Paeds · haematology-oncology-and-transfusion
Bone and soft-tissue sarcomas
Also known as Paediatric sarcoma · Childhood bone tumour · Osteosarcoma · Ewing sarcoma · Ewing family of tumours · Rhabdomyosarcoma · Soft-tissue sarcoma of childhood · Non-rhabdomyosarcoma soft-tissue sarcoma · Small round blue-cell tumour of bone
Fellowship guide to bone and soft-tissue sarcomas in children. Covers osteosarcoma as the commonest primary malignant bone tumour of adolescence arising in the metaphysis around the knee with a sunburst and Codman triangle periosteal reaction and osteoid production, Ewing sarcoma as the small round blue-cell tumour of bone carrying the EWSR1-FLI1 fusion and the onion-skin periosteal reaction in the diaphysis and flat bones, rhabdomyosarcoma as the commonest soft-tissue sarcoma of childhood split into the favourable embryonal and botryoid subtypes of the head and neck and genitourinary sites and the unfavourable PAX-FOXO1-fusion alveolar subtype, the red flags of persistent non-mechanical limb pain and a palpable mass that declare a sarcoma, the never-incisional-biopsy-without-planning rule that protects limb salvage, the staging pathway of magnetic resonance imaging of the whole compartment with computed tomography of the chest and an isotope bone scan or positron-emission tomography and marrow sampling, the neoadjuvant and adjuvant chemotherapy backbones of methotrexate-doxorubicin-cisplatin for osteosarcoma, vincristine-doxorubicin-cyclophosphamide alternating with ifosfamide-etoposide for Ewing sarcoma, and vincristine-actinomycin-cyclophosphamide for rhabdomyosarcoma, the place of limb-salvage surgery and radiotherapy, and the survivorship burden of cardiotoxicity, second malignancy and late effects across the life of the cured child.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A thirteen-year-old boy presents with three months of deep aching pain in the distal thigh, worse at night and unrelated to football, and his mother now feels a firm swelling just above the knee. The single decision that decides whether this child keeps his limb and his life is whether the clinician recognises the pattern of a malignant bone tumour and reaches the scan and the biopsy through the right service, rather than labelling it growing pains or a sports injury and sending him to physiotherapy. Bone and soft-tissue sarcomas are a heterogeneous family of mesenchymal malignancies of childhood and adolescence, and they are rare enough that the general paediatrician will meet only a few in a career but common enough that a delay in their recognition is one of the most heavily weighted errors in the fellowship examination. [1][4]
A sarcoma is a malignant tumour arising from the connective and supportive tissues, the bone, the cartilage, the muscle, the fat and the fibrous tissue, and it differs from the carcinoma in being a tumour of mesenchymal rather than epithelial origin. The paediatric sarcomas cluster into two groups. The bone sarcomas are led by osteosarcoma, the commonest primary malignant bone tumour of the young, and Ewing sarcoma, the small round blue-cell tumour of bone. The soft-tissue sarcomas are led by rhabdomyosarcoma, the commonest soft-tissue sarcoma of childhood, with a tail of the non-rhabdomyosarcoma soft-tissue sarcomas that are individually rare. The two bone tumours and the one dominant soft-tissue tumour are the three this page owns, because they are the three the boards examine and the three the general paediatrician must learn to recognise. [1][5][7]
The task at the bedside is not to name the tumour on the first visit but to judge whether the child is in danger, to image the lesion, and to place the child into the specialist sarcoma pathway before any tissue is removed. The gravity of the biopsy is the reason this topic rewards the careful clinician and punishes the hasty one, because a biopsy that is not planned with the future resection in mind contaminates the compartment and turns a limb-salvage operation into an amputation. The fellow who carries the red flags of persistent non-mechanical pain and a palpable mass, who reaches the magnetic resonance scan, and who refuses the unplanned biopsy is demonstrating exactly the reasoning the examination tests. [2][4]
Classification
The most useful way to classify a paediatric sarcoma at the bedside is by the tissue of origin and by whether it arises in bone or in soft tissue, because the site predicts the radiographic pattern, the differential and the treatment. The bone sarcomas hold osteosarcoma and Ewing sarcoma, and they declare themselves on the radiograph through the periosteal reaction and the matrix they produce. The soft-tissue sarcomas hold rhabdomyosarcoma and the non-rhabdomyosarcoma group, and they declare themselves as a deep mass in the head and neck, the genitourinary tract, the extremity or the trunk. The first branch the fellow draws at the bedside is therefore bone versus soft tissue, and the second is the named tumour within the branch. [4][5]
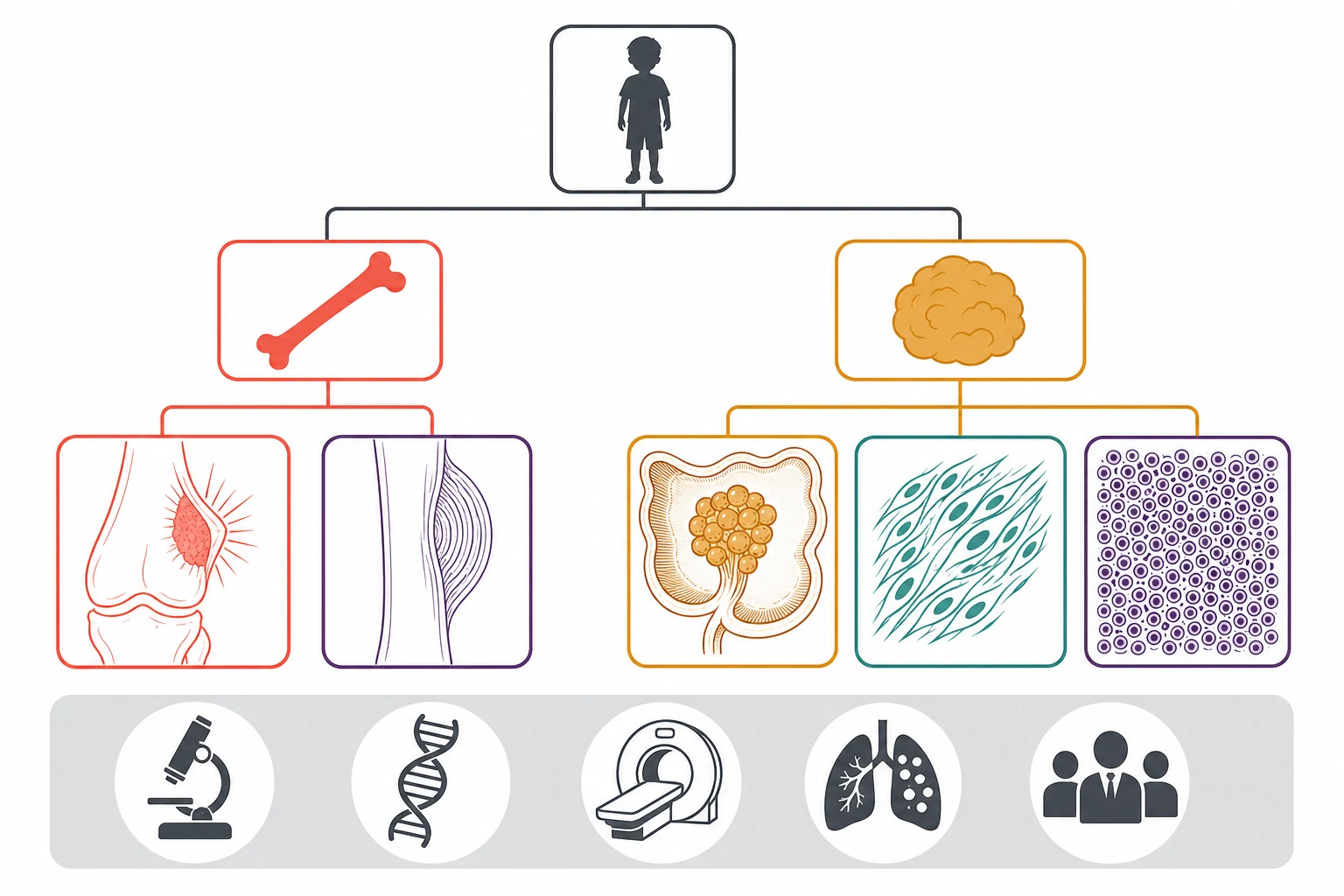
A parallel classification runs through the histology and the defining molecular alteration, and it is the layer that drives the modern diagnosis. Osteosarcoma is defined by the malignant osteoblasts that produce osteoid, the pink seams of bone matrix that are the histological hallmark regardless of the subtype. Ewing sarcoma is defined by the sheets of small round blue cells and by the pathognomonic translocation, the t(11;22) that fuses the EWSR1 gene on chromosome twenty-two with the FLI1 gene on chromosome eleven to form the EWSR1-FLI1 fusion in roughly four out of five cases. Alveolar rhabdomyosarcoma is defined by the PAX3-FOXO1 or the PAX7-FOXO1 fusion, which separates it from the fusion-negative embryonal subtype and carries the worse prognosis. The molecular layer matters because it confirms the diagnosis, it refines the risk group, and it is increasingly the target of the new therapies. [5][8]
Osteosarcoma
bone-forming
- Commonest primary malignant bone tumour of childhood and adolescence
- Metaphysis of long bones, especially around the knee and the proximal humerus
- Produces osteoid, the histological hallmark
- Sunburst and Codman triangle periosteal reaction on the radiograph
- Treated with neoadjuvant and adjuvant MAP chemotherapy and wide surgical resection
Ewing sarcoma
small round blue cell
- Second commonest paediatric bone tumour
- Diaphysis of long bones and the flat bones, pelvis, ribs, scapula, spine
- EWSR1-FLI1 fusion from the t(11;22) translocation
- Onion-skin or sunburst periosteal reaction with a large soft-tissue mass
- Treated with VDC/IE chemotherapy, surgery and selected radiotherapy
Rhabdomyosarcoma
soft-tissue
- Commonest soft-tissue sarcoma of childhood
- Head and neck and genitourinary sites predominate
- Embryonal and botryoid subtypes favourable, alveolar with PAX-FOXO1 unfavourable
- Presents as a painless mass, sometimes with obstruction or discharge
- Treated with VAC chemotherapy, surgery and radiotherapy, risk-stratified
Epidemiology & Risk Factors
Osteosarcoma and Ewing sarcoma together account for roughly six per cent of all childhood cancers, and rhabdomyosarcoma accounts for a further three per cent, so the sarcomas sit behind the leukaemias, the lymphomas and the central nervous system tumours in overall frequency but ahead of the other solid tumours in adolescence. Osteosarcoma has a sharp peak in the adolescent growth spurt, between ten and twenty years, because the rapidly dividing osteoblasts of the growing physis are the substrate for the malignant transformation, and it shows a slight male predominance. Ewing sarcoma peaks in the same adolescent window but occurs slightly younger in some series and is strikingly rare in children of African, East Asian and South Asian ancestry, with a marked predominance in European-derived populations. [1][5]
Rhabdomyosarcoma has a bimodal age distribution, with a peak in early childhood between one and four years and a second peak in adolescence, and it is the commonest soft-tissue sarcoma of children under the age of fifteen. The embryonal subtype dominates the younger child and the head-and-neck and genitourinary sites, while the alveolar subtype clusters in the older child and the extremities and the trunk. Roughly fifteen to twenty per cent of children with a sarcoma present with metastatic disease, and the lung is the single commonest site of haematogenous spread, with bone and bone marrow added in Ewing sarcoma and rhabdomyosarcoma. The burden of metastatic disease at the diagnosis is one of the strongest determinants of the outcome. [7][8]
The inherited cancer predisposition syndromes are the risk factors worth naming, because they change the counselling and the surveillance of the family. Li-Fraumeni syndrome, with its germline TP53 mutation, raises the risk of osteosarcoma and other sarcomas across the lifespan, and a child with a sarcoma and a family history of early-onset breast cancer, brain tumour or sarcoma is offered the genetic assessment. Hereditary retinoblastoma, with the germline RB1 mutation, raises the risk of osteosarcoma within and outside the radiation field. Rothmund-Thomson syndrome and Werner syndrome, the RECQL helicase disorders, raise the risk of osteosarcoma. Prior therapeutic radiation and the rare Paget disease of bone in the older patient complete the list, while the bulk of paediatric sarcomas arise without an identifiable cause. [1][2]
Pathophysiology
The pathophysiology of the bone sarcoma begins in the bone-forming cell and the marrow cavity, and it explains both the radiographic pattern and the metastatic behaviour. Osteosarcoma arises from the malignant osteoblast, the cell that should lay down healthy bone, and the tumour produces its own disordered osteoid that appears as the cloudlike density and the sunburst spicules on the radiograph. As the tumour breaks through the cortex it lifts the periosteum, and the reactive bone laid down at the angled edge of the lifted periosteum forms the Codman triangle, while the perpendicular reactive bone forms the sunburst. The mixed lytic and sclerotic appearance reflects the balance between the bone destruction and the osteoid production. [2]
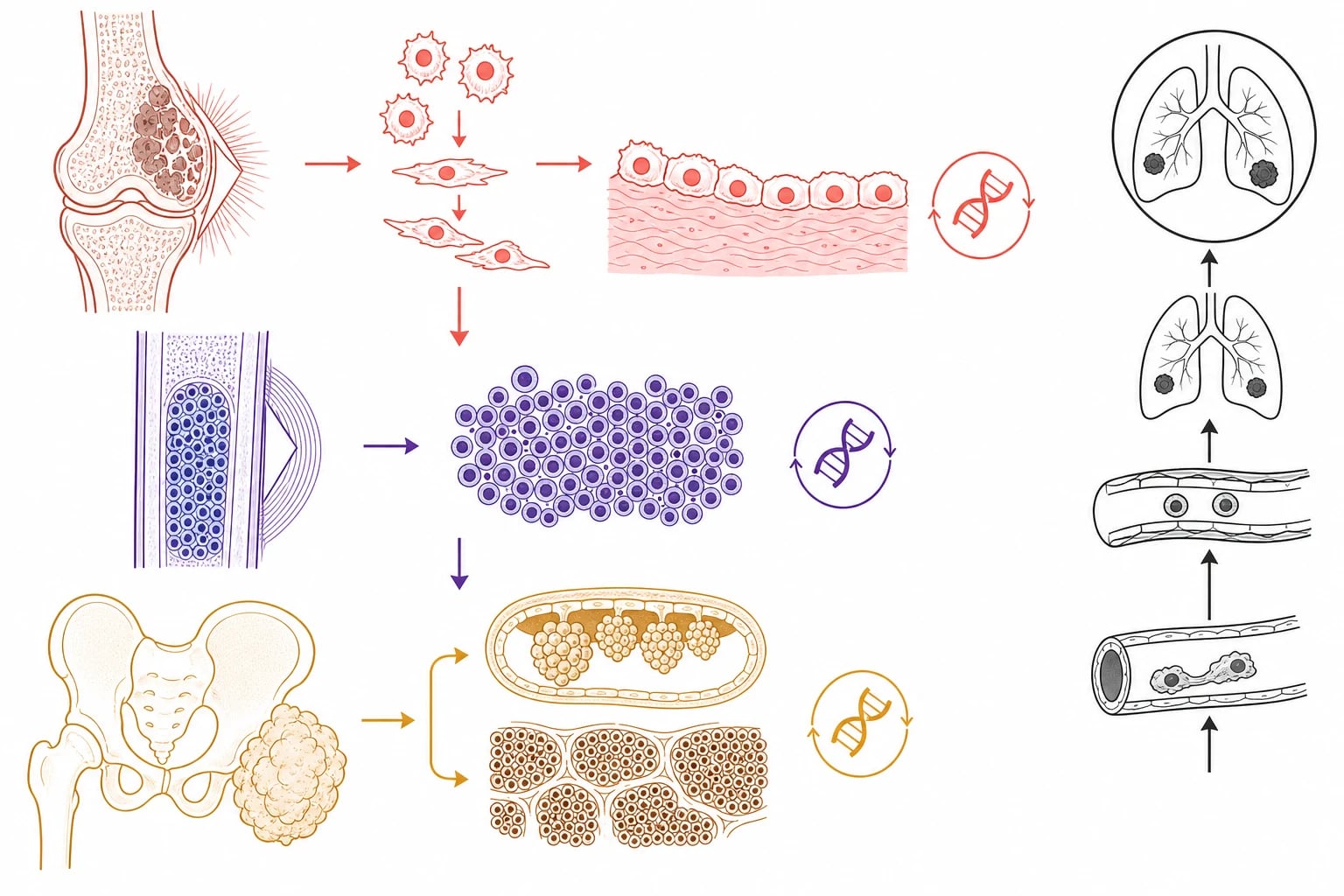
Ewing sarcoma arises from a different cell, the primitive neuroectodermal or mesenchymal stem cell, and it carries a defining chromosomal translocation that is both the diagnostic marker and the oncogenic driver. The t(11;22) translocation fuses the EWSR1 gene with the FLI1 gene to produce the EWSR1-FLI1 fusion protein, an aberrant transcription factor that switches on a programme of undifferentiated proliferation. The tumour fills the medullary cavity of the diaphysis with sheets of small round blue cells, it permeates the Haversian systems to break through the cortex, and it provokes the layered onion-skin and the sunburst periosteal reactions as it elevates the periosteum layer by layer. The large extraosseous soft-tissue mass is a hallmark that often dwarfs the bone lesion. [5]
Rhabdomyosarcoma arises from the primitive mesenchymal cell that is committed to the skeletal-muscle lineage, and its biology turns on whether the myogenic differentiation is arrested early or late. The embryonal subtype, fusion-negative, retains some capacity to differentiate along the muscle pathway and behaves more favourably, and its botryoid variant grows as a grape-like cluster inside a hollow viscus such as the bladder, the vagina or the nasopharynx. The alve subtype, carrying the PAX3-FOXO1 or the PAX7-FOXO1 fusion, is driven by an aberrant transcription factor that locks the cell in a proliferative state, and it behaves aggressively with a higher rate of metastasis and a worse outcome. The haematogenous spread to the lung, and the lymphatic spread to the regional nodes, are common to all three tumours and they shape the staging. [8][9]
Clinical Presentation
The child with a bone sarcoma presents most often with pain, and the character of the pain is the clue that separates the tumour from the ordinary injury. The pain of a bone sarcoma is deep, it is unrelated to activity, it is often worse at night, and it progresses over weeks rather than resolving with rest. A child or adolescent with persistent limb pain that wakes them from sleep, that does not settle, and that is accompanied by a limp or a reluctance to bear weight has a bone tumour until imaging proves otherwise, however normal the early radiograph may appear. A firm, deep, fixed mass becomes palpable as the tumour enlarges, and a pathological fracture through the weakened bone is occasionally the first presentation. [2][4]
Ewing sarcoma adds the systemic features that can mislead the clinician, because the fever, the malaise, the weight loss and the anaemia can mimic an infection and the local warmth and swelling can mimic osteomyelitis. The pelvic and the axial lesions present with vague pain that is slow to localise, and the chest-wall Ewing sarcoma, the classic Askin tumour, presents with a pleural effusion and dyspnoea. The lesson is that a diaphyseal bone lesion with a large soft-tissue mass and systemic symptoms is Ewing sarcoma until the biopsy and the fusion settle it, and the broad-spectrum antibiotics that treat the mimicked osteomyelitis must not delay the imaging. [5][6]
Rhabdomyosarcoma presents as a painless mass whose location dictates the syndrome, and the head-and-neck and genitourinary sites dominate. An orbital rhabdomyosarcoma presents with proptosis and a swollen eyelid. A parameningeal rhabdomyosarcoma, in the nasopharynx, the paranasal sinuses or the middle ear, presents with nasal obstruction, discharge, a facial swelling, or a cranial nerve palsy, and it can extend through the skull base to cause cord or cranial-nerve compression. A bladder or prostate rhabdomyosarcoma presents with urinary obstruction or a pelvic mass, and a vaginal rhabdomyosarcoma presents with a grapelike mass protruding through the introitus, the botryoid presentation of the embryonal subtype. The paratesticular rhabdomyosarcoma presents as a painless scrotal swelling. [7][8]
The non-rhabdomyosarcoma soft-tissue sarcomas present as a deep, firm, slowly enlarging mass in the soft tissues, often greater than five centimetres, and they are individually rare in children. The lesson for the general paediatrician is the same for all the soft-tissue masses, which is that a deep mass greater than five centimetres, a mass that is enlarging, or a mass that is not the simple superficial lesion it appears to be is referred for imaging and specialist review before any excision. The size and the depth are the two features that flag the soft-tissue mass as suspicious, and the rule of referring the suspicious mass before the biopsy applies to the soft-tissue sarcoma as much as to the bone sarcoma. [4][10]
Differential Diagnosis
The differential of the child with a painful bone lesion is built around the radiographic pattern and the tempo, and the magnetic resonance imaging resolves the majority. The benign bone tumours, the simple bone cyst, the aneurysmal bone cyst, the non-ossifying fibroma and the osteoid osteoma, present with pain or a pathological fracture and a characteristic radiograph, and they are separated from the malignant lesion by the well-defined margins and the absence of the aggressive periosteal reaction. The osteomyelitis and the septic arthritis present with acute pain, fever and the systemic features, and they mimic the Ewing sarcoma with the diaphyseal pain and the swelling, which is why the blood cultures and the inflammatory markers are sent alongside the scan. [4][5]
The Langerhans cell histiocytosis, the eosinophilic granuloma, produces a lytic lesion that can mimic a tumour, and the leukaemia and the metastatic neuroblastoma produce the diffuse bone pain and the marrow infiltration that must be excluded in the younger child with the systemic symptoms. The differential of the soft-tissue mass is narrower, and it holds the benign lesions such as the haemangioma, the lymphangioma and the fibromatosis alongside the malignant rhabdomyosarcoma and the non-rhabdomyosarcoma sarcoma. The magnetic resonance imaging and the biopsy resolve the soft-tissue differential, and the rule of never excising a suspicious mass without the prior imaging and the histology protects the child from the inadequate excision of an unsuspected sarcoma. [4][7]
The chief diagnostic pitfalls are the cases in which the diagnosis is delayed because the pattern is misread. The child whose limb pain is labelled a sports injury or growing pains, and whose night pain and progression are missed, is the classic delayed diagnosis of osteosarcoma. The child whose Ewing sarcoma is treated as osteomyelitis with antibiotics, and whose radiograph is not pursued to the magnetic resonance scan, is the second. The child whose orbital swelling is treated as a cellulitis, and whose proptosis is missed, is the third. The single message for the exam is that the persistent non-mechanical pain and the unexplained mass are imaged, and the normal early radiograph is never the end of the workup. [2][8]
Clinical & Bedside Assessment
The bedside assessment of the child with a suspected sarcoma is a search for the local signs of the tumour, the regional signs of the spread, and the systemic features that declare the metastatic or the advanced disease. The assessment begins with the general inspection for the pallor, the weight loss and the systemic illness, because the fever and the malaise of the Ewing sarcoma and the anaemia of the marrow disease are detected at the bedside. The vital signs and the pain score are recorded, and the child who is in severe pain or who shows the signs of cord compression is moved to the emergency imaging and the analgesia. [2]
The local examination of the limb or the mass is systematic and takes only a few minutes, but each finding carries weight. The mass is measured and its size, its depth, its consistency and its fixity to the underlying structures are recorded, because a deep mass greater than five centimetres is a sarcoma until proven otherwise. The overlying skin is inspected for the warmth, the erythema and the venous dilatation that suggest a highly vascular lesion, and the regional lymph nodes are palpated because the lymphatic spread is significant in rhabdomyosarcoma and the epithelioid sarcoma. The joint above and below the lesion is examined for the effusion and the range of movement, and the neurovascular status of the limb distal to the lesion is documented because the tumour can compress the nerve or the vessel. [7][10]
The focused assessment of the child with a suspected bone or soft-tissue sarcoma
Assess the airway, breathing and circulation and the pain, and look for the pallor, the weight loss and the systemic illness
Inspect and palpate the mass, recording the size, the depth, the consistency and the fixity, and the overlying skin changes
Examine the regional lymph nodes, because the lymphatic spread is significant in rhabdomyosarcoma
Test the neurovascular status of the limb distal to the lesion, looking for the nerve or the vessel compression
Examine the joint above and below for the effusion and the movement, and observe the gait for the antalgic limp
Look for the cord compression in any child with a parameningeal or a spinal lesion, examining for the weakness, the sensory level and the sphincter tone
Request the plain radiograph and the magnetic resonance imaging of the whole compartment, and refer to the specialist sarcoma service before any biopsy
The recognition of the oncologic emergency at the bedside is the skill that changes the outcome, because the cord compression and the tumour lysis syndrome are the two complications that demand the immediate action. A child with a parameningeal or a spinal tumour who develops the back pain, the progressive limb weakness, the sensory level or the new sphincter disturbance has spinal cord compression until imaging proves otherwise, and the dexamethasone and the emergency magnetic resonance imaging are given without delay. A child with a bulky chemosensitive tumour at the start of the treatment is at risk of the tumour lysis syndrome, and the hydration and the urate-lowering prophylaxis begin before the first dose of the chemotherapy. [4][11]
Investigations
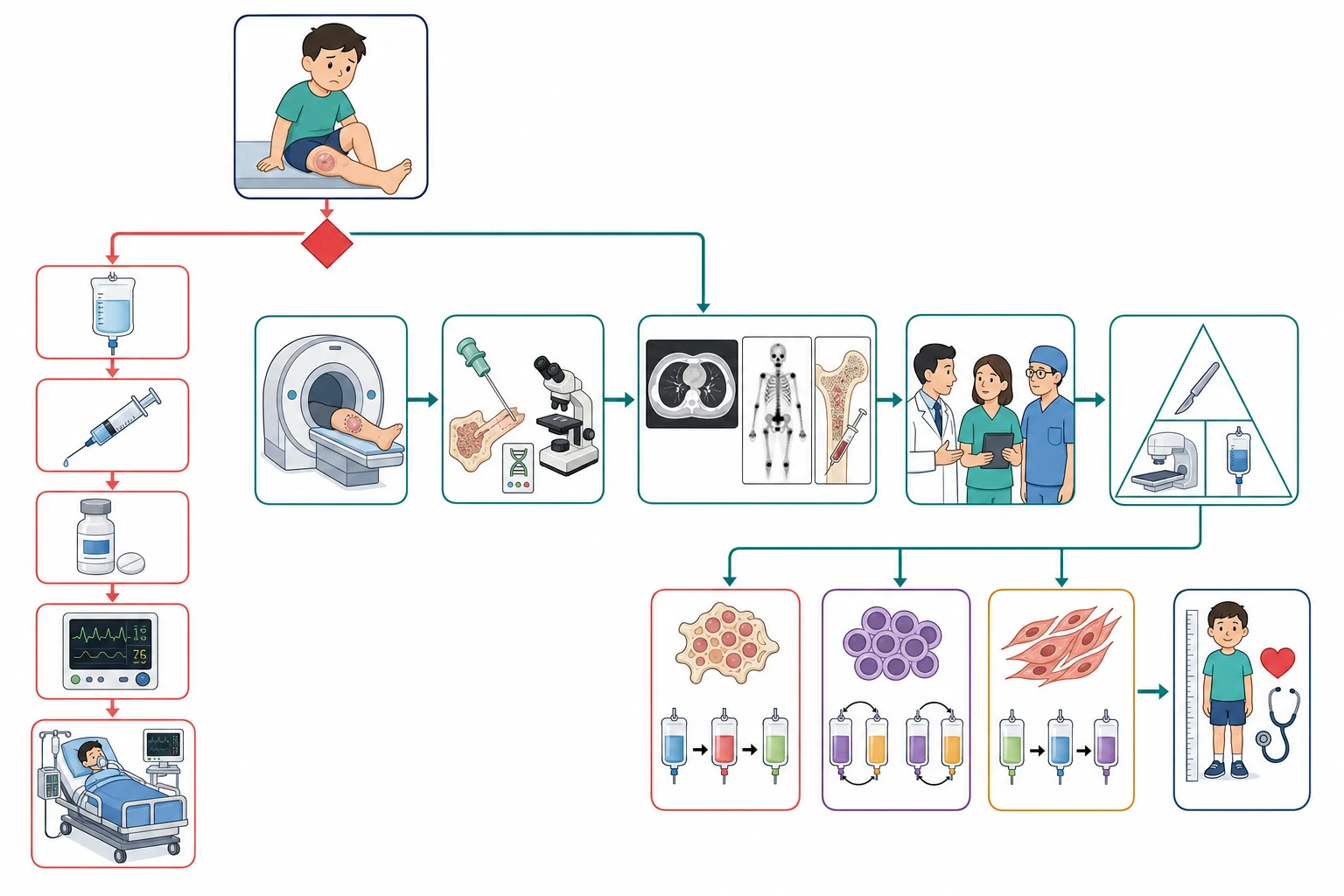
The investigation of a suspected sarcoma moves in three steps, the imaging that defines the local tumour, the biopsy that names it, and the staging that maps the distant disease, and the order matters because the biopsy must come after the complete imaging and through the right service. The first test for a suspected bone sarcoma is the plain radiograph of the limb in two planes, because the radiograph often shows the aggressive pattern of the permeative destruction, the cortical breach, the sunburst or the Codman triangle and the onion-skin that flag the malignant lesion. The early radiograph can be subtle, and a normal radiograph never excludes a bone sarcoma in the child with the persistent pain. [2][4]
The magnetic resonance imaging of the whole compartment, from joint to joint, is the local staging test, because it defines the tumour extent within the bone, the soft-tissue mass, the involvement of the neurovascular bundle and the skip lesions within the same bone. The magnetic resonance imaging is the study that decides whether the limb-salvage surgery is possible, because the encasement of the major vessel or the nerve is the finding that turns the plan toward the amputation. The computed tomography of the chest is the test for the pulmonary metastases, the commonest site of the haematogenous spread, and the isotope bone scan is the test for the skeletal metastases in osteosarcoma. The positron-emission tomography combined with the computed tomography is increasingly used in the Ewing sarcoma and the rhabdomyosarcoma to detect the distant disease and to assess the response to the treatment. [4][6]
Across Australia, Aotearoa New Zealand, the United Kingdom, the United States and Canada, the magnetic resonance imaging of the whole compartment, the computed tomography of the chest, the isotope bone scan or the positron-emission tomography, and the bone-marrow sampling for the Ewing sarcoma and the rhabdomyosarcoma form the standard staging panel. The biopsy of a suspected sarcoma is performed at the specialist centre that will deliver the definitive surgery, because the tract must be planned to lie within the future resection. The fellow should know the local sarcoma referral pathway and the nearest paediatric oncology centre.
[4][10]The bone-marrow aspirate and trephine are sent in the Ewing sarcoma and the rhabdomyosarcoma because both tumours can infiltrate the marrow, and they are not required in the osteosarcoma. The biopsy is the diagnostic test, and it is performed as an image-guided core needle biopsy or an incisional biopsy at the specialist centre, never as an unplanned excision. The histology names the tumour, and the molecular studies add the defining fusion, the EWSR1-FLI1 for the Ewing sarcoma and the PAX-FOXO1 for the alveolar rhabdomyosarcoma. The blood tests are sent for the baseline and the prognostic markers, including the full blood count, the lactate dehydrogenase that reflects the tumour burden and the turnover, and the electrolytes and the renal function before the chemotherapy. [5][8]
Management — Resuscitation
The resuscitation of the child with a sarcoma rests on three legs, the relief of the pain, the management of the oncologic emergency, and the prophylaxis of the tumour lysis syndrome at the start of the chemotherapy. The analgesia is built around the World Health Organization analgesic ladder, escalating from the paracetamol and the non-steroidal anti-inflammatory drug to the opioid for the severe bone pain, and the pain of the bone sarcoma often requires the regular opioid until the chemotherapy and the surgery reduce the tumour burden. The pathological fracture through the tumour is splinted and stabilised, and the child is made non-weight-bearing on the affected limb. [2]
The tumour lysis syndrome is the metabolic emergency that follows the rapid release of the intracellular contents from the killed tumour cells, and the bulky chemosensitive sarcoma at the start of the treatment is the high-risk setting. The Cairo-Bishop classification defines the laboratory tumour lysis syndrome as two or more abnormalities, within three days before to seven days after the start of the therapy, of the raised uric acid, the raised potassium, the raised phosphate and the lowered calcium, each by an absolute threshold or a twenty-five per cent change from the baseline. The clinical tumour lysis syndrome adds the acute kidney injury, the cardiac arrhythmia and the seizure to the laboratory derangement. The prophylaxis rests on the intravenous hydration and the urate lowering, with the allopurinol for the intermediate-risk and the rasburicase for the high-risk or the established syndrome. [11]
The spinal cord compression is the emergency that does not wait, and the child with the back pain, the progressive limb weakness, the sensory level or the new sphincter disturbance is given the dexamethasone and the emergency magnetic resonance imaging. The cord does not recover once it is compressed, and the window for the functional recovery is short, which is why the recognition of the progressive paraparesis and the emergency imaging are so heavily weighted in the exam. The definitive treatment of the cord compression is the surgery or the urgent radiotherapy, depending on the tumour and the stability, and the parameningeal rhabdomyosarcoma and the spinal Ewing sarcoma are the two tumours that most often produce it. [4][6]
Management — Definitive & Stepwise
The definitive management of the paediatric sarcoma is built around the three modalities of the chemotherapy, the surgery and the radiotherapy, and the combination is tailored to the tumour, the site, the metastatic status and the response. The overarching principle is that the surgery is the foundation of the local control for the resectable tumour, that the chemotherapy is given both before and after the surgery for the high-grade bone and soft-tissue sarcomas, and that the radiotherapy is reserved for the unresectable, the incompletely resected and the chemoresponsive lesions such as the Ewing sarcoma and the rhabdomyosarcoma. The histologic response to the neoadjuvant chemotherapy, the proportion of the tumour that is killed, is the prognostic marker that refines the postoperative treatment. [2][3]
[2] [6]Osteosarcoma is the paradigm of the neoadjuvant chemotherapy, and the regimen the fellow must know is the MAP backbone of the high-dose methotrexate, the doxorubicin and the cisplatin, given before the surgery to shrink the tumour and to treat the micrometastatic disease. The definitive surgery is the wide local resection with the limb-salvage reconstruction whenever it is oncologically safe, and the amputation is reserved for the tumour that cannot be resected with the clear margins because it encases the neurovascular bundle. The postoperative chemotherapy is continued, and the histologic response guides the intensity, because the good responder, with at least ninety per cent tumour necrosis, carries the better prognosis. The EURAMOS-1 trial tested the addition of the maintenance pegylated interferon to the MAP regimen in the good responders, and it refined the modern approach. [2][3]
Risk-adapted therapy for osteosarcoma
Dose
Neoadjuvant MAP chemotherapy with high-dose methotrexate, doxorubicin and cisplatin, followed by the wide local resection with limb-salvage reconstruction or amputation, and the adjuvant MAP chemotherapy guided by the histologic response, with the maintenance pegylated interferon added in selected good responders on the EURAMOS-1 schema
Ewing sarcoma is managed with the alternating chemotherapy regimen of the VDC and the IE, the vincristine, the doxorubicin and the cyclophosphamide alternating with the ifosfamide and the etoposide, given before the local control. The local control is the surgery wherever it is feasible, because the complete resection offers the best local control and the best survival, and the radiotherapy is added for the incompletely resected, the unresectable, and the lesions with the poor histologic response. The pelvis and the spine, where the surgery is difficult, are the sites that most often rely on the radiotherapy. The metastatic Ewing sarcoma is treated with the intensified chemotherapy and the local control to all the sites of the disease, including the lungs, and the prognosis is guarded. [5][6]
Rhabdomyosarcoma is managed with the VAC backbone of the vincristine, the actinomycin D and the cyclophosphamide, and the treatment is risk-stratified by the pretreatment stage, the clinical group and the histology. The surgery is the primary local control for the resectable lesion, and the radiotherapy is added for the group-two to group-four disease, the residual tumour and the alveolar subtype, because the rhabdomyosarcoma is a radiosensitive tumour. The low-risk embryonal rhabdomyosarcoma of a favourable site, such as the orbit or the vagina, is treated with the less intensive chemotherapy and carries the excellent prognosis, while the metastatic alveolar rhabdomyosarcoma carries the poor prognosis and is treated on the high-risk protocols. The lymph-node sampling and the sentinel-node biopsy guide the regional staging, following the contemporary consensus. [7][10]
Specific Subtypes & Scenarios
Osteosarcoma and the adolescent with the painful knee
Osteosarcoma is the commonest primary malignant bone tumour of childhood and adolescence, and the paradigmatic presentation is the adolescent with the deep pain around the knee and the firm mass of the distal femur or the proximal tibia. The tumour arises in the metaphysis, and the commonest sites are the distal femur, the proximal tibia and the proximal humerus, the bones that grow fastest in the adolescent spurt. The radiograph shows the mixed lytic and sclerotic lesion with the cortical destruction, the sunburst spicules and the Codman triangle, and the magnetic resonance imaging defines the medullary extent and the soft-tissue mass. The lung is the dominant site of the metastasis, and the computed tomography of the chest is mandatory at the staging. The treatment is the neoadjuvant MAP chemotherapy, the wide resection and the adjuvant chemotherapy, and the localised disease carries the five-year survival of about seventy per cent. [1][2]
Ewing sarcoma and the diaphyseal lesion with the systemic illness
Ewing sarcoma is the second commonest paediatric bone tumour, and it differs from the osteosarcoma in the site, the histology and the molecular driver. The tumour arises in the diaphysis of the long bones and the flat bones, with the pelvis, the ribs and the femur the commonest sites, and it presents with the pain, the swelling and the systemic features that can mimic the infection. The radiograph shows the permeative lytic diaphyseal lesion with the onion-skin or the sunburst periosteal reaction and the large soft-tissue mass. The defining feature is the EWSR1-FLI1 fusion of the t(11;22) translocation, and the molecular study confirms the diagnosis. The treatment is the VDC/IE chemotherapy, the surgery and the selected radiotherapy, and the disease is radiosensitive in contrast to the osteosarcoma. [5][6]
SARCOMA
Rhabdomyosarcoma and the head-and-neck or genitourinary mass
Rhabdomyosarcoma is the commonest soft-tissue sarcoma of childhood, and the histology drives the prognosis. The embryonal subtype, common in the young child, arises in the head and neck and the genitourinary tract, and it carries the favourable prognosis, with the botryoid variant presenting as the grapelike mass in the bladder, the vagina or the nasopharynx. The alveolar subtype, common in the adolescent, arises in the extremities and the trunk, and it carries the PAX3-FOXO1 or the PAX7-FOXO1 fusion and the unfavourable prognosis. The staging combines the pretreatment TNM, based on the site, the size, the nodal status and the metastasis, with the clinical group, based on the extent of the resection and the residual disease. The treatment is the risk-stratified VAC chemotherapy, the surgery and the radiotherapy, and the localised embryonal disease carries the survival of around eighty per cent while the metastatic alveolar disease carries the survival of under thirty per cent. [7][8]
The non-rhabdomyosarcoma soft-tissue sarcomas
The non-rhabdomyosarcoma soft-tissue sarcomas are individually rare in children, and they include the synovial sarcoma, the malignant peripheral nerve sheath tumour, the infantile fibrosarcoma and the alveolar soft-part sarcoma. The unifying presentation is the deep, firm, slowly enlarging mass, often greater than five centimetres, and the management rests on the wide surgical resection with the adjuvant radiotherapy for the high-grade or the incompletely resected lesion. The infantile fibrosarcoma is the exception, because it carries the favourable prognosis and the defining ETV6-NTRK3 fusion that responds to the targeted therapy with the TRK inhibitors. The fellow should know the group exists and that the deep mass greater than five centimetres is the red flag that drives the referral. [4][8]
Complications & Pitfalls
The complications of the paediatric sarcoma divide into the disease-related and the treatment-related, and the fellow must hold both because the iatrogenic harm can rival the disease. The disease-related complications are the pathological fracture, the spinal cord compression, the cord and the cranial-nerve compromise of the parameningeal lesion, and the metastatic disease to the lung, the bone and the marrow. The oncologic emergencies of the cord compression and the tumour lysis syndrome are the complications that drive the resuscitation and that are anticipated from the moment the tumour is seen on the scan. [4][11]
The surgical complications centre on the limb-salvage reconstruction and its failure, and the infection, the loosening of the prosthesis, the fracture and the leg-length discrepancy are the long-term burdens of the endoprosthetic reconstruction. The amputation, where it is unavoidable, carries its own burden of the phantom pain and the prosthetic fitting. The radiotherapy complications are the growth arrest of the irradiated physis, the soft-tissue fibrosis, the contracture and the radiation-induced second malignancy in the field years later. The pelvic and the spinal radiotherapy add the gonadal damage and the infertility, and the fertility preservation is offered before the gonadotoxic chemotherapy in every adolescent. [4][12]
The chemotherapy complications are the costs of the cure, and they shape the survivorship. The anthracycline cardiotoxicity of the doxorubicin is cumulative and dose-dependent, and the cardiac surveillance with the echocardiography is lifelong. The cisplatin produces the sensorineural hearing loss and the renal toxicity, and the audiometry and the renal monitoring are part of the follow-up. The alkylating agents, the cyclophosphamide and the ifosfamide, produce the gonadal damage, the haemorrhagic cystitis and the second malignancy, and the ifosfamide adds the encephalopathy. The secondary malignancy, the myelodysplasia and the leukaemia, and the radiation-induced sarcoma, are the late costs that the survivor carries for life. [12]
Prognosis & Disposition
The prognosis of a child with a sarcoma spans the range from the cure of the localised embryonal rhabdomyosarcoma to the guarded outlook of the metastatic alveolar disease, and it is determined by the tumour type, the site, the metastatic status, the histologic response and the completeness of the resection. The localised osteosarcoma carries the five-year survival of about seventy per cent, the localised Ewing sarcoma of about seventy per cent, and the localised embryonal rhabdomyosarcoma of about eighty per cent. The metastatic disease cuts the survival sharply, to under thirty per cent for the metastatic osteosarcoma and the alveolar rhabdomyosarcoma, and the lung metastases that are resectable carry a better outlook than the widespread disease. [1][8]
The disposition of the child is the specialist paediatric oncology centre, and the management is delivered through the multidisciplinary team. The child with the newly diagnosed sarcoma is transferred to the tertiary centre with the paediatric oncology, the orthopaedic oncology, the radiation oncology, the radiology and the pathology, and the regional or the rural hospital is responsible for the recognition and the stabilisation before the retrieval. The surgery is performed at the centre that can deliver the limb-salvage reconstruction or the amputation, and the radiotherapy is delivered at the specialised paediatric radiation oncology service. The fellow who builds the multidisciplinary plan demonstrates the care that the complex disease demands. [2][6]
In Australia and Aotearoa New Zealand, the child with a newly diagnosed sarcoma is managed in a tertiary paediatric oncology centre with the paediatric retrieval services transferring the unstable child from the regional or the rural hospital. The orthopaedic oncology and the radiation oncology are delivered in the few specialised centres, and the family is supported by the social work, the educational liaison and the psychology. The long distances and the retrieval times are the reason the early recognition and the stabilisation in the referring hospital are so heavily weighted in the exam, and the Australia and New Zealand Sarcoma Association provides the referral and the clinical-trial framework.
[4]The long-term surveillance of the survivor is the reward and the burden of the cure, because the late effects of the therapy are common and they accumulate with the time. The survivor has the imaging for the local recurrence and the lung metastases, the echocardiography for the anthracycline cardiotoxicity, the audiometry for the cisplatin hearing loss, and the renal and the endocrine surveillance for the gonadal damage and the growth effects. The secondary malignancy surveillance, the fertility counselling, and the vocational and the educational support are addressed before the transition to the adult late-effects service. The fellow who builds the survivorship plan demonstrates the care that extends beyond the cure. [12]
Special Populations
The adolescent at the peak age of the osteosarcoma and the Ewing sarcoma is the special population that holds the additional burden of the growth, the body image and the fertility. The limb-salvage surgery in the growing adolescent carries the leg-length discrepancy and the need for the expandable prosthesis, and the amputation carries the body-image and the prosthetic burden. The gonadotoxic chemotherapy places the fertility at risk, and the fertility preservation, the sperm banking in the male and the oocyte or the ovarian-tissue cryopreservation in the female, is offered before the gonadotoxic therapy. The transition to the adult care is prepared in the late adolescence with the counselling, the documentation and the survivorship plan. [12]
The child with the inherited cancer predisposition syndrome is the special population that changes the counselling and the surveillance. The Li-Fraumeni syndrome, with its germline TP53 mutation, raises the risk of the osteosarcoma and the other sarcomas, and the child with a sarcoma and the suggestive family history is offered the genetic assessment that informs the screening for the breast cancer, the brain tumour and the second sarcoma across the lifespan. The hereditary retinoblastoma and the Rothmund-Thomson syndrome are the other predispositions that are sought in the child with the osteosarcoma. These syndromes are identified because they change the plan and the surveillance of the family. [1][2]
Socioeconomic disadvantage, remoteness and the migrant or refugee status shape the access to the diagnosis and the treatment, and they are the reason the early recognition in the primary care and the regional hospital is so heavily emphasised. A child far from a specialist centre may first present to a clinician who sees few such cases, and the persistent limb pain or the unexplained mass that flags the tumour is the bridge to the retrieval and the specialist care. The language and the cultural barriers are addressed with the interpreter and the cultural support, and the family is supported through the long and the unfamiliar treatment, with the accommodation and the educational liaison for the child who travels for the therapy. [4]
The very young child with a sarcoma holds a special position, because the infantile tumours are biologically distinct and the therapy is built around the avoidance of the long-term harm. The infantile fibrosarcoma carries the ETV6-NTRK3 fusion and the favourable response to the targeted therapy, and the congenital and the infantile rhabdomyosarcoma carry a distinct biology. The radiotherapy is avoided where possible in the very young child because of the growth and the developmental injury, and the chemotherapy and the surgery are tailored. The fellow should know the young child is biologically distinct and that the infant protocols differ from the adolescent ones. [8][9]
Evidence, Guidelines & Regional Differences
The landmark evidence that underpins the modern treatment of the paediatric sarcoma is the product of successive international collaborative trials, and it is the reason the survival of the localised disease has risen while the metastatic disease remains the challenge. The EURAMOS-1 collaboration defined the role of the maintenance pegylated interferon in the good responders to the MAP chemotherapy for the osteosarcoma, and it established the histologic response as the marker that guides the postoperative intensity. The European and the American Ewing sarcoma trials refined the VDC/IE backbone and the local control, and the Intergroup Rhabdomyosarcoma Studies and the Children's Oncology Group trials defined the VAC backbone and the risk stratification for the rhabdomyosarcoma. [3][7]
The contemporary evidence is built around the biology and the targeted therapy. The molecular classification of the Ewing sarcoma by the EWSR1 fusion and the rhabdomyosarcoma by the PAX-FOXO1 fusion has refined the diagnosis and the risk group, and the targeted agents against the fusions and the receptor pathways are the research frontier. The proton therapy is increasingly used to deliver the radiotherapy with the reduced exit dose and the lower late-effect burden, especially for the spinal and the pelvic Ewing sarcoma and the parameningeal rhabdomyosarcoma. The international consensus on the lymph-node staging and the treatment of the paediatric soft-tissue sarcoma has unified the approach across the study groups. [5][10]
Across Australia, Aotearoa New Zealand, the United Kingdom, the United States and Canada, the MAP backbone for the osteosarcoma, the VDC/IE backbone for the Ewing sarcoma and the VAC backbone for the rhabdomyosarcoma are the consistent standard, with the risk-adapted intensity. The radiotherapy practice and the proton versus the photon modality vary across the regions, with the proton therapy increasingly preferred for the paediatric sarcoma to reduce the late effects. The local control, the surgery versus the radiotherapy for the pelvic and the spinal Ewing sarcoma, follows the regional protocol, and the fellow should know the local guidance.
[4][6]The controversies and the open questions are the live ones. The place of the maintenance therapy and the immunotherapy in the osteosarcoma is being defined by the contemporary trials, and the metastatic osteosarcoma remains the disease in which the survival has barely moved. The optimal local control for the pelvic Ewing sarcoma, the surgery versus the radiotherapy, is a continuing debate. The role of the targeted agents and the immunotherapy in the relapsed rhabdomyosarcoma is the research frontier, and the de-escalation of the therapy in the favourable-risk disease to reduce the late effects is balanced against the recurrence risk. The survivorship burden of the evolving regimens is now quantified by the Childhood Cancer Survivor Study, and it shapes the contemporary risk-benefit counselling. [8][12]
Exam Pearls
The high-yield facts for the exam are the ones that change a decision at the bedside, and they are worth carrying as sharp statements. Persistent non-mechanical limb pain that wakes the child at night and a palpable mass is a sarcoma until imaging proves otherwise, and the early radiograph can be normal. The three tumours to know are osteosarcoma (metaphyseal, osteoid, sunburst and Codman triangle), Ewing sarcoma (diaphyseal and flat bones, small round blue cell, EWSR1-FLI1 fusion, onion-skin) and rhabdomyosarcoma (embryonal favourable with botryoid, alveolar unfavourable with PAX-FOXO1). The biopsy is never unplanned, because the tract must be excised with the tumour. [2][5]
The staging pathway is the magnetic resonance imaging of the whole compartment, the computed tomography of the chest, the isotope bone scan or the positron-emission tomography, and the bone-marrow sampling for the Ewing sarcoma and the rhabdomyosarcoma. The chemotherapy backbones are the MAP for the osteosarcoma, the VDC/IE for the Ewing sarcoma and the VAC for the rhabdomyosarcoma, alongside the surgery and the radiotherapy, the last reserved for the unresectable and the chemoresponsive lesions. The localised osteosarcoma and the Ewing sarcoma carry the five-year survival of about seventy per cent, and the metastatic disease cuts it to under thirty per cent. [3][6]
The final pearls are the ones that catch the candidate who has learned the headline and forgotten the corner. The Ewing sarcoma mimics the osteomyelitis, and the antibiotics must not delay the imaging. The parameningeal rhabdomyosarcoma extends through the skull base to cause the cord and the cranial-nerve compromise. The chest-wall Ewing sarcoma, the Askin tumour, presents with the pleural effusion. The infantile fibrosarcoma carries the ETV6-NTRK3 fusion and the response to the TRK inhibitors. The message for the exam is that the corners are where the marks are won, and the reasoning that holds the red flags and the biopsy principle at the centre is the one the boards reward. [5][8]
References
- [1]Mirabello L, Troisi RJ, Savage SA Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer, 2009.PMID 19197972
- [2]Isakoff MS, Bielack SS, Meltzer P, Gorlick R Osteosarcoma: Current Treatment and a Collaborative Pathway to Success. J Clin Oncol, 2015.PMID 26304877
- [3]Bielack SS, Smeland S, Whelan JS, Marina N, et al. Methotrexate, Doxorubicin, and Cisplatin (MAP) Plus Maintenance Pegylated Interferon Alfa-2b Versus MAP Alone in Patients With Resectable High-Grade Osteosarcoma and Good Histologic Response to Preoperative MAP: First Results of the EURAMOS-1 Good Response Randomized Controlled Trial. J Clin Oncol, 2015.PMID 26033801
- [4]Strauss SJ, Frezza AM, Abecassis N, Bajpai J, et al. Bone sarcomas: ESMO-EURACAN-GENTURIS-ERN PaedCan Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol, 2021.PMID 34500044
- [5]Grunewald TGP, Cidre-Aranaz F, Surdez D, Tomazou EM, et al. Ewing sarcoma. Nat Rev Dis Primers, 2018.PMID 29977059
- [6]Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J Clin Oncol, 2015.PMID 26304893
- [7]Malempati S, Hawkins DS Rhabdomyosarcoma: review of the Children's Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer, 2012.PMID 22378628
- [8]Gartrell J, Pappo A Recent advances in understanding and managing pediatric rhabdomyosarcoma. F1000Res, 2020.PMID 32695311
- [9]Hettmer S, Li Z, Billin AN, Barr FG, et al. Rhabdomyosarcoma: current challenges and their implications for developing therapies. Cold Spring Harb Perspect Med, 2014.PMID 25368019
- [10]Terwisscha van Scheltinga S, Schoot RA, Routh JC, Seitz G, et al. Lymph Node Staging and Treatment in Pediatric Patients With Soft Tissue Sarcomas: A Consensus Opinion From the Children's Oncology Group, European paediatric Soft Tissue Sarcoma Study Group, and the Cooperative Weichteilsarkom Studiengruppe. Pediatr Blood Cancer, 2025.PMID 39844722
- [11]Cairo MS, Bishop M Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol, 2004.PMID 15384972
- [12]Ramsey DC, Shulman DS, Zhou GC, Cameron D, et al. Long-term outcomes of evolving treatment regimens in Ewing sarcoma survivors diagnosed 1970-1999: A report from the Childhood Cancer Survivor Study. Cancer, 2026.PMID 42390889