Paeds · haematology-oncology-and-transfusion
G6PD deficiency and enzymopathies
Also known as Glucose-6-phosphate dehydrogenase deficiency · Favism · G6PD deficiency · Red cell enzymopathy · Oxidative haemolysis
Fellowship guide to glucose-6-phosphate dehydrogenase deficiency and the red-cell enzymopathies in children. Covers the X-linked loss of the pentose phosphate pathway enzyme that leaves red cells unable to regenerate NADPH and reduced glutathione, the WHO classification of variants by enzyme activity from Class I severe to Class V increased, the global prevalence of roughly 330 to 400 million people concentrated across sub-Saharan Africa, the Mediterranean, the Middle East, South and Southeast Asia and linked to historic malaria protection, the acute haemolytic crisis triggered by infection, fava beans and oxidant drugs such as primaquine, tafenoquine, rasburicase, methylene blue and dapsone, the neonatal jaundice that carries a high kernicterus risk, the diagnostic blood film with bite cells, blister cells and Heinz bodies on a supravital stain with a negative direct antiglobulin test, the critical pitfall that the G6PD assay is falsely normal during acute haemolysis and must be repeated two to three months later, the supportive management with trigger withdrawal and transfusion of leucodepleted packed red cells 10 to 20 mL per kilogram for a haemoglobin under 70 g/L or symptomatic anaemia, and the lifelong trigger-avoidance counselling and family screening under the 2023 Clinical Pharmacogenetics Implementation Consortium guideline.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A previously well boy of Mediterranean ancestry eats a plate of fava beans on a Sunday. By Monday he is pale and his urine is the colour of strong tea. This is the textbook face of glucose-6-phosphate dehydrogenase deficiency, the most common inherited red-cell enzyme disorder in humans and the archetype of a red-cell enzymopathy. The defect lies in the enzyme that lets a red cell defend itself against oxidant stress, and without it a burst of oxidation shatters the cell from within whenever a trigger arrives. [1]
Glucose-6-phosphate dehydrogenase, or G6PD, is the first enzyme of the pentose phosphate pathway. It oxidises glucose-6-phosphate and in doing so reduces NADP to NADPH. That NADPH is the only way a mature red cell keeps its glutathione in the reduced, protective form that neutralises the hydrogen peroxide generated constantly inside the cell. Because a red cell has no nucleus and cannot make new enzyme, its survival depends entirely on the G6PD it was born with. When the gene is defective, the cell loses this defence, and any oxidant load, from an infection, a fava bean, or a drug, can denature the haemoglobin and rupture the membrane. [1]
The disease is X-linked, carried on the long arm of the X chromosome at Xq28, so it manifests fully in males and travels through carrier females. It affects an estimated 330 to 400 million people, roughly four to five per cent of the world's population, with the highest frequencies across sub-Saharan Africa, the Mediterranean, the Middle East, South and Southeast Asia, and parts of Latin America. The distribution mirrors the historical reach of falciparum malaria, because carrying one copy of the deficient gene protects partially against severe malaria, the classic balanced polymorphism. [2][3][4]
Three ideas make this topic central to the fellowship examination. The mechanism is examinable and elegant: the loss of NADPH and reduced glutathione explains every clinical and laboratory feature. The diagnosis has a single trap that examiners love, namely that the enzyme assay is falsely normal during an acute crisis and must be repeated weeks later. And the management is almost entirely preventive: identify and avoid the triggers, counsel the family, and screen the relatives, because most patients are perfectly well between crises. The 2023 Clinical Pharmacogenetics Implementation Consortium guideline gives the current prescribing standard. [5][6]
Classification
The disease classifies along two axes that the candidate must hold in mind at once: the enzyme-activity class set by the World Health Organization, and the clinical expression that the child actually shows. The enzyme-activity class is the deeper framework because it predicts severity, and it runs from the rare severe Class I variants that cause continuous haemolysis, through the common Class II and Class III variants that cause episodic crises, to the normal and increased-activity classes. [1][2]

The class that matters most in the African child is G6PD A-minus, a Class III variant with moderate enzyme activity of ten to sixty per cent. Its defining feature is that the enzyme activity decays faster than normal as the red cell ages, so the oldest cells carry the least enzyme and are lysed first. This is why a crisis in G6PD A-minus is usually self-limited: once the deficient old-cell cohort is destroyed, the haemolysis stops, because the surviving younger cells still carry enough enzyme to cope. The Mediterranean and Canton variants are Class II, with severe deficiency under ten per cent activity, and their crises are more profound because even younger cells are vulnerable. [1]
The clinical classification that the bedside demands is simpler, because it sorts children by what they actually present with. Most patients fall into one of four expressions: episodic acute haemolytic anaemia triggered by an oxidant, neonatal jaundice, the rare chronic non-spherocytic haemolytic anaemia of Class I variants, and the asymptomatic carrier discovered only by screening. The candidate who names the clinical expression and the WHO class together demonstrates the full grasp of the disease. [2]
Epidemiology & Risk Factors
The single most important epidemiological fact is the sheer scale of G6PD deficiency. The systematic review and meta-analysis of Nkhoma and colleagues put the global prevalence at roughly 330 to 400 million people, around four to five per cent of humanity. The burden is concentrated where the gene is protective, namely the malaria belt, and it falls off sharply outside it. Sub-Saharan Africa carries the G6PD A-minus variant at high frequency, the Mediterranean basin carries the Mediterranean variant, and South and Southeast Asia carry Canton and related variants. [3]
The geographic distribution is the clearest example in medicine of a balanced polymorphism. Carrying one copy of a G6PD-deficient allele partially protects a red cell against invasion by Plasmodium falciparum, which is why the gene has persisted at high frequency across exactly the regions where malaria has killed the most children over millennia. Howes and colleagues mapped the spatial distribution of the variants across the malaria-endemic regions and confirmed the tight link between parasite pressure and gene frequency. This is also why the disease is highly relevant to the vivax malaria pathway: primaquine and tafenoquine, the only drugs that kill the liver hypnozoite and prevent relapse, are themselves oxidant triggers, so curing the malaria can trigger the haemolysis. [4]
The risk of an acute crisis is governed by the precipitant, and the precipitants fall into four families. Infection is the commonest trigger worldwide, from a viral upper respiratory illness to hepatitis or a bacterial sepsis. Fava beans, the broad bean Vicia faba, cause favism and are especially potent in Class II variants. Oxidant drugs form the most examinable list, led by primaquine, tafenoquine, rasburicase, methylene blue, dapsone, the sulphonamides, nitrofurantoin, nalidixic acid and niridazole. Chemical exposure to naphthalene in mothballs and to henna completes the set. Diabetic ketoacidosis is a recognised metabolic trigger. [5]
In the neonate, G6PD deficiency raises the risk of severe hyperbilirubinaemia roughly two- to threefold, and it is a leading cause of kernicterus in regions of high prevalence. Kaplan and colleagues showed the importance of the deficiency in African American male neonates with significant hyperbilirubinaemia, and the lesson for the fellowship candidate is that a G6PD neonate can develop bilirubin neurotoxicity at a level lower than the standard exchange threshold. [8][7]
Pathophysiology
The mechanism of G6PD deficiency is the mechanism of oxidative haemolysis, and it is worth following step by step because it explains every clinical and laboratory finding. The story begins with a peculiar vulnerability of the red cell: it has no nucleus, no mitochondria, and no ribosomes, so once it leaves the marrow it cannot make a single new protein. Every enzyme it will ever need, it must carry from birth. Its only source of NADPH is the G6PD step of the pentose phosphate pathway, which makes G6PD uniquely indispensable in the red cell even though it is redundant elsewhere. [1]
G6PD oxidises glucose-6-phosphate to six-phosphogluconolactone and in doing so reduces NADP to NADPH. The NADPH is then used by glutathione reductase to keep glutathione in its reduced form, reduced glutathione or GSH. Reduced glutathione, together with the enzyme glutathione peroxidase, neutralises the hydrogen peroxide and other oxidants that are generated constantly as the red cell carries oxygen. In a normal red cell this cycle runs quietly in the background. In a G6PD-deficient red cell, the moment an oxidant load arrives, the limited NADPH and reduced glutathione are consumed faster than they can be regenerated, and the defence collapses. [1][2]
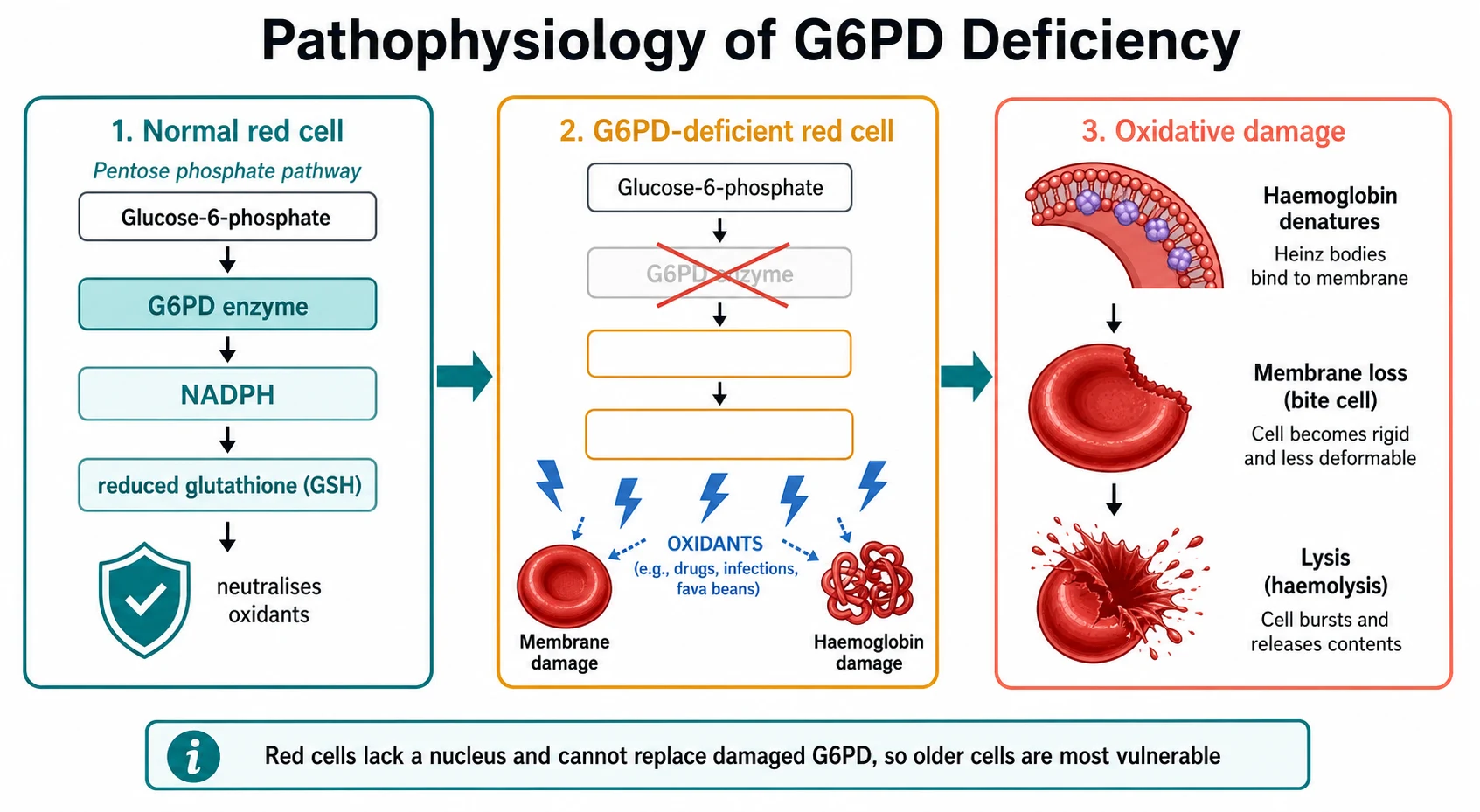
With the defences gone, the oxidants attack two targets. They oxidise the haemoglobin, which denatures and precipitates on the inner surface of the membrane as the clumps called Heinz bodies. These are invisible on a routine stain and need a supravital stain such as crystal violet to be seen. They also oxidise the membrane lipids and proteins, making the cell rigid and leaky. As these damaged cells pass through the spleen, the macrophages pit the Heinz bodies, tearing away bites of membrane and producing the characteristic bite cells and blister cells on the blood film. The membrane eventually fragments beyond repair and the cell lyses, releasing free haemoglobin into the plasma and causing the intravascular haemolysis that colours the urine dark. [1][2]
The age of the red cell is the key to understanding both the self-limited course and the diagnostic trap. Because the deficient enzyme, particularly in G6PD A-minus, decays as the cell ages, the oldest cells carry the least enzyme and are destroyed first. The haemolysis therefore targets a defined cohort and stops when that cohort is gone, which is why an African-variant crisis is usually self-limited. The same fact explains why an enzyme assay done during the crisis can read falsely normal: the deficient old cells have already been destroyed, and the surviving cells are the young reticulocytes, which carry the highest enzyme activity. This is the single most testable point in the topic. [2]
Clinical Presentation
The archetype is unmistakable once seen. A previously well boy, often of African, Mediterranean, Middle Eastern or Asian ancestry, develops sudden jaundice, dark tea- or cola-coloured urine, pallor, and weakness over hours to a day or two. There is often a recognisable trigger in the preceding day or week: a febrile illness, a meal of fava beans, a newly started drug, or exposure to naphthalene mothballs or henna. Abdominal or back pain is common, generated by the intravascular haemolysis. The tempo is fast enough that families often notice the dark urine before anything else. [1][9]
On examination the child is pale, with scleral icterus and visibly dark urine. There may be a tachycardia and a flow murmur from the anaemia, and a mild splenomegaly. The child is usually alert unless the haemolysis is massive or the anaemia profound. In acute massive haemolysis the picture can escalate to haemoglobinuria with acute kidney injury, haemodynamic instability, and a falling haemoglobin that needs urgent transfusion, as Lau and colleagues described in the Hong Kong series of children with severe crises. [9]
The neonate presents differently and more dangerously. The hallmark is early and often severe unconjugated hyperbilirubinaemia, frequently with no obvious oxidant trigger at all. The danger is that G6PD-deficient neonates can progress to acute bilirubin encephalopathy and kernicterus at bilirubin levels lower than the standard thresholds would predict, because the haemolysis compounds the immature blood-brain barrier and hypoalbuminaemia. Watchko emphasised that G6PD deficiency remains a refractory cause of kernicterus in developed countries precisely because it can strike at unexpectedly low bilirubin. Early bilirubin encephalopathy shows as lethargy, hypotonia, poor suck and a high-pitched cry, and it is a neurological emergency. [7]
A few presentations are deliberately tested because they catch the unwary. The first is the mild chronic anaemia that has been attributed to iron deficiency or another cause for years, until a crisis finally reveals the deficiency. The second is the symptomatic heterozygous female, made clinically affected by skewed X-inactivation, who presents with a typical crisis but is missed because the family history is male-line and the candidate assumes only males are affected. The third is the rare Class I chronic non-spherocytic haemolytic anaemia, which presents from infancy with persistent anaemia, splenomegaly and pigment gallstones rather than episodic crises. [2]
Differential Diagnosis
The differential of acute jaundice with anaemia in a child is broad, but the discriminating question is whether the haemolysis is immune or non-immune, and whether it is intravascular or extravascular. The direct antiglobulin test divides the field cleanly: G6PD deficiency is direct-antiglobulin-test-negative, which immediately separates it from autoimmune haemolysis and from ABO or rhesus haemolytic disease of the newborn. The blood film then refines the direct-antiglobulin-test-negative group. [1]
Three categories complete the differential. The other inherited haemolytic anaemias include hereditary spherocytosis with its spherocytes and family history of splenectomy or gallstones, sickle cell disease and the thalassaemias with their characteristic films and electrophoresis, and pyruvate kinase deficiency, the second commonest enzymopathy, which causes chronic direct-antiglobulin-test-negative haemolysis without spherocytes and without oxidant dependence. The immune causes, autoimmune haemolytic anaemia and haemolytic disease of the newborn, are direct-antiglobulin-test-positive. Finally, the non-haemolytic causes of jaundice must not be forgotten: sepsis, hepatitis, and obstructive causes can all present with jaundice, but they will not show the falling haemoglobin, raised reticulocytes, raised lactate dehydrogenase and low haptoglobin of haemolysis. [1]
The recurring errors are the ones the examiner probes. The first is attributing dark urine and jaundice to hepatitis when the unconjugated fraction and the falling haemoglobin point to haemolysis. The second is labelling a direct-antiglobulin-test-negative haemolysis as idiopathic and never measuring G6PD. The third is measuring G6PD during the crisis, accepting a falsely normal result, and falsely reassuring the family. A candidate who names these three traps demonstrates real understanding. [2]
Clinical & Bedside Assessment
The assessment begins with an overall sick-or-well judgement and a paediatric assessment of airway, breathing and circulation. In the stable child the history is where the diagnosis is won. Ask about ethnicity and family origin, because the gene clusters by ancestry across the malaria belt. Ask about a family history of anaemia, gallstones, splenectomy, jaundice after fava beans, or reactions to antimalarials, because the X-linked pattern often surfaces as a male relative who was told to avoid certain foods or drugs. Then take a precise inventory of every medicine, food and chemical exposure in the preceding week, naming the high-risk drugs by name. [5]
The focused examination looks for pallor, scleral icterus and jaundice, dark urine, tachycardia, a flow murmur, and mild splenomegaly. Look deliberately for the signs of an underlying infection, because infection is the commonest trigger and may itself need treatment. In the neonate, assess feeding, tone and the cephalocaudal level of the jaundice, and actively seek the early signs of bilirubin encephalopathy: lethargy, hypotonia, poor suck and a high-pitched cry. The neonatal assessment is never complete without a plan to plot the bilirubin on an hour-specific nomogram and to treat at a lower threshold given the raised neurotoxicity risk. [7]
[1] [5]The bedside synthesis is what the examiner rewards. The candidate who frames the case as a sudden direct-antiglobulin-test-negative intravascular haemolysis in a boy of at-risk ancestry after an oxidant trigger has already made the diagnosis before the assay returns. The skill is to hold that synthesis while sending the confirmatory tests and, critically, while planning to repeat the assay weeks later if the first one is normal. [1]
Investigations
The core panel confirms a haemolytic anaemia and defines it as non-immune. The full blood count shows a haemoglobin that may be mildly or profoundly low. The reticulocyte count is raised as the marrow responds. The peripheral blood film shows bite cells, blister cells, and Heinz bodies, though Heinz bodies are seen only on a supravital stain such as crystal violet and are absent on a routine stain. The unconjugated bilirubin and lactate dehydrogenase are raised, the haptoglobin is low, and the direct antiglobulin test is negative, confirming a non-immune intravascular haemolysis. A urine dipstick shows blood on the pad with no red cells on microscopy, the signature of haemoglobinuria. [1]
The definitive test is a quantitative G6PD enzyme assay on a red cell lysate, measuring the rate of NADPH generation spectrophotometrically. A reduced activity confirms the diagnosis. The trap, which the examiner will probe, is that the assay may be falsely normal during an acute crisis, because the enzyme-deficient old cells have already lysed and the surviving cells are young reticulocytes rich in enzyme. A normal result during a crisis never excludes the diagnosis; the assay is repeated two to three months later, once the red cell population has returned to its normal age distribution. [2]
[2] [1]Genotyping identifies the specific variant, such as G6PD A-minus, Mediterranean, or Canton, and is increasingly used, especially before primaquine or tafenoquine therapy for vivax malaria where knowing the variant refines the risk. Point-of-care quantitative G6PD tests are now validated and are transforming screening in malaria-endemic regions, as the individual participant data meta-analysis of Sadhewa and colleagues confirmed; they bring rapid, field-deployable quantitation that qualitative tests cannot match. In the neonate, send the routine haemolytic workup and a G6PD assay alongside the bilirubin plotted on the hour-specific nomogram. [11][6]
Management — Resuscitation
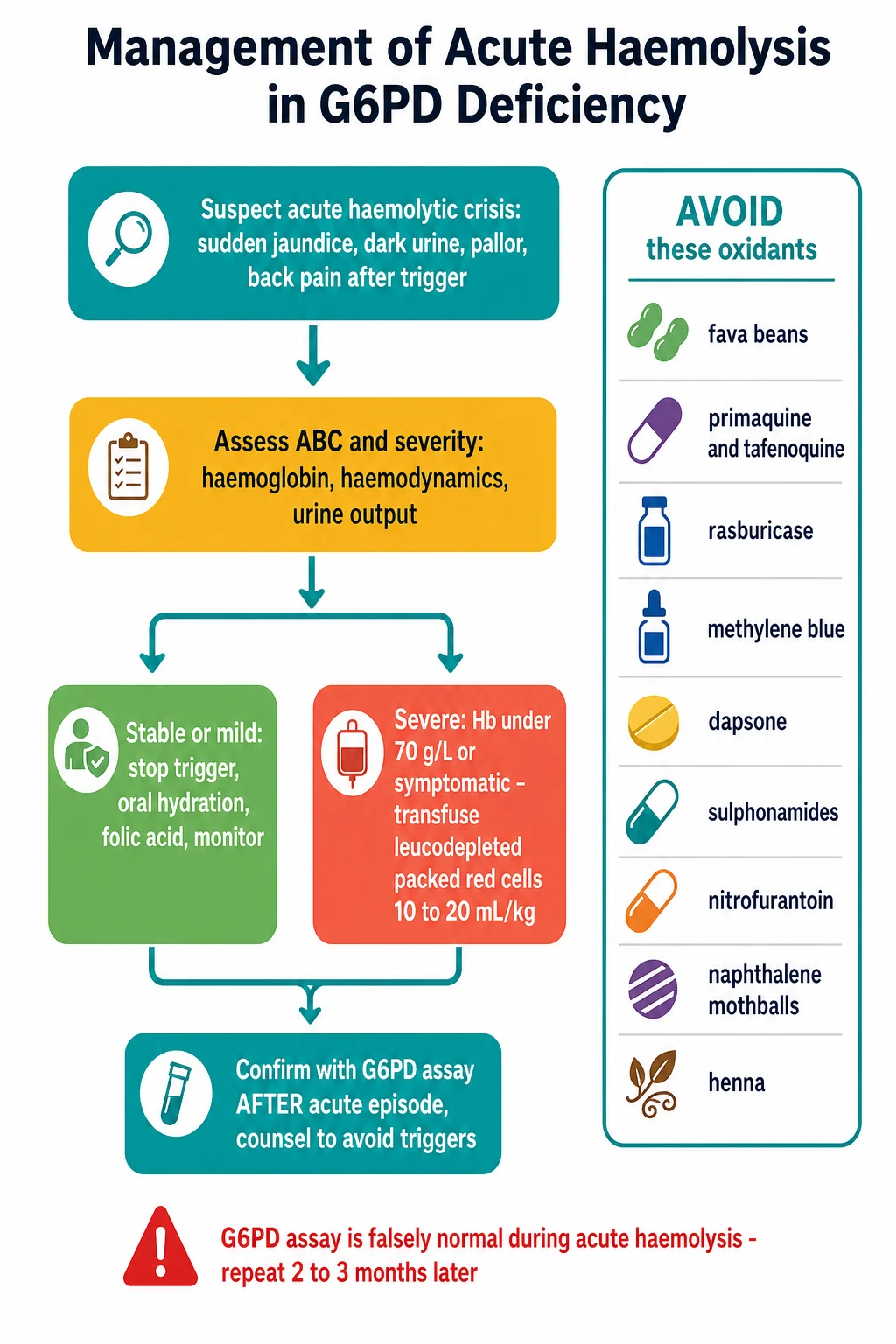
The first principle is reassuring: most acute G6PD crises are self-limited and need supportive care alone. The immediate priorities are to find and remove the trigger, to protect the kidney, and to watch the haemoglobin. Identify the precipitant and act on it: withdraw the offending drug, stop the fava beans, treat the underlying infection, and remove the naphthalene or henna source. Ensure adequate hydration, oral where possible and intravenous where the child is vomiting or unwell, to maintain a good urine output and protect the kidney from the haemoglobin load. Give folic acid to support the marrow recovery. [9]
Blood transfusion is reserved for the severe end of the spectrum. The threshold is a haemoglobin under 70 g per litre, or any symptomatic anaemia with haemodynamic compromise or a haemoglobin that is falling rapidly. The product is leucodepleted packed red cells given at 10 to 20 mL per kilogram, crossmatched in the usual way. Lau and colleagues described the children in the Hong Kong series who presented with acute massive haemolysis and required urgent transfusion, and the fellowship candidate should be ready to name the dose and the threshold. [9]
The indications for paediatric intensive care are acute kidney injury from haemoglobinuria, deepening or uncontrolled haemolysis, haemodynamic instability, and severe symptomatic anaemia. In the neonate, intensive phototherapy is the first-line treatment, using high-intensity blue light at 430 to 490 nanometres with maximal skin exposure, and exchange transfusion is used at the threshold determined by the hour-specific nomogram. Because G6PD raises the neurotoxicity risk, a lower threshold for both phototherapy and exchange is wise, and any sign of early bilirubin encephalopathy is treated as an emergency. [7]
The resuscitation of an acute G6PD crisis, step by step
Identify and stop the trigger: withdraw the drug, stop the fava beans, treat the infection, remove naphthalene or henna
Assess airway, breathing and circulation, and check the haemoglobin, urine output and haemodynamics
Hydrate to protect the kidney from haemoglobinuria, oral or intravenous, and give folic acid to support marrow recovery
Transfuse leucodepleted packed red cells 10 to 20 mL per kilogram if the haemoglobin is under 70 g per litre or the anaemia is symptomatic with compromise
Escalate to paediatric intensive care for acute kidney injury, uncontrolled haemolysis, or haemodynamic instability
In the neonate, give intensive phototherapy and use exchange transfusion at a lower threshold given the raised kernicterus risk
Management — Definitive & Stepwise
[1] [6]The definitive management is preventive, and it is built in five steps. The first, and by far the most important, is trigger avoidance for life. Hand the family a written list of the drugs to avoid and advise them to avoid fava beans, naphthalene mothballs, and henna. The list is not optional: Youngster and colleagues built the evidence-based drug review that underpins it, and the 2023 Clinical Pharmacogenetics Implementation Consortium guideline of Gammal and colleagues is the current prescribing standard that stratifies the drugs by risk. [5][6]
The Clinical Pharmacogenetics Implementation Consortium guideline divides the oxidant drugs by the strength of the evidence. Rasburicase, methylene blue, primaquine and tafenoquine, and dapsone are the strong triggers that a deficient patient should avoid. Nitrofurantoin, the sulphonamides, and several others carry weaker or uncertain risk and may be used with caution and monitoring when no alternative exists. The candidate who can separate the strong from the weak triggers demonstrates command of the guideline. [6]
The second step is folic acid supplementation during and after a crisis to support the marrow. The third step is transfusion for severe or symptomatic anaemia, already described. The fourth step is screening and counselling of first-degree relatives, because the X-linked inheritance means the maternal line and male siblings are at risk and deserve an assay. The fifth step is special prescribing caution in the situations where the deficiency changes a standard treatment decision. [6][5]
The three prescribing cautions that the examiner rewards are crisp. Never give primaquine or tafenoquine for the radical cure of vivax or ovale malaria without first confirming a normal G6PD status, because these drugs are strong oxidant triggers. Never use methylene blue to treat methaemoglobinaemia in a G6PD-deficient patient, because it both fails to reduce the methaemoglobin and precipitates haemolysis. And check G6PD status before using rasburicase for tumour lysis prophylaxis. Routine splenectomy is not part of G6PD management, except in the rare severe Class I chronic non-spherocytic haemolytic anaemia where it is occasionally considered. [6]
Specific Subtypes & Scenarios
The commonest subtype worldwide is G6PD A-minus, the Class III African variant. Its enzyme activity runs at ten to sixty per cent, and because the activity decays faster than normal as the red cell ages, the oldest cells are selectively lysed. The clinical consequence is that a crisis is usually self-limited: the haemolysis stops once the deficient old-cell cohort is cleared, and the baseline haemoglobin is normal between crises. This is the variant the fellowship candidate will most often see in a child of African ancestry in Australia, New Zealand, the United Kingdom, the United States or Canada. [1]
The Mediterranean and Canton variants are Class II, with severe deficiency under ten per cent activity. Here the crisis can be profound, because even younger cells carry too little enzyme to cope. Favism, the haemolytic crisis after ingesting fava beans, is especially characteristic of the severe variants, and it can be severe enough to threaten the child. Prashanth and colleagues compared fava-bean-induced crises with non-fava-bean crises in children and found the fava-bean crises tended to be more severe, reinforcing the need for firm dietary counselling in affected families. [10]
Neonatal G6PD jaundice is a subtype in its own right and the one with the greatest potential for permanent harm. It can present without any oxidant exposure, is often severe, and carries the kernicterus risk that justifies a lower treatment threshold. The chronic non-spherocytic haemolytic anaemia of the Class I variants is rare; it presents from infancy with persistent anaemia, splenomegaly and pigment gallstones, and is distinguished by continuous haemolysis rather than episodic crises. Finally, the symptomatic heterozygous female, made clinically affected by skewed X-inactivation, can present with a typical crisis and is easily missed; she needs a quantitative assay or genotyping when suspected. [2][7]
Complications & Pitfalls
The acute complications follow directly from the intravascular haemolysis. Haemoglobinuria can cause acute kidney injury as the free haemoglobin and haemosiderin damage the renal tubule. Massive haemolysis can cause haemodynamic collapse and a falling haemoglobin that demands urgent transfusion. In the neonate, the gravest complication is kernicterus, with its permanent sequelae of athetoid cerebral palsy, sensorineural hearing loss, and upward gaze palsy. Over the longer term, repeated severe crises can lead to pigment gallstones from the chronic bilirubin load. [9][7]
The pitfalls are the heart of the examination, and the candidate should be able to name each one and how to avoid it. The classic and most-tested pitfall is measuring G6PD during an acute crisis, accepting the falsely normal result, and falsely reassuring the family. The defence is to know that the surviving cells are young reticulocytes rich in enzyme, and to repeat the assay two to three months later. The second is giving primaquine, tafenoquine, methylene blue, or rasburicase without checking G6PD status first; the defence is to treat these drugs as requiring a confirmed normal assay. [2][6]
The third pitfall is attributing a direct-antiglobulin-test-negative haemolysis to idiopathic causes and never measuring G6PD; the defence is to include G6PD in every direct-antiglobulin-test-negative haemolysis workup. The fourth is forgetting that females can be symptomatic heterozygotes; the defence is to test symptomatic females with a quantitative assay or genotyping. The fifth is failing to counsel the whole family because of the X-linked pattern; the defence is to screen first-degree relatives. The sixth, and the most dangerous in the neonate, is underestimating the kernicterus risk and using standard rather than lower bilirubin thresholds; the defence is to treat the G6PD neonate as high neurotoxicity risk. [7][2]
Prognosis & Disposition
The prognosis of G6PD deficiency is excellent once the diagnosis is made and the triggers are avoided. The vast majority of patients are completely asymptomatic between crises, and life expectancy is normal. A single acute crisis resolves as the deficient old-cell cohort is cleared and the marrow replaces the loss over two to three weeks, with the reticulocyte count rising first and the haemoglobin following. The disease becomes a matter of informed avoidance rather than illness. [1]
The disposition from an acute crisis is governed by the haemoglobin and the haemodynamics. A well child with mild anaemia and a clear, removed trigger can be managed as an outpatient, with close follow-up, a repeat full blood count, and a clear safety-net to return. Severe anaemia, haemoglobinuria with kidney injury, or haemodynamic instability require admission, transfusion, and often intensive care. The neonate is admitted for phototherapy and observation at the appropriate bilirubin threshold, with a low threshold for exchange transfusion if the bilirubin rises or signs of encephalopathy appear. [9][7]
Every patient and family leave with three things. A written trigger-avoidance list, naming the drugs and the foods and the chemicals to avoid, so that it can be shown to every future prescriber. A plan for family screening, because the X-linked inheritance puts the maternal line and male siblings at risk. And a clear safety-net, so that the family knows to return at once if jaundice, dark urine, or pallor recur. The combination of informed avoidance, family screening, and a safety-net is what turns a potentially dangerous enzyme defect into a safely managed condition. [5]
Special Populations
Neonates are the highest-risk group for harm from G6PD deficiency, because the condition is a leading cause of severe hyperbilirubinaemia and kernicterus in regions of high prevalence. The lesson from Kaplan and colleagues, who showed the importance of the deficiency in African American male neonates, is that the neonate needs a low threshold for phototherapy and exchange, and active surveillance for early bilirubin encephalopathy. Targeted or universal G6PD screening of neonates is practised in many high-prevalence countries, and the case for wider screening is actively debated. [8][7]
Symptomatic heterozygous females are a population that is easily missed. Skewed X-inactivation can make a carrier clinically affected, and she can present with a typical crisis. Because the family history is male-line, the diagnosis is often delayed. The defence is to test symptomatic females with a quantitative enzyme assay or genotyping, recognising that a single assay can be normal in a heterozygote with two red-cell populations. [2]
Children returning from or living in vivax-endemic regions, such as Papua New Guinea, need a confirmed normal G6PD status before any primaquine or tafenoquine radical cure is given. The radical cure is essential to prevent relapse, but in a deficient child it can trigger a severe haemolysis, so the assay is a non-negotiable prerequisite. Indigenous and migrant families from sub-Saharan Africa, the Mediterranean, the Middle East, and South and Southeast Asia are the populations in whom the diagnosis must be actively considered, and culturally appropriate counselling about diet and medicines, with written and translated trigger lists, improves outcomes. Children on long-term oxidant drugs for other conditions, such as dapsone for dermatitis herpetiformis or nitrofurantoin for prophylaxis, should be screened before the drug is started. [6][4]
Evidence, Guidelines & Regional Differences
The evidence base rests on a set of landmark papers that the fellowship candidate should be able to name. The classic biochemistry and clinical review of Cappellini and Fiorelli in the Lancet remains the foundation. The pharmacogenetic review of Luzzatto and Seneca frames G6PD deficiency as the textbook example of pharmacogenetics with on-going clinical implications. The global prevalence meta-analysis of Nkhoma and colleagues quantified the burden, and the spatial mapping of Howes and colleagues tied the variants to the malaria-endemic regions. [1][2][3][4]
The prescribing evidence rests on the drug review of Youngster and colleagues and on the 2023 Clinical Pharmacogenetics Implementation Consortium guideline of Gammal and colleagues, which is the current standard for medication use in the context of G6PD genotype. The guideline stratifies the oxidant drugs by the strength of the evidence into strong triggers to avoid and weaker triggers to use with caution, and it is the reference the candidate should cite when discussing prescribing decisions. The neonatal risk is grounded in the work of Kaplan and colleagues and the kernicterus review of Watchko. [5][6][8][7]
The regional differences are real and examinable. Many Mediterranean, Middle Eastern and Asian countries screen neonates universally or by risk, and G6PD deficiency is a routine part of the newborn panel in several high-prevalence health systems. Australia, New Zealand, the United Kingdom, the United States and Canada generally target at-risk groups rather than screen universally, though the debate over universal neonatal screening continues. The point-of-care quantitative tests validated by Sadhewa and colleagues are reshaping the field, because they make rapid, field-deployable quantitation possible, which is especially important before primaquine radical cure in malaria-endemic regions. [11]
In Australia and Aotearoa New Zealand, G6PD deficiency is a targeted rather than universal screen. The diagnosis is most often made after an acute crisis in a child of African, Mediterranean, Middle Eastern, South Asian or Southeast Asian ancestry, or in a returned traveller or a child from the Papua New Guinea pathway who needs vivax radical cure. The Clinical Pharmacogenetics Implementation Consortium 2023 guideline applies, and every primaquine or tafenoquine decision for a child with vivax malaria from Papua New Guinea is preceded by a confirmed normal G6PD assay. Neonatal management follows the 2022 American Academy of Pediatrics hyperbilirubinaemia thresholds with a deliberately lower treatment threshold in the confirmed or suspected G6PD neonate.
Exam Pearls
G6PD deficiency is X-linked, carried at Xq28, and is the most common red-cell enzyme defect and the most common human enzyme deficiency, affecting roughly 330 to 400 million people. Males are affected; females are carriers or symptomatic heterozygotes by skewed X-inactivation. The mechanism is the loss of NADPH and reduced glutathione, so oxidants denature haemoglobin into Heinz bodies and the spleen produces bite cells and blister cells. The direct antiglobulin test is negative. These six facts open the viva. [1][3]
IFOND
The four triggers are infection, fava beans, oxidant drugs, and naphthalene or henna. The drugs to never give without checking G6PD are primaquine, tafenoquine, rasburicase, methylene blue, and dapsone. The single most-tested pitfall is that the G6PD assay is falsely normal during an acute crisis because the surviving cells are young reticulocytes, so repeat it two to three months later. G6PD neonates are at high risk of kernicterus at lower-than-expected bilirubin. The blood film shows bite cells, blister cells, and Heinz bodies on a supravital stain. Transfuse for severe anaemia under 70 g per litre or symptomatic anaemia, with leucodepleted packed red cells 10 to 20 mL per kilogram; most crises need only trigger withdrawal and hydration. Always counsel the family and screen the relatives. [6][2][7]
[2] [6] [5]References
- [1]Cappellini MD, Fiorelli G Glucose-6-phosphate dehydrogenase deficiency. Lancet, 2008.PMID 18177777
- [2]Luzzatto L, Seneca E G6PD deficiency: a classic example of pharmacogenetics with on-going clinical implications. Br J Haematol, 2014.PMID 24372186
- [3]Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol Dis, 2009.PMID 19233695
- [4]Howes RE, Dewi M, Piel FB, Monteiro WM, et al Spatial distribution of G6PD deficiency variants across malaria-endemic regions. Malar J, 2013.PMID 24228846
- [5]Youngster I, Arcavi L, Schechmaster R, Akayzen Y, et al Medications and glucose-6-phosphate dehydrogenase deficiency: an evidence-based review. Drug Saf, 2010.PMID 20701405
- [6]Gammal RS, Pirmohamed M, Somogyi AA, Morris SA, et al Expanded Clinical Pharmacogenetics Implementation Consortium Guideline for Medication Use in the Context of G6PD Genotype. Clin Pharmacol Ther, 2023.PMID 36049896
- [7]Watchko JF Refractory Causes of Kernicterus in Developed Countries: Can We Eradicate G6PD Deficiency Triggered and Low-Bilirubin Kernicterus? Curr Pediatr Rev, 2017.PMID 28721814
- [8]Kaplan M, Herschel M, Hammerman C, Hoyer JD, Stevenson DK Neonatal hyperbilirubinemia in African American males: the importance of glucose-6-phosphate dehydrogenase deficiency. J Pediatr, 2006.PMID 16860133
- [9]Lau HK, Li CH, Lee AC Acute massive haemolysis in children with glucose-6-phosphate dehydrogenase deficiency. Hong Kong Med J, 2006.PMID 16603783
- [10]Prashanth GP, Al-Shafey M, Tandon A, Ismail S Fava Bean- Versus Non-Fava Bean-Induced Acute Hemolytic Crisis in Children With Glucose-6-Phosphate Dehydrogenase Deficiency: A Prospective Comparative Study. Pediatr Blood Cancer, 2025.PMID 39956941
- [11]Sadhewa A, Satyagraha AW, Alam MS, Adissu W, et al Performance of quantitative point-of-care tests to measure G6PD activity: An individual participant data meta-analysis. PLoS Negl Trop Dis, 2025.PMID 40132008