Paeds · haematology-oncology-and-transfusion
Brain and spinal tumours
Also known as Paediatric central nervous system tumours · Childhood brain tumour · Posterior fossa tumour · Medulloblastoma · Diffuse intrinsic pontine glioma · DIPG · Diffuse midline glioma H3 K27-altered · Pilocytic astrocytoma · Spinal cord tumour
Fellowship guide to brain and spinal tumours in children. Covers the central nervous system as the second commonest site of childhood cancer and the leading cause of cancer death in children, the predominance of the posterior fossa in the young brain with medulloblastoma, cerebellar pilocytic astrocytoma, ependymoma and brainstem glioma, the supratentorial lesions of pilocytic astrocytoma and craniopharyngioma, the clinical red flags of raised intracranial pressure with early morning headache and vomiting and papilloedema, the triad of cranial nerve palsy, long tract signs and ataxia that declares the diffuse intrinsic pontine glioma, the urgent imaging pathway with magnetic resonance imaging of the brain and the whole neuraxis, the perioperative stabilisation with dexamethasone for vasogenic oedema and the management of hydrocephalus, the molecular classification that has reshaped medulloblastoma into WNT, SHH, Group 3 and Group 4 subgroups, the surgical, radiotherapy and chemotherapy principles, the devastating prognosis of the diffuse intrinsic pontine glioma and the curative potential of the pilocytic astrocytoma, and the long term late effects that shape survivorship.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A child is brought in with three weeks of worsening headache on waking, vomiting that relieves the headache, and a new squint, and the question at the bedside is whether this is a benign tension headache or the first sign of a tumour inside a rigid box. Brain and spinal tumours are the commonest solid tumours of childhood and, after infancy, the leading cause of cancer death in children, and the single thing that separates the child who does well from the child who does not is how fast the clinician recognises the pattern of raised pressure and the focal deficit and reaches the scan. The central nervous system is a closed space, the tumour adds volume that the skull cannot expand in the older child, and the symptoms of raised intracranial pressure follow a logic that every paediatrician must own. [1][2]
A central nervous system tumour in a child is a mass of abnormal cells arising within the brain, the spinal cord, or their coverings, and it behaves very differently from its adult counterpart. The young brain is dominated by the posterior fossa, so that roughly half of all paediatric brain tumours sit below the tentorium, in the cerebellum, the fourth ventricle, or the brainstem, which is the reverse of the adult pattern. The tumours are also biologically distinct, with the embryonal tumours such as medulloblastoma, the grade one pilocytic astrocytoma, and the diffuse midline gliomas that have no adult equivalent. The molecular era has rewritten the classification, so that the diagnosis now pairs the histology with a defining molecular alteration that drives both the prognosis and the treatment. [2][3]
The first task at the bedside is not to name the tumour but to judge whether the child is in danger from raised pressure, from hydrocephalus, or from a focal deficit that threatens a critical pathway. The second task is to image the brain, and the third is to build the multidisciplinary plan that runs paediatric oncology, neurosurgery, radiation oncology, endocrinology, and neuropsychology together. The gravity of the topic is the reason it sits at the heart of the fellowship examination, because a candidate who can read the pattern, reach the scan, and chart a safe path through the surgery, the radiotherapy and the late effects is demonstrating exactly the reasoning the boards test. [1][12]
Classification
The most useful way to classify a paediatric brain tumour at the bedside is by where it sits, because the location predicts the histology, the presenting deficit, and the surgical approach. The posterior fossa holds the cerebellum, the fourth ventricle and the brainstem, and it is the home of the four tumours that dominate childhood, the embryonal medulloblastoma, the grade one cerebellar pilocytic astrocytoma, the ependymoma of the fourth ventricle, and the brainstem glioma. The supratentorial compartment holds the cerebral hemispheres, the diencephalon and the sellar region, and it gives rise to the hemispheric pilocytic astrocytoma and ganglioglioma, the craniopharyngioma, the germ cell tumours of the pineal and suprasellar region, and the choroid plexus tumours. The spinal axis holds the intramedullary astrocytoma and ependymoma and the extramedullary lesions of the nerve sheath and the meninges. [2]
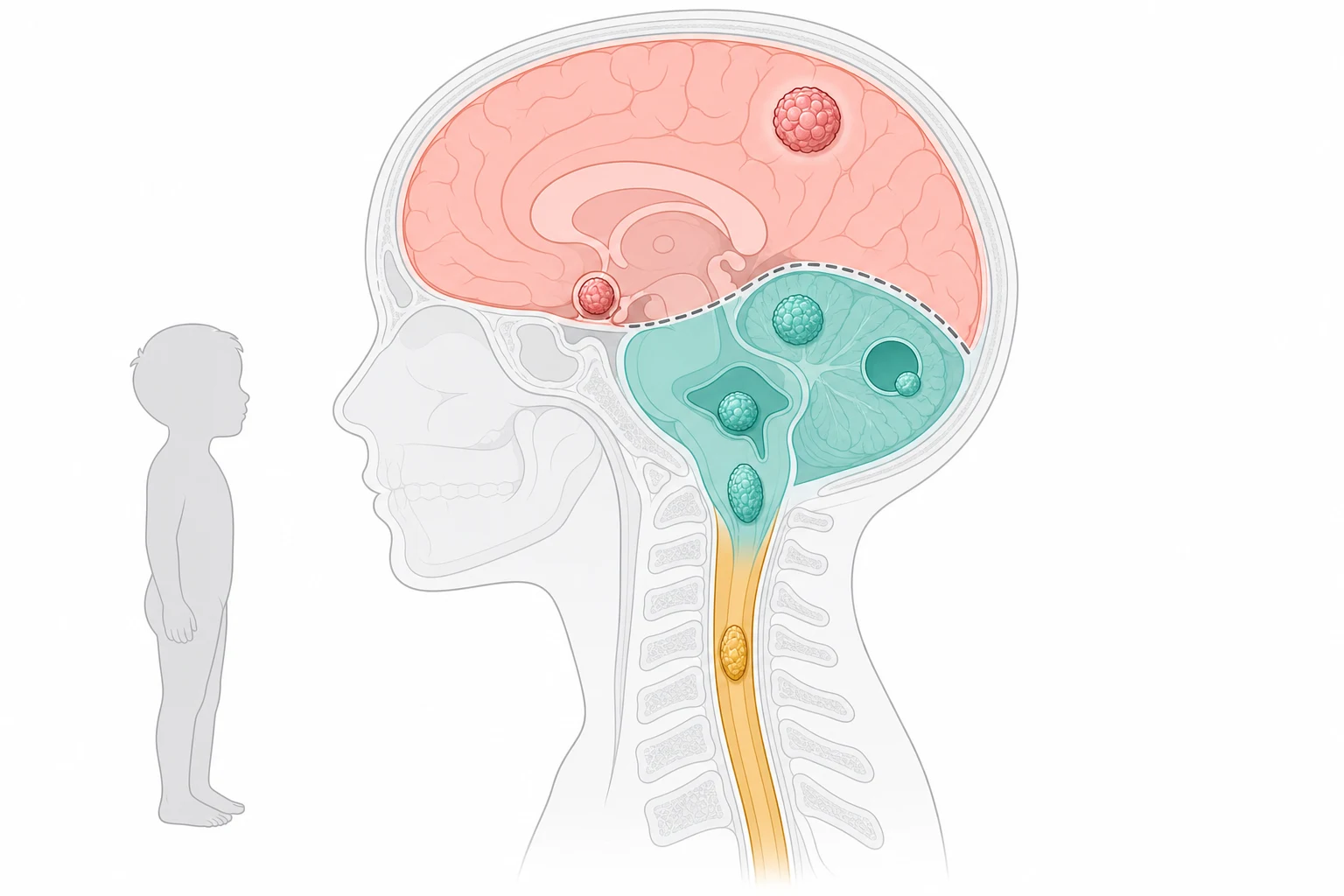
A parallel classification runs through the histology and the behaviour, and it sorts the tumours into the benign and the malignant in a way that changes everything. The grade one pilocytic astrocytoma is the paradigm of the curable lesion, slow growing and well circumscribed and cured by complete surgical resection. The embryonal tumours, led by medulloblastoma, are the malignant, fast growing and cerebrospinal-fluid-disseminating lesions that demand multimodal therapy. The ependymomas sit in between, with the extent of resection and the molecular subgroup driving the behaviour. The diffuse midline gliomas are the grade four lesions that defy cure, and their place in the classification is defined by the H3 K27M mutation rather than by the histology alone. [3][9]
The molecular classification is the modern layer that sits on top of the histology, and nowhere is it more important than in medulloblastoma. The 2012 consensus divided medulloblastoma into four molecular subgroups, the WNT, the SHH, the Group 3 and the Group 4, each with a distinct cell of origin, a distinct cytogenetic driver, a distinct age and sex distribution, and a distinct prognosis. The same principle now orders the ependymomas by their location and fusion, and the low-grade gliomas by their BRAF alteration. The molecular layer matters because it changes the risk group and the intensity of the therapy, and the fellow who knows it can explain why two children with the same histology receive very different treatment. [3][4]
Medulloblastoma
embryonal, malignant
- Four molecular subgroups: WNT, SHH, Group 3, Group 4
- Arises in the cerebellar vermis, fills the fourth ventricle
- Spreads through the cerebrospinal fluid, needs craniospinal imaging
- Treated with surgery, craniospinal irradiation and chemotherapy
Cerebellar pilocytic astrocytoma
grade one, benign
- The classic cyst with a mural nodule on imaging
- BRAF fusion, the KIAA1549-BRAF, is common
- Gross total resection is curative
- Excellent prognosis, survival over ninety five percent
Ependymoma
fourth ventricle
- Arises from the ependymal lining, often the fourth ventricle
- Extent of resection drives the outcome
- Posterior fossa group A carries the worse prognosis
- Treated with surgery and focal radiotherapy
Diffuse intrinsic pontine glioma
brainstem, grade four
- H3 K27M mutation, diffuse midline glioma
- Triad of cranial nerve palsy, long tract signs and ataxia
- Diagnosis is radiographic, biopsy increasingly performed
- Focal radiotherapy is the standard, near uniform fatal outcome
Epidemiology & Risk Factors
The epidemiology of childhood central nervous system tumours is the story of a common and lethal disease. Central nervous system tumours are the commonest solid tumour of childhood and the second commonest childhood malignancy overall, behind only the leukaemias, and they are the leading cause of cancer death in children after infancy. The annual incidence sits around three per one hundred thousand children in the reports of the Central Brain Tumor Registry of the United States, and the incidence is broadly similar across the high-income regions, with a slight male predominance for medulloblastoma and the germ cell tumours. The peak ages are spread across childhood, with the embryonal tumours clustering in the young child and the low-grade gliomas spanning the whole of childhood. [1]
The posterior fossa is the single most important epidemiological fact about the paediatric brain tumour, because it holds roughly half of all the tumours of childhood. This is the reverse of the adult pattern, in which the supratentorial glioblastoma and the metastases dominate, and it is the reason a child with a brain tumour so often presents with hydrocephalus and ataxia rather than with a seizure and a hemianopia. Medulloblastoma is the commonest malignant brain tumour of childhood, the pilocytic astrocytoma is the commonest overall, and the diffuse intrinsic pontine glioma is the commonest brainstem tumour, each with its own age peak and its own behaviour. [2][3]
The risk factors cluster around the inherited cancer predisposition syndromes and the prior exposure to radiation. The neurofibromatosis type one predisposes to the optic pathway and the low-grade gliomas, the tuberous sclerosis complex to the subependymal giant cell astrocytoma, the Li-Fraumeni syndrome to a range of gliomas, and the Gorlin basal cell nevus syndrome to the SHH medulloblastoma. Prior therapeutic irradiation of the head raises the risk of a second meningioma or glioma years later, and it is the reason the craniospinal irradiation that cures the medulloblastoma also seeds the late effects that follow. These predispositions are sought in every child with a brain tumour, because they change the counselling and the surveillance of the family. [2]
The environmental and infectious risk factors are weak and inconsistent, and the fellow should not overstate them. The reported associations with parental pesticide exposure, with maternal diet, and with the common infections have not held up in the larger studies, and the bulk of childhood brain tumours arise without an identifiable cause. The one strong and modifiable exposure is the therapeutic radiation, which is why the contemporary radiotherapy is delivered in the most conformal way and avoided altogether in the youngest children. The lesson for the exam is that the inherited syndromes are the risk factors worth naming, and the environmental causes are the ones worth questioning. [1]
Pathophysiology
The pathophysiology of the paediatric brain tumour is best understood as the collision between a growing mass and a rigid, fluid-filled box. The skull and the dura form a closed container of fixed volume in the older child, holding the brain, the blood and the cerebrospinal fluid in equilibrium, so that any added mass must displace one of the three. A tumour grows, it swells with oedema, and it obstructs the cerebrospinal fluid pathway, and the intracranial pressure rises once the compensatory displacement of the venous blood and the cerebrospinal fluid is exhausted. The result is the gradient that drives the headache, the papilloedema, and the herniation that kills the child if the pressure is not relieved. [2]
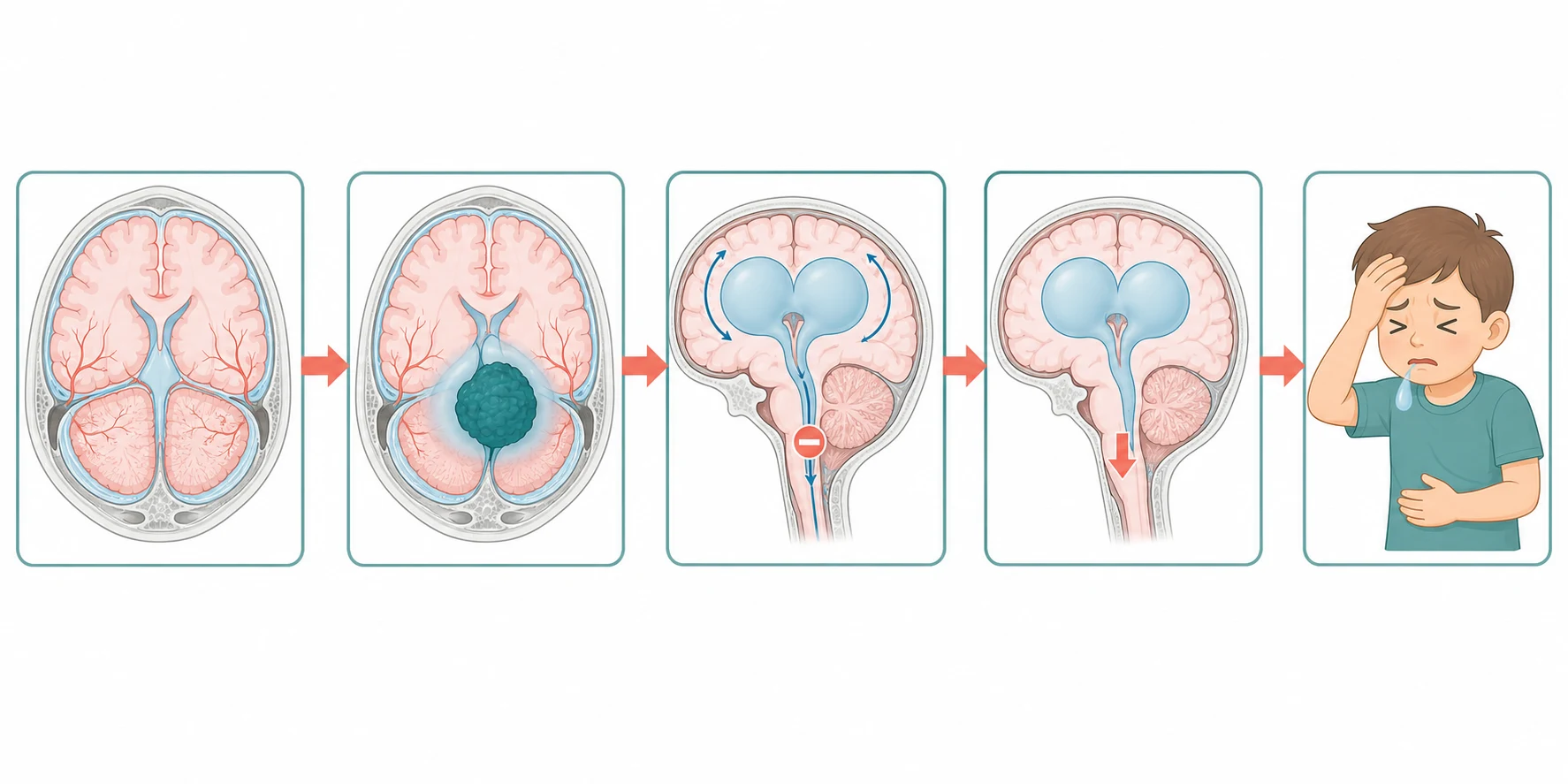
A second mechanism explains why the symptoms of a posterior fossa tumour differ so sharply from those of a supratentorial lesion. A tumour in the cerebellum or the fourth ventricle sits directly on the cerebrospinal fluid outflow, so that it obstructs the aqueduct or the fourth ventricle early and produces an obstructive hydrocephalus that declares as the headache and the vomiting of raised pressure. A tumour in the brainstem, by contrast, infiltrates the cranial nerve nuclei and the long tracts directly, so that it declares as the cranial nerve palsy and the pyramidal weakness rather than as raised pressure. A tumour in the supratentorial cortex irritates the grey matter and declares as a seizure, and a tumour in the sellar region compresses the optic chiasm and the hypothalamic-pituitary axis and declares as the visual field defect and the endocrine failure. The location is the pathophysiology. [2]
The molecular pathophysiology is the modern layer that explains the behaviour of the tumour at the cellular level, and it is the reason the classification has moved beyond the histology. The medulloblastoma subgroups arise from distinct cells of origin in the developing cerebellum, the WNT from the lower rhombic lip and the SHH from the external granule cell layer, and they carry distinct driver alterations that mark them out. The diffuse midline glioma carries the lysine twenty seven to methionine substitution at position twenty seven of the histone H3 tail, the H3 K27M mutation, which reprograms the epigenome and locks the cell in a proliferative, undifferentiated state. These molecular events are not academic, because they define the disease and they are the targets of the new therapies. [3][7]
The pathophysiology of the late effects is the cost of the therapy that saves the child, and the fellow must hold it alongside the cure. The craniospinal irradiation injures the developing white matter and the vasculature and produces the neurocognitive decline that worsens with time, the younger age, and the higher dose. The radiation to the hypothalamic-pituitary axis produces the growth hormone deficiency, the hypothyroidism and the pubertal disturbance. The cisplatin and the radiotherapy together produce the sensorineural hearing loss. Understanding these mechanisms is what allows the survivorship plan to anticipate and to mitigate them rather than to react after the harm is done. [12]
Clinical Presentation
The child with a brain tumour presents in one of three ways, and the recognition of the pattern at the bedside is the skill that decides the outcome. The first is the syndrome of raised intracranial pressure, the progressive early morning headache and the vomiting, worse on waking and often relieved by vomiting, with the lethargy and the behavioural change that the parents notice first. The second is the focal neurological deficit, the ataxia, the squint, the hemiparesis or the visual loss, that points to the site of the tumour. The third is the seizure, which points to the supratentorial cortex. The infant adds a fourth, the enlarging head circumference, the bulging fontanelle and the sunset sign, because the open skull absorbs the pressure at the cost of the head growth. [2]
The history holds the diagnosis more often than the examination, and the tempo is the single most important feature. A headache that is progressive, that wakes the child from sleep, that is worse in the early morning, and that is associated with vomiting is a tumour headache until imaging proves otherwise, however normal the examination. A new squint, especially the abducens palsy that is a false-localising sign of raised pressure, an ataxia or a deterioration in school performance are the subtle presentations that precede the overt deficit. The message for the clinician is to image the progressive headache and not to be reassured by the normal examination, because the focal deficit is a late sign. [2]
The posterior fossa tumour presents with the combination of raised pressure and the cerebellar signs. The medulloblastoma, arising in the vermis, fills the fourth ventricle, obstructs the cerebrospinal fluid and presents with the headache and vomiting of hydrocephalus alongside the truncal ataxia and the wide-based gait. The cerebellar pilocytic astrocytoma, arising in the hemisphere, presents with the ipsilateral dysmetria and the intention tremor of a cerebellar hemisphere lesion, often with less pressure because it obstructs later. The ependymoma of the fourth ventricle presents like the medulloblastoma but may also produce the neck pain and the head tilt of the foramen magnum extension. [3][8]
The supratentorial tumour presents with the seizure, the hemiparesis or the visual and endocrine disturbance, and the location again predicts the pattern. The hemispheric pilocytic astrocytoma and ganglioglioma present with the seizure or the focal deficit of the cortex. The craniopharyngioma, sitting above the sella, compresses the optic chiasm to produce the bitemporal hemianopia and the hypothalamic-pituitary axis to produce the growth failure, the delayed puberty and the diabetes insipidus. The germ cell tumours of the pineal and the suprasellar region present with the diabetes insipidus, the Parinaud syndrome of the upgaze palsy, and the precocious puberty. [10]
The spinal tumour presents with the back pain, the progressive weakness, the sensory level and the sphincter disturbance, and it is the one presentation that is a true emergency because the cord does not recover once it is compressed. The intramedullary astrocytoma produces the localised pain and the progressive myelopathy, the extramedullary lesion produces the radicular pain and the earlier block, and the metastatic drop from a medulloblastoma can produce an acute cord syndrome. Any child with progressive limb weakness, a sensory level or new bladder or bowel dysfunction has spinal cord compression until imaging proves otherwise, and the dexamethasone and the emergency scan are given without delay. [2]
Differential Diagnosis
The differential of the child with a suspected brain tumour is built around the syndrome that brought the child in, and the imaging is the test that resolves it. The progressive headache and vomiting of raised pressure sit on a differential that includes the idiopathic intracranial hypertension, the chronic meningitis, the subdural collection and the obstructive hydrocephalus from any cause, and the magnetic resonance scan separates the tumour from the rest. The ataxia sits on a differential that includes the acute cerebellitis, the posterior fossa abscess, the drug ingestion and the inherited ataxias, and again the scan is decisive. The focal deficit sits on a differential that includes the stroke, the demyelination and the vascular malformation. [2]
The benign causes that mimic the tumour must be excluded, because treating them as a tumour is as harmful as missing a tumour. The idiopathic intracranial hypertension produces the headache and the papilloedema with a normal scan and a high opening pressure, and it is excluded by the imaging and the lumbar puncture. The acute cerebellitis and the postinfectious demyelination produce the ataxia and the cerebellar swelling that can look like a tumour on the first scan, and the short history and the resolution on the follow-up imaging settle the question. The arachnoid cyst and the choroid plexus tumour can produce a ventricular expansion that mimics the obstructive hydrocephalus. [2]
The chief diagnostic pitfalls for the fellow are the cases in which the diagnosis is delayed because the pattern is misread. The child whose headache is labelled a migraine or a tension headache, and whose progressive and early-morning character is missed, is the classic delayed diagnosis, and the lesson is to image any progressive headache or any headache with the red flag features. The child whose ataxia is attributed to a viral infection or a drug, and whose persistence is not acted upon, is the second. The child whose new squint is treated as a benign sixth nerve palsy without the scan, and whose underlying raised pressure is missed, is the third. [2]
The differential of the brainstem lesion is narrow and high-stakes, because the diffuse intrinsic pontine glioma is so distinctive that the radiographic pattern is itself the diagnosis. The one mimic that must be excluded is the brainstem abscess or the demyelinating lesion, which can enlarge the pons and produce the cranial nerve signs, and which demand a different and urgent treatment. The focal brainstem glioma, by contrast, is the low-grade tectal or cervicomedullary glioma that is cystic or focal, that grows slowly, and that carries a far better prognosis, which is why the radiographic pattern rather than the histology alone is what separates the curable from the incurable brainstem lesion. [7]
Clinical & Bedside Assessment
The bedside assessment of the child with a suspected brain tumour is a search for the signs of raised pressure, the focal deficit and the unstable child who needs resuscitation before the scan. The assessment begins with the airway, the breathing and the circulation, and with the level of consciousness, because a child with a falling Glasgow coma score, an irregular breathing pattern or a bradycardia with hypertension is herniating and needs the emergency measures. Once the child is safe, the focused history turns to the tempo of the symptoms, the headache character, the visual and the endocrine symptoms, the developmental and the family history, and the inherited cancer predisposition syndromes. [2]
The examination is systematic and takes only a few minutes, but each finding carries weight. The head circumference is measured and plotted in every child, because the crossing of the centiles is a sign of the raised pressure in the infant. The fundi are examined for the papilloedema, which is the single most important bedside sign of raised pressure and which may be absent in the young infant. The cranial nerves are tested for the false-localising sixth palsy, the seventh palsy of the brainstem lesion, and the upgaze palsy of the Parinaud syndrome. The limbs are tested for the tone, the power, the reflexes and the plantar responses, and the gait is observed for the ataxia. [2]
The focused examination of the child with a suspected brain tumour
Assess the airway, breathing, circulation and the level of consciousness first, looking for the bradycardia with hypertension and the irregular breathing of herniation
Measure and plot the head circumference, and palpate the fontanelle for fullness in the infant
Examine the fundi for papilloedema, the single most important bedside sign of raised intracranial pressure
Test the cranial nerves for the false-localising sixth palsy, the brainstem seventh palsy and the Parinaud upgaze palsy
Examine the limbs for the tone, power, reflexes and plantar responses, looking for the pyramidal long tract signs
Observe the gait for the truncal ataxia and the wide-based cerebellar gait
Arrange the urgent magnetic resonance imaging of the brain and alert the neurosurgical and oncology teams
The severity of the raised pressure and the danger of the herniation are judged at the bedside, and they decide the speed of the scan and the start of the dexamethasone. The child with the falling consciousness, the asymmetric pupils, the posturing or the Cushing response of the bradycardia and the hypertension is moved immediately to the resuscitation, the head elevation, the dexamethasone and the emergency scan, with the neurosurgery and the intensive care alerted. The child with the stable consciousness and the progressive symptoms is imaged urgently and admitted, and the family is counselled on the plan. The teaching of the family is part of the assessment, because a child sent home while awaiting the scan must be told to return at once with any deterioration. [11]
Investigations
The investigation of a suspected brain tumour moves in two steps, the imaging that confirms and localises the tumour, and the histological and molecular studies that name it. The first and the decisive test is the magnetic resonance imaging of the brain with and without contrast, because it shows the tumour, the oedema, the hydrocephalus and the relationship to the critical structures in a way the computed tomography cannot. The computed tomography is reserved for the emergency setting where the magnetic resonance is not available, and it shows the hydrocephalus and the calcification but misses the posterior fossa detail. [2]
The whole neuraxis is imaged at the diagnosis, because the embryonal tumours disseminate through the cerebrospinal fluid and the staging changes the treatment. The spinal magnetic resonance imaging is performed to look for the drop metastases, and the lumbar puncture for the cerebrospinal fluid cytology is performed once the raised pressure is relieved, to look for the leptomeningeal spread. The medulloblastoma and the other embryonal tumours are staged by the Chang criteria, and the presence of the metastatic disease moves the child from the average-risk to the high-risk category and intensifies the craniospinal irradiation. [3][5]
Across Australia, Aotearoa New Zealand, the United Kingdom, the United States and Canada, the magnetic resonance imaging of the brain and the whole spine with and without contrast is the standard diagnostic test for a suspected paediatric brain tumour. The molecular panel sent on the tumour tissue, and the specific fusion and methylation tests that drive the modern risk stratification, differ between the international study groups, and the fellow should know the local protocol. In the well-resourced centres the diffusion tensor imaging and the functional mapping are added to plan the safe surgical approach to the eloquent cortex.
[3][9]The histological and the molecular diagnosis is made on the tumour tissue, obtained at the surgery or, increasingly in the diffuse midline glioma, at the stereotactic biopsy. The histology grades the tumour by the World Health Organization criteria, and the molecular studies add the defining alteration, the H3 K27M mutation for the diffuse midline glioma, the BRAF fusion for the pilocytic astrocytoma, and the subgroup and the cytogenetic markers for the medulloblastoma. The methylation profiling is now the contemporary standard for the classification of the paediatric brain tumours, because it resolves the cases that the histology alone cannot. [3][7]
The preoperative blood and the endocrine tests are requested once the tumour is localised, and they anticipate the surgical and the endocrine complications. The full blood count, the coagulation and the electrolytes are sent before the surgery. The pituitary and the hypothalamic axis are tested in every child with a sellar or suprasellar tumour, because the cortisol deficiency and the diabetes insipidus are dangerous at the surgery and must be corrected beforehand. The tumour markers, the alpha-fetoprotein and the beta-human chorionic gonadotropin, are sent in the suspected germ cell tumour, because a raised marker is itself diagnostic and changes the management away from the surgery. [10]
Management — Resuscitation
The unstable child with a brain tumour is resuscitated before the definitive plan is pursued, and the resuscitation rests on three legs: the relief of the raised pressure, the control of the oedema with the dexamethasone, and the management of the hydrocephalus. The child with the falling consciousness or the signs of the herniation is managed with the head elevation, the isotonic fluids, the gentle hyperventilation in the intubated child, and the urgent neurosurgical referral. The emergency measures buy the time to reach the scan and the surgery, and they are not a substitute for either. [11]
The dexamethasone is the cornerstone of the control of the vasogenic tumour oedema, and it is started at the first sign of the pressure or the deficit that the oedema is driving. A typical paediatric regimen is the dexamethasone at zero point one to zero point two milligrams per kilogram per dose, given every six hours, tapered as the definitive therapy relieves the mass effect. The dexamethasone reduces the oedema within hours, it improves the consciousness and the deficit, and it is tapered rapidly after the surgery to avoid the steroid toxicity. The fellow must know the indications, the dose and the taper, because the dexamethasone is one of the few interventions that changes the child before the scan is read. [11]
The hydrocephalus from the posterior fossa tumour is managed before or at the surgery, because the obstructed cerebrospinal fluid pathway is the immediate threat to the child. The options are the emergency external ventricular drain, the endoscopic third ventriculostomy, and the shunt, and the choice depends on the anatomy and the tumour. The external ventricular drain is placed at the surgery or before it, and it is removed once the tumour is resected and the pathway is restored. The endoscopic third ventriculostomy avoids the lifelong shunt in the selected child, and it is preferred where the anatomy allows. [2]
The seizure in the child with a supratentorial tumour is managed with the antiseizure medicine, and the choice is built around the lesion and the electroencephalogram. The levetiracetam is the common first-line agent for the tumour-related seizure, because of its favourable profile and the absence of the enzyme interactions, and it is loaded intravenously for the ongoing seizure and converted to the oral route. The prophylactic antiseizure medicine is not given routinely before the surgery, and it is reserved for the child with the established seizure or the high-risk lesion. The temperature, the glucose and the electrolytes are checked and corrected, because the metabolic disturbance can worsen the seizure and the pressure. [2]
Management — Definitive & Stepwise
The definitive management of the paediatric brain tumour is built around the three modalities of the surgery, the radiotherapy and the chemotherapy, and the combination is tailored to the tumour, the location, the molecular subtype, and the age of the child. The overarching principle is that the maximal safe resection is the foundation of the cure for the resectable tumours, that the radiotherapy is reserved for the malignant and the incompletely resected lesions, and that the chemotherapy is added for the embryonal tumours and used in place of the radiotherapy in the youngest children. The age of three years is the threshold below which the craniospinal irradiation is avoided because of the devastating neurocognitive injury, and a chemotherapy-first strategy is used instead. [3]
[3] [12]Medulloblastoma is the paradigm of the risk-adapted multimodal therapy, and it is the tumour the fellow must know in detail. The treatment begins with the maximal safe resection, and the residual tumour is measured on the postoperative scan, because a residual of under one point five square centimetres with no metastatic disease defines the average risk and the larger residual or the metastatic disease defines the high risk. The average-risk child receives the craniospinal irradiation at twenty three point four gray to the neuraxis with a boost to the posterior fossa, combined with the chemotherapy. The high-risk child receives the higher craniospinal irradiation dose of thirty six gray and the more intensive chemotherapy. The molecular subgroup refines the risk further, with the WNT carrying the best and the Group 3 the worst prognosis. [3][5]
Risk-adapted therapy for medulloblastoma
Dose
Average risk: maximal safe resection to a residual under one point five square centimetres, craniospinal irradiation at twenty three point four gray with a posterior fossa boost, and concomitant and maintenance chemotherapy. High risk: residual over one point five square centimetres or metastatic disease, craniospinal irradiation at thirty six gray with the more intensive chemotherapy
The diffuse intrinsic pontine glioma is the opposite of the medulloblastoma, because it is the one paediatric brain tumour for which no curative therapy exists. The standard treatment is the focal radiotherapy to the pons at fifty four to fifty nine gray, delivered over six weeks, which improves the symptoms and the quality of life and extends the median survival to the nine to eleven months, but which does not cure. The chemotherapy, the targeted agents and the immunotherapies have all failed to improve the survival in the trials, and the contemporary research is built around the oncosphere drugs such as the ONC201, the dordaviprone, and the targeted approaches against the H3 K27M alteration. The palliative care is involved early, and the family is counselled honestly on the prognosis. [7]
The cerebellar pilocytic astrocytoma is the curable lesion, and its management rests on the surgery alone. The gross total resection is curative, and no further therapy is given for the completely resected tumour. The subtotal resection is followed with the surveillance imaging, because the residual may remain stable for years, and the chemotherapy with the carboplatin and the vincristine or the targeted BRAF inhibitor is reserved for the progressive or the unresectable disease. The ependymoma is managed with the maximal safe resection and the focal radiotherapy, and the extent of the resection is the single strongest determinant of the outcome. The craniopharyngioma is managed with the surgery that balances the resection against the hypothalamic injury, with the radiotherapy for the residual, and the lifelong endocrine replacement. [8][9][10]
Specific Subtypes & Scenarios
Medulloblastoma and its molecular subgroups
Medulloblastoma is the commonest malignant brain tumour of childhood, and it is the tumour that has been transformed by the molecular classification. The four subgroups carry distinct cells of origin, distinct drivers, and distinct outcomes, and the fellow must know the summary. The WNT subgroup carries the best prognosis, with a survival over ninety percent, and it arises from the lower rhombic lip with the CTNNB1 mutation. The SHH subgroup arises from the cerebellar granule cell precursor, it has a bimodal age distribution in the infant and the adult, and it carries the TP53-mutant high-risk variant that worsens the prognosis. The Group 3 carries the worst prognosis, with the MYC amplification and the frequent metastasis, and the Group 4 is the commonest, with an intermediate prognosis. [3][4]
MSHAR
The treatment is the risk-adapted combination of the surgery, the craniospinal irradiation and the chemotherapy, and the molecular subgroup refines the intensity. The contemporary trials test the de-escalation of the therapy in the favourable WNT subgroup to reduce the late effects, and the intensification in the high-risk Group 3. The carboplatin and the isotretinoin added to the standard therapy improved the event-free survival in the high-risk medulloblastoma in the Children's Oncology Group trial, and the targeted agents against the SHH pathway, the smoothened inhibitors, have a role in the relapsed disease. The lesson is that the molecular subgroup is no longer a research interest but a treatment decision. [5][6]
The diffuse intrinsic pontine glioma and the diffuse midline glioma
The diffuse intrinsic pontine glioma is the most devastating paediatric brain tumour, and its biology is now defined by the H3 K27M mutation that reclassifies it as the diffuse midline glioma, the H3 K27-altered, the World Health Organization grade four. The tumour arises in the pons of the school-age child, it infiltrates the cranial nerve nuclei and the long tracts, and it produces the classical triad of the cranial nerve palsy, the pyramidal signs and the ataxia. The magnetic resonance imaging shows the diffusely enlarged pons that encases the basilar artery, with the T2 and the fluid-attenuated inversion recovery hyperintensity and the variable enhancement. [7]
The diagnosis is radiographic, and the biopsy is increasingly performed for the molecular profiling that the clinical trials demand. The standard treatment is the focal radiotherapy to the pons at fifty four to fifty nine gray, which relieves the symptoms and extends the median survival to the nine to eleven months, but which does not cure. The numerous chemotherapy and the targeted therapy trials have failed to improve the survival, and the contemporary research is built around the oncosphere drug ONC201, the dordaviprone, and the immunotherapeutic approaches. The fewer than ten percent two-year survival and the near uniform fatality are the reason the honest family counselling and the early palliative care involvement are so central. [7]
The cerebellar pilocytic astrocytoma and the low-grade glioma
The cerebellar pilocytic astrocytoma is the paradigm of the curable paediatric brain tumour, and it is the tumour the fellow must place at the opposite end of the spectrum from the diffuse intrinsic pontine glioma. It is the World Health Organization grade one lesion, it grows slowly, and it presents with the classic cyst with the mural nodule on the imaging and the ipsilateral cerebellar signs. The BRAF fusion, the KIAA1549-BRAF, is the common molecular alteration, and it carries a favourable prognosis and a target for the inhibitor therapy in the unresectable disease. [8]
The management rests on the maximal safe resection, and the gross total resection is curative, with no further therapy given. The subtotal resection is followed with the surveillance imaging, because the residual may remain stable for years, and the chemotherapy with the carboplatin and the vincristine or the targeted BRAF inhibitor is reserved for the progressive, the recurrent or the unresectable disease, especially in the optic pathway glioma of the neurofibromatosis type one. The survival exceeds ninety five percent for the completely resected cerebellar lesion, and the fellow must hold this against the diffuse intrinsic pontine glioma to appreciate the range of the paediatric brain tumour. [8]
The craniopharyngioma and the sellar region
The craniopharyngioma is the sellar and the suprasellar tumour of the child, arising from the remnants of the Rathke pouch, and it presents with the visual disturbance, the endocrine failure and the raised pressure. The adamantinomatous type of the child is the cystic and the calcified lesion, sitting above the sella and compressing the optic chiasm to produce the bitemporal hemianopia and the hypothalamic-pituitary axis to produce the growth failure, the delayed puberty and the diabetes insipidus. The management balances the resection against the hypothalamic injury, because the aggressive resection that achieves the cure can also produce the devastating hypothalamic obesity and the panhypopituitarism. [10]
The contemporary strategy is the safer subtotal resection with the radiotherapy or the cyst instillation, designed to spare the hypothalamus and to reduce the endocrine and the metabolic morbidity. The proton therapy is increasingly used to deliver the radiotherapy with the reduced exit dose. The lifelong endocrine replacement, the growth hormone, the hydrocortisone, the levothyroxine and the desmopressin, is the rule, and the hypothalamic obesity, the sleep disturbance and the neurocognitive decline are the long-term morbidities that dominate the survivorship. The fellow who understands that the cure is not the end of the disease demonstrates the reasoning the boards reward. [10]
Complications & Pitfalls
The complications of the paediatric brain tumour divide into the disease-related and the treatment-related, and the fellow must hold both because the iatrogenic harm can rival the disease. The disease-related complications are the hydrocephalus, the herniation, the focal deficit and the spinal cord compression, and they are the complications that drive the resuscitation and that are anticipated from the moment the tumour is seen on the scan. The leptomeningeal and the drop metastases of the embryonal tumours are the disease complications that demand the whole-neuraxis imaging and the staging. [2]
The treatment-related complications begin at the surgery, and the most distinctive is the posterior fossa syndrome, the cerebellar mutism. The cerebellar mutism declares itself twenty four to seventy two hours after the posterior fossa surgery, with the mutism, the emotional lability, the hypotonia and the ataxia, and it arises from the injury to the dentato-thalamo-cortical pathway. The mutism is usually transient, but the long-term neurocognitive and the behavioural deficits persist in many, and it is one of the most distressing complications of the medulloblastoma surgery. The other surgical complications are the new focal deficit, the cerebrospinal fluid leak and the infection. [8]
The radiotherapy and the chemotherapy complications are the costs of the cure, and they shape the survivorship. The craniospinal irradiation produces the neurocognitive decline that worsens with the time, the younger age and the higher dose, and it is the reason the dose is de-escalated in the favourable-risk medulloblastoma and avoided in the under-three. The endocrine late effects, the growth hormone deficiency, the hypothyroidism and the pubertal disturbance, follow the radiation to the hypothalamic-pituitary axis. The sensorineural hearing loss follows the cisplatin and the radiotherapy, and the secondary malignancy follows the radiation and the chemotherapy years later. [12]
Prognosis & Disposition
The prognosis of a child with a brain tumour spans the full range from the cure of the pilocytic astrocytoma to the near uniform fatality of the diffuse intrinsic pontine glioma, and it is determined by the tumour type, the location, the molecular subtype, the extent of the resection and the age of the child. The cerebellar pilocytic astrocytoma has a survival over ninety five percent after the complete resection, the WNT medulloblastoma over ninety percent, and the average-risk medulloblastoma around seventy to eighty percent. The Group 3 medulloblastoma and the high-risk disease carry the worse prognosis, and the diffuse intrinsic pontine glioma carries the near uniform fatality within the year. [3][7]
The disposition of the child is determined by the diagnosis and the stability, and the management is delivered in the specialist paediatric neuro-oncology centre. The child with the newly diagnosed brain tumour is transferred to the tertiary centre with the paediatric neurosurgery, the radiation oncology and the paediatric intensive care, and the regional or the rural hospital is responsible for the recognition and the stabilisation before the retrieval. The child with the palliative disease, such as the diffuse intrinsic pontine glioma, is managed with the focal radiotherapy and the early palliative care, with the local services supported by the specialist centre. [2]
In Australia and Aotearoa New Zealand, the child with a newly diagnosed brain tumour is managed in a tertiary paediatric neuro-oncology centre, with the paediatric retrieval services transferring the unstable child from the regional or the rural hospital. The radiation oncology is delivered in the few specialised centres, and the family is supported by the social work, the educational liaison and the neuropsychology. The long distances and the retrieval times are the reason the early recognition and the stabilisation in the referring hospital are so heavily weighted in the exam.
[2]The long-term surveillance of the survivor is the reward and the burden of the cure, because the late effects of the therapy are common and they accumulate with the time. The survivor has the annual magnetic resonance imaging for the recurrence, the annual endocrine assessment for the growth hormone deficiency and the hypothyroidism, the audiology for the hearing loss, and the neuropsychology for the cognitive decline. The transition to the adult late-effects service is prepared in the adolescence, with the reproductive and the genetic counselling, the secondary malignancy surveillance and the vocational support. The fellow who builds the survivorship plan demonstrates the care that extends beyond the cure. [12]
Special Populations
The infant and the very young child hold a special position in this topic, because the developing brain is uniquely vulnerable to the therapy and the treatment is built around the avoidance of the radiotherapy. The craniospinal irradiation is avoided under three years of age because of the devastating neurocognitive injury, and a chemotherapy-first strategy is used to defer or to replace the radiotherapy. The infant brain tumour is also biologically distinct, with the congenital tumours and the atypical teratoid rhabdoid tumour that dominate the very young, and the management is delivered on the tailored infant protocols that the fellow should know exist. [3]
The child with the inherited cancer predisposition syndrome is the special population that changes the counselling and the surveillance. The neurofibromatosis type one carries the optic pathway glioma and the low-grade glioma risk, and the magnetic resonance surveillance is built into the care of the affected child. The tuberous sclerosis complex carries the subependymal giant cell astrocytoma risk. The Li-Fraumeni syndrome and the Gorlin syndrome carry the glioma and the SHH medulloblastoma risks respectively, and the family is counselled on the inheritance and the surveillance. These syndromes are sought in every child with a brain tumour because they change the plan. [2]
Socioeconomic disadvantage, remoteness and the migrant or refugee status shape the access to the diagnosis and the treatment, and they are the reason the early recognition in the primary care and the regional hospital is so heavily emphasised. A child far from a specialist centre may first present to a clinician who sees few such cases, and the progressive headache or the ataxia that flags the tumour is the bridge to the retrieval and the specialist care. The language and the cultural barriers are addressed with the interpreter and the cultural support, and the family is supported through the long and the unfamiliar treatment. [1]
The adolescent and the young adult with a brain tumour is prepared for the transition to the adult service with the counselling and the documentation that make it safe. The reproductive and the genetic counselling, the fertility preservation before the gonadotoxic therapy, the endocrine and the neuropsychological late effects, and the vocational and the educational support are addressed before the handover. The young person leaves the paediatric service with a survivorship plan and a named adult provider, and the transition is a clinical act as important as the diagnosis. [12]
Evidence, Guidelines & Regional Differences
The landmark evidence that underpins the modern treatment of the paediatric brain tumour is the product of successive international collaborative trials, and it is the reason the survival has risen for the resectable tumours while it has barely moved for the diffuse intrinsic pontine glioma. The medulloblastoma trials refined the craniospinal irradiation dose from the thirty six gray to the twenty three point four gray for the average-risk disease, with the chemotherapy added to maintain the survival while reducing the late effects. The molecular subgroups were defined in the 2012 consensus and confirmed in the subsequent trials, and they now drive the risk stratification. [3][4]
The contemporary medulloblastoma evidence tests the de-escalation in the favourable WNT subgroup and the intensification in the high-risk Group 3. The Children's Oncology Group trial of the carboplatin and the isotretinoin added to the standard therapy showed an improved event-free survival in the high-risk medulloblastoma, and the targeted smoothened inhibitors have a role in the relapsed SHH disease. The proton therapy is increasingly used to deliver the craniospinal irradiation with the reduced exit dose and the lower late-effect burden, and it is the contemporary standard in the well-resourced centres. [5][6]
The radiotherapy thresholds and the proton versus the photon practice vary across Australia, Aotearoa New Zealand, the United Kingdom, the United States and Canada, with the proton therapy increasingly preferred for the craniospinal irradiation to reduce the late effects. The management of the diffuse intrinsic pontine glioma is broadly consistent, with the focal radiotherapy at fifty four to fifty nine gray the universal standard and the chemotherapy reserved for the trials. The fellow should know the local protocol and the regional radiation oncology guidance.
[3][7]The controversies and the open questions are the live ones. The place of the biopsy in the diffuse intrinsic pontine glioma, once avoided and now increasingly performed for the molecular profiling, is one. The optimal de-escalation of the craniospinal irradiation in the favourable-risk medulloblastoma, balanced against the recurrence risk, is another. The role of the oncosphere drug ONC201, the dordaviprone, in the diffuse midline glioma is being defined by the contemporary trials, and the targeted agents against the H3 K27M alteration are the research frontier. The fellow holds these as the open questions and cites the trials and the guidelines rather than the dogma. [7]
Exam Pearls
The high-yield facts for the exam are the ones that change a decision at the bedside, and they are worth carrying as sharp statements. Brain tumours are the commonest solid tumour of childhood and the leading cause of cancer death in children after infancy, and the posterior fossa holds roughly half of them. The progressive early morning headache and vomiting with the papilloedema is raised pressure from a tumour until imaging proves otherwise. The triad of the cranial nerve palsy, the pyramidal signs and the ataxia is the diffuse intrinsic pontine glioma, and the magnetic resonance scan is the diagnosis. [1][7]
The medulloblastoma has the four molecular subgroups, the WNT with the best and the Group 3 with the worst prognosis, and it is treated with the maximal safe resection, the craniospinal irradiation and the chemotherapy. The average-risk disease is defined by the residual under one point five square centimetres and the absence of the metastasis, and it receives the twenty three point four gray craniospinal irradiation. The high-risk disease receives the thirty six gray and the more intensive chemotherapy. The craniospinal irradiation is avoided under three years of age. The pilocytic astrocytoma is cured by the complete resection, and the diffuse intrinsic pontine glioma is treated with the focal radiotherapy alone and carries the near uniform fatality. [3][5]
The final pearls are the ones that catch the candidate who has learned the headline and forgotten the corner. The computed tomography misses the posterior fossa detail and the small tumour, and the magnetic resonance is the standard. The cerebellar mutism declares twenty four to seventy two hours after the posterior fossa surgery. The craniopharyngioma survivor needs the lifelong endocrine replacement and the hypothalamic obesity watch. The optic pathway glioma of the neurofibromatosis type one is often observed, not operated, because many remain stable. The message for the exam is that the corners are where the marks are won, and the reasoning that holds the location and the molecular classification at the centre is the one the boards reward. [8][10]
References
- [1]Ostrom QT, Price M, Ryan K CBTRUS Statistical Report: Pediatric Brain Tumor Foundation Childhood and Adolescent Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014-2018 Neuro Oncol, 2022.PMID 36066969
- [2]Pollack IF, Agnihotri S, Broniscer A Childhood brain tumors: current management, biological insights, and future directions J Neurosurg Pediatr, 2019.PMID 30835699
- [3]Northcott PA, Robinson GW, Kratz CP Medulloblastoma Nat Rev Dis Primers, 2019.PMID 30765705
- [4]Taylor MD, Northcott PA, Korshunov A Molecular subgroups of medulloblastoma: the current consensus Acta Neuropathol, 2012.PMID 22134537
- [5]Ramaswamy V, Remke M, Bouffet E Risk stratification of childhood medulloblastoma in the molecular era: the current consensus Acta Neuropathol, 2016.PMID 27040285
- [6]Leary SES, Packer RJ, Li Y Efficacy of Carboplatin and Isotretinoin in Children With High-risk Medulloblastoma: A Randomized Clinical Trial From the Children's Oncology Group JAMA Oncol, 2021.PMID 34292305
- [7]van den Bent M, Saratsis AM, Geurts M H3 K27M-altered glioma and diffuse intrinsic pontine glioma: Semi-systematic review of treatment landscape and future directions Neuro Oncol, 2024.PMID 38102230
- [8]Bornhorst M, Frappaz D, Packer RJ Pilocytic astrocytomas Handb Clin Neurol, 2016.PMID 26948364
- [9]Boop SH, Shimony N, Boop FA Review and update on pediatric ependymoma Childs Nerv Syst, 2023.PMID 37493720
- [10]Gan HW, Morillon P, Albanese A National UK guidelines for the management of paediatric craniopharyngioma Lancet Diabetes Endocrinol, 2023.PMID 37549682
- [11]Malbari F, Staggers KA, Minard CG Provider views on perioperative steroid use for patients with newly diagnosed pediatric brain tumors J Neurooncol, 2020.PMID 32026434
- [12]Rey-Casserly C, Diver T Late effects of pediatric brain tumors Curr Opin Pediatr, 2019.PMID 31693589