platelet morphology (giant platelets in Bernard-Soulier syndrome), and flags blast cells or fragmented schistocytes that change the diagnosis entirely. The prothrombin time tests the extrinsic and common pathways (factor VII, then X, V, II, I), while the activated partial thromboplastin time tests the intrinsic and common pathways (factors XII, XI, IX, VIII, then X, V, II, I). [2]
Every coagulation value in a child must be interpreted against age-specific paediatric reference ranges. Neonates and young infants have physiologically lower vitamin-K-dependent and contact-pathway factors, so their prothrombin and activated partial thromboplastin times are naturally longer than adult values. Reading an infant's results against adult ranges generates false positives and false diagnoses. The bleeding time is no longer recommended, because it is poorly reproducible and adds nothing that specific assays do not provide better. [9]
Tier two is targeted by the screening pattern and the bleeding type. A prolonged activated partial thromboplastin time with a deep-bleeding pattern demands factor VIII and IX assays, and the answer is usually haemophilia A or B. A normal activated partial thromboplastin time with a convincing mucocutaneous pattern demands Von Willebrand disease testing, which is Von Willebrand factor antigen, a functional assay such as ristocetin cofactor activity or a glycoprotein Ib-binding assay, and factor VIII, with blood-group-adjusted interpretation and multimer analysis to subtype. A bleeding assessment tool score should precede tier two in a child with vague symptoms, because it quantifies whether the bleeding is even abnormal enough to justify the assays. [5]
Tier three is specialised testing. Light transmission aggregometry or a flow-cytometric platelet screen is used for suspected inherited platelet function disorders, and molecular genetic testing is increasingly used to confirm the diagnosis and enable family counselling. The 2021 Von Willebrand disease diagnosis guidelines stress that Von Willebrand factor levels are variable and stress-responsive, so a borderline low value should be repeated before a diagnosis is made or excluded. [5]
[2]Management — Resuscitation
A child with active, severe, or life-threatening bleeding is managed with airway-breathing-circulation first, with investigation running in parallel rather than in series. Establish reliable intravenous access, take the tier-one samples before any blood product is given, and control local bleeding with firm sustained pressure. A bedside glucose and blood gas help to exclude hypoglycaemia and acidosis. [1]
The treatment of active bleeding follows the suspected defect. For severe thrombocytopenia with active bleeding, platelet transfusion is indicated, but in immune thrombocytopenia transfused platelets are destroyed within minutes, so platelet transfusion is reserved for life-threatening bleeding and is paired with intravenous immunoglobulin or corticosteroids to raise the count. For a known or suspected factor deficiency with a major bleed, factor replacement is the definitive treatment and must not wait for a complete workup if the bleed is life-threatening; a life-threatening haemophilia bleed is treated presumptively to a factor level of 100 per cent. [2]
Avoid intramuscular injections, arterial puncture, and invasive procedures in any child with a suspected coagulopathy until the workup is complete, because a muscle bleed in haemophilia can cause a compartment syndrome. A child with suspected non-accidental injury must be safeguarded concurrently. The threshold for red-cell transfusion rests on haemodynamic compromise and the rate of haemoglobin fall, not on a single haemoglobin value. [1]
Intravenous immunoglobulin (immune thrombocytopenia)
Dose
0.8 to 1 g per kg
Management — Definitive & Stepwise
The definitive management is cause-specific and is covered in the disease-specific topics, but the diagnostic-approach task is to deliver the clinician to the correct diagnosis safely and to start the right interim care. Once the tier-one tests return, the pathway branches by the pattern and the platelet count. [1]
Isolated thrombocytopenia in a well child is managed as suspected immune thrombocytopenia. A child with no bleeding or only skin bleeding and a count above a safe threshold can be observed with a clear safety-net and early review, because most acute immune thrombocytopenia resolves spontaneously. Treatment with corticosteroids or intravenous immunoglobulin is reserved for significant bleeding, a very low count with risk features, or an urgent need to raise the count. A bone marrow aspirate is considered before steroid therapy if any feature suggests malignancy, to avoid masking leukaemia. [3]
For the inherited disorders, the workup triggers long-term care. Von Willebrand disease is managed with desmopressin for type 1, which releases stored Von Willebrand factor and factor VIII from endothelium, and with Von Willebrand factor-containing concentrates for type 3 or major bleeding; desmopressin is avoided in type 2B because it can cause transient thrombocytopenia. Haemophilia is managed with factor replacement and prophylaxis. Every newly diagnosed inherited disorder needs referral to a comprehensive haemophilia or haematology service, family screening, genetic counselling, and a written emergency plan. [5]
PLAN the bleeding-child workup
Two adjuncts deserve a place in the algorithm. Antifibrinolytics such as tranexamic acid are useful for mucosal bleeding, particularly dental, nasal, and menstrual bleeding, and they are safe across most disorders. Heavy menstrual bleeding is managed alongside the workup with combined hormonal contraception or a levonorgestrel intrauterine system, so the adolescent girl is not left bleeding while the diagnosis is pursued. [1]
Specific Subtypes & Scenarios
Immune thrombocytopenia is the prototype thrombocytopenic presentation and the one most likely to appear in a written stem. The classic picture is sudden petechiae and bruising in an afebrile, otherwise well two-to-five-year-old, one to two weeks after a viral illness, with an isolated platelet count often under 20 times 10 to the 9 per litre and a normal haemoglobin and white cell count. The blood film confirms isolated thrombocytopenia and excludes leukaemia. The danger is intracranial haemorrhage, which is rare but catastrophic, so the family must be safety-netted to present immediately with head injury, severe headache, or any significant bleeding. [3]
Von Willebrand disease is the prototype mucocutaneous disorder and the commonest inherited bleeding disorder. It presents with mucocutaneous bleeding from childhood, menorrhagia in adolescent girls, and a normal or mildly prolonged activated partial thromboplastin time with a normal platelet count. The diagnosis requires specific Von Willebrand factor assays, and the level is strongly influenced by blood group, with group O giving lower levels, so a borderline result must be interpreted in that context and repeated. The 2021 international guidelines emphasise that the diagnosis should never rest on a single low value. [5]
Haemophilia is the prototype coagulation disorder. It is X-linked recessive, so it presents in boys, with cephalohaematoma or bleeding after circumcision in infancy, and with joint and muscle bleeds once the child is mobile. The screen shows a prolonged activated partial thromboplastin time with a normal prothrombin time and platelet count, and the factor assay shows a low factor VIII (haemophilia A) or factor IX (haemophilia B). Modern prophylaxis and non-factor agents have transformed the outlook, but the diagnosis still triggers comprehensive care, family screening, and genetic counselling. [2]
[5]Neonatal bleeding deserves a separate mention because the causes and the reference ranges differ. Vitamin K deficiency bleeding appears in breastfed infants who did not receive prophylaxis, with early, classic, or late presentations. Neonatal alloimmune thrombocytopenia causes petechiae in an otherwise well neonate while the mother's platelet count is normal, distinguishing it from autoimmune thrombocytopenia where the mother is also thrombocytopenic. Haemophilia can present as cephalohaematoma or circumcision bleeding in a male infant. All neonatal coagulation results must be read against neonatal reference ranges. [9]
Complications & Pitfalls
The principal pitfall is misclassifying the bleeding, in either direction. Over-investigating normal childhood bruising wastes resources and anxiety, while missing a serious disorder or non-accidental injury can be fatal. The bleeding assessment tool exists to make the first call objective rather than impressionistic, and it should be used before expensive tier-two tests are ordered. [6]
A normal activated partial thromboplastin time does not exclude mild haemophilia, Von Willebrand disease, or a platelet function disorder. A convincing bleeding history with normal screening tests warrants tier-two testing, because the screening tests are insensitive to mild and moderate defects. Von Willebrand disease is the classic miss, because the level is borderline, blood-group dependent, and stress-responsive, so a child tested during an intercurrent illness can return a falsely reassuring normal level and needs repeat testing. [5]
The diagnostic pitfalls cluster around three errors. The first is interpreting an infant's coagulation times against adult reference ranges, which labels a healthy baby as coagulopathic. The second is sending tier-one samples after a transfusion, which confounds interpretation. The third is treating suspected immune thrombocytopenia with steroids before a blood film has excluded leukaemia, which can mask the diagnosis and delay definitive care. [3]
Missing non-accidental injury is the final and most dangerous pitfall. Attributing unexplained or patterned bruises to a bleeding disorder, without a concurrent safeguarding assessment, has caused repeated harm. The rule is simple: every non-mobile infant with bruising, and every child with patterned, symmetric, or multi-site bruising, is assessed for non-accidental injury in parallel with any haematological workup. A bleeding disorder and non-accidental injury can coexist. [1]
Prognosis & Disposition
The prognosis depends almost entirely on the underlying cause. Acute immune thrombocytopenia in a young child resolves spontaneously in the majority within six months and carries an excellent prognosis; chronic or persistent disease needs haematology follow-up, but serious bleeding becomes uncommon once the count recovers and the family is safety-netted. [3]
The inherited disorders are lifelong but highly manageable. Von Willebrand disease is usually mild and managed with desmopressin or concentrates as needed. Haemophilia is severe but has been transformed by prophylaxis and non-factor agents, so a child diagnosed today expects a near-normal life with modern care. The inherited platelet function disorders vary by severity, from mild bruising to a haemophilia-like picture. [2]
Disposition is dictated by the bleeding severity and the count or factor level. A well child with immune thrombocytopenia, a count above a safe threshold, and no significant bleeding can be managed as an outpatient with a clear safety-net and early review. Any active bleeding, a very low count with risk features, or a suspected severe factor deficiency belongs in hospital for monitoring and treatment. Every child diagnosed with an inherited bleeding disorder needs referral to a comprehensive haemophilia or haematology service, family screening, a written emergency plan, and education about avoiding triggers and presenting early. [4]
Special Populations
Adolescent girls with heavy menstrual bleeding deserve a low threshold for a bleeding-disorder workup. Heavy menstrual bleeding is the most common presentation of Von Willebrand disease and the inherited platelet function disorders in females, and it is routinely dismissed as a normal variant. Any girl whose menstrual bleeding interferes with daily life, or who develops iron deficiency anaemia from menarche, should have a bleeding assessment and a Von Willebrand disease workup rather than reassurance alone. [5]
Neonates bleed for distinct reasons, and their reference ranges differ. Vitamin K deficiency bleeding appears in breastfed infants without prophylaxis. Neonatal alloimmune thrombocytopenia causes petechiae in an otherwise well neonate with a normal maternal platelet count, in contrast to autoimmune thrombocytopenia where the mother is thrombocytopenic. Haemophilia can present as cephalohaematoma or circumcision bleeding in a male infant. The neonatal haemostatic system is physiologically immature, so all coagulation results must be read against neonatal ranges. [9]
Children with chronic liver disease have a combined defect from synthetic failure of clotting factors and thrombocytopenia from hypersplenism, so they bleed from both pathways. Immunocompromised and oncology children bleed mainly from thrombocytopenia and the effects of treatment. Indigenous, migrant, and refugee families may carry bleeding disorders and haemoglobinopathies at different frequencies and need culturally safe counselling and reliable access to specialist follow-up. [2]
Every non-mobile infant with bruising must be assessed for non-accidental injury regardless of a possible bleeding disorder, and every safeguarding concern must run in parallel with the haematological workup rather than waiting for it to finish. A bleeding disorder and maltreatment can coexist, and the safest practice is to consider both from the first encounter. [1]
Evidence, Guidelines & Regional Differences
The diagnostic framework rests on international consensus. The International Society on Thrombosis and Haemostasis standardised the bleeding assessment tool for inherited bleeding disorders, and it was validated in children by the prospective paediatric bleeding questionnaire studies, which showed that a quantified bleeding score distinguishes children with a bleeding disorder from those without, and that it performs better than clinical impression alone. A separate standardised bleeding assessment tool exists for immune thrombocytopenia. [7]
The American Society of Hematology, the International Society on Thrombosis and Haemostasis, the National Hemophilia Foundation, and the World Federation of Hemophilia jointly produced the 2021 Von Willebrand disease diagnosis guidelines. These define the modern testing algorithm, caution against diagnosing Von Willebrand disease on a single low value given the variability of the factor, and recommend repeating borderline levels before a diagnosis is made or excluded. [5]
The American Society of Hematology 2019 immune thrombocytopenia guidelines, updated in 2022, define when to treat, observe, and investigate in children. They emphasise observation with a safety-net for the well child with little or no bleeding, the importance of a normal blood film to exclude leukaemia before steroid therapy, and the selection of corticosteroids, intravenous immunoglobulin, or anti-D according to the clinical context. [4]
[4]Regional differences reflect both practice and access. Universal vitamin K prophylaxis at birth has made vitamin K deficiency bleeding uncommon where it is routine, so its occurrence should prompt a search for an underlying cause. Access to specialised haemophilia comprehensive care, paediatric reference-range coagulation testing, and platelet function and genetic testing is uneven between metropolitan and remote services. Ongoing controversies include the role of global haemostasis assays and next-generation sequencing panels in the unexplained-bleeding child, and whether to screen all adolescent girls with heavy menstrual bleeding for Von Willebrand disease. [10]
Exam Pearls
PETECHIAE means a platelet problem
References
- [1]van Ommen CH, Peters M The bleeding child. Part I: primary hemostatic disorders. Eur J Pediatr, 2012.PMID 21800040
- [2]van Herrewegen F, Meijers JC, Peters M, et al Clinical practice: the bleeding child. Part II: disorders of secondary hemostasis and fibrinolysis. Eur J Pediatr, 2012.PMID 21922352
- [3]Neunert C, Terrell DR, Arnold DM, et al American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv, 2019.PMID 31794604
- [4]Neunert CE, Arnold DM, Grace RF, et al The 2022 review of the 2019 American Society of Hematology guidelines on immune thrombocytopenia. Blood Adv, 2024.PMID 38608258
- [5]James PD, Connell NT, Ameer B, et al ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv, 2021.PMID 33570651
- [6]Bidlingmaier C, Grote V, Budde U, et al Prospective evaluation of a pediatric bleeding questionnaire and the ISTH bleeding assessment tool in children and parents in routine clinical practice. J Thromb Haemost, 2012.PMID 22578063
- [7]Rodeghiero F, Tosetto A, Abshire T, et al ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost, 2010.PMID 20626619
- [8]Rodeghiero F, Michel M, Gernsheimer T, et al Standardization of bleeding assessment in immune thrombocytopenia: report from the International Working Group. Blood, 2013.PMID 23361904
- [9]Andrew M, Vegh P, Johnston M, et al Maturation of the hemostatic system during childhood. Blood, 1992.PMID 1391957
- [10]Tosetto A, Eikenboom J Clinical and laboratory diagnosis of von Willebrand disease. Haematologica, 2026.PMID 41496702