Paeds · haematology-oncology-and-transfusion
Haemolytic anaemia: diagnostic approach
Also known as Haemolytic anaemia · Haemolysis workup · Coombs test interpretation · Direct antiglobulin test approach · Autoimmune haemolytic anaemia workup
Fellowship guide to the systematic diagnostic approach to haemolytic anaemia in infants, children and adolescents. Covers recognising haemolysis from reticulocytes, lactate dehydrogenase, haptoglobin and unconjugated bilirubin; splitting immune from non-immune causes with the direct antiglobulin test; and the morphological and confirmatory tests that distinguish hereditary spherocytosis, glucose-6-phosphate dehydrogenase deficiency, sickle cell disease, thalassaemia and microangiopathic haemolysis.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
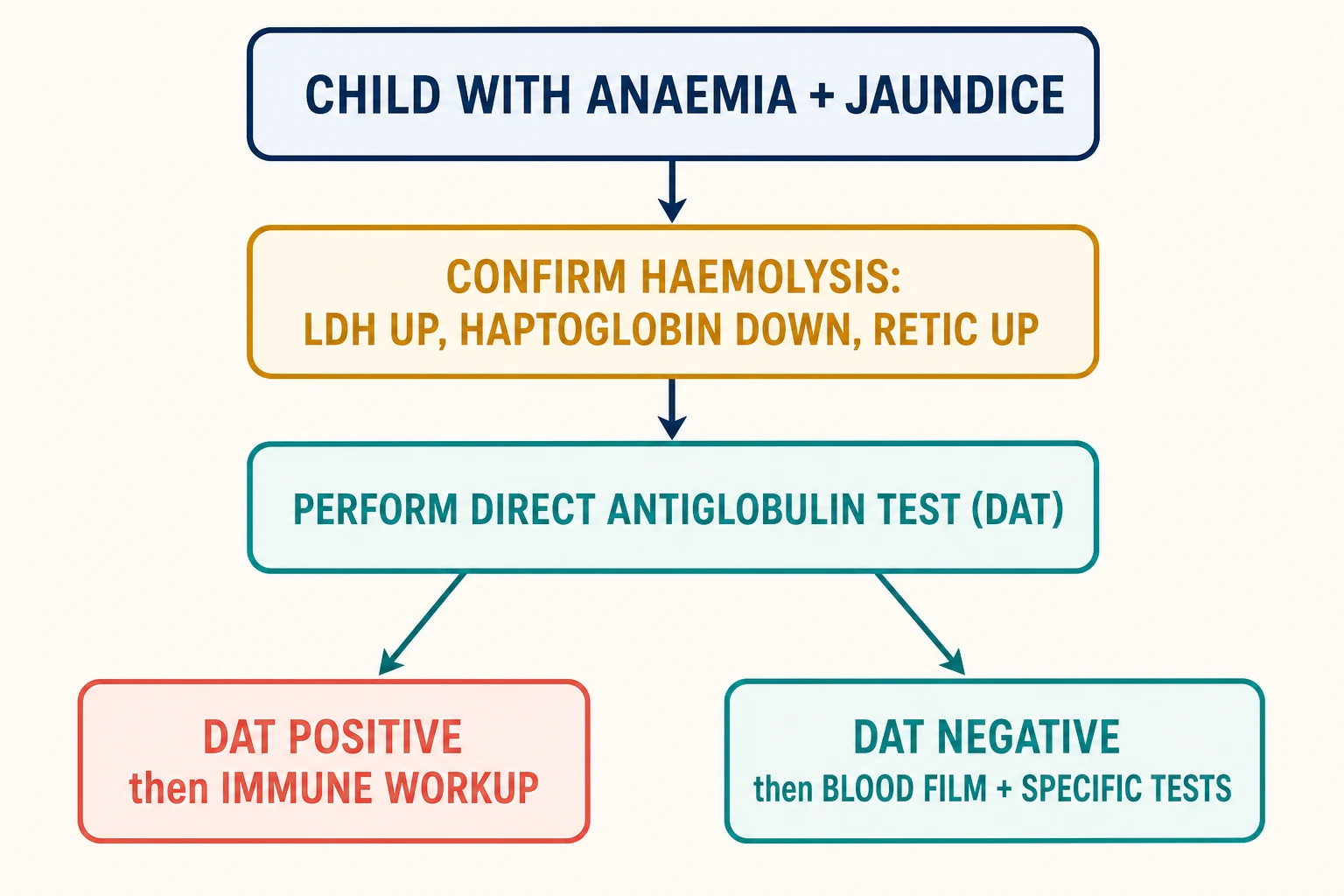
Haemolytic anaemia is anaemia produced by a shortened red cell lifespan: the bone marrow cannot keep up with the rate of destruction. The central diagnostic task is not to name every cause at once, but to move through three ordered questions. Is the anaemia really haemolytic? Is it immune or non-immune? And which specific mechanism is responsible? Answering these in sequence is what separates a focused workup from a scattergun battery of tests. [2]
The first question is settled by a small panel of indirect haemolysis markers. A raised reticulocyte count, a raised lactate dehydrogenase, a low or absent haptoglobin, and a raised unconjugated bilirubin together confirm that red cells are being destroyed faster than they are made. These markers are cheap, rapid, and available in every hospital laboratory, and they anchor every subsequent decision. [2]
The second question is settled by a single test, the direct antiglobulin test. A positive result points the clinician toward antibody-mediated destruction and a search for an immune trigger. A negative result redirects the workup toward the inherited and microangiopathic causes, guided by the blood film. The third question is settled by morphology and a handful of targeted confirmatory assays. [3]
Classification
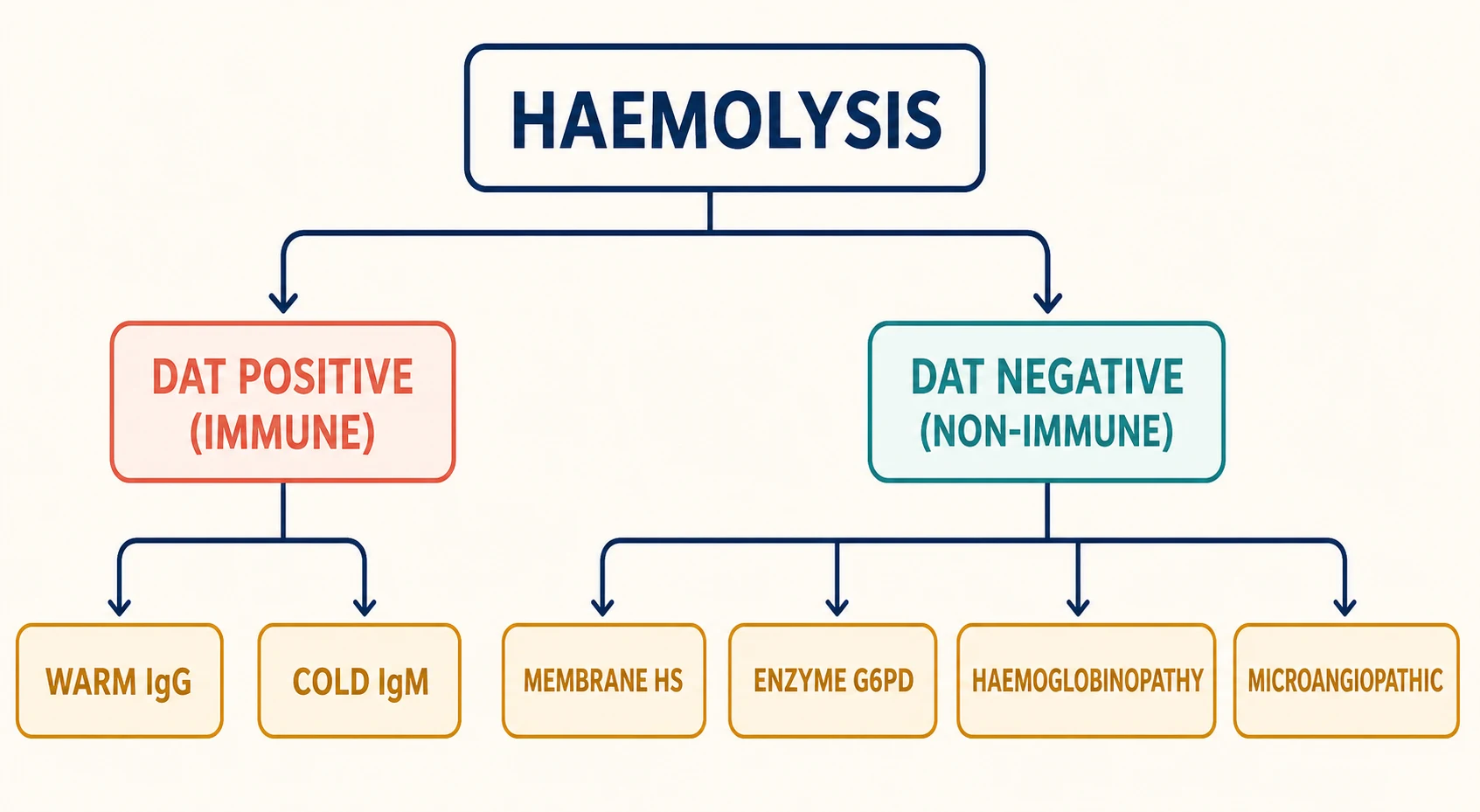
The most useful single branch point is the direct antiglobulin test. Splitting haemolysis into direct-antiglobulin-positive and direct-antiglobulin-negative groups immediately halves the differential and tells the clinician whether to chase an immune process or an intrinsic red cell defect. This division is the backbone of the diagnostic algorithm and should be taught to trainees before any finer subclassification. [3]
[4]Within the immune group, the antibody class and its thermal optimum matter because they change both the film and the management. Warm autoimmune haemolytic anaemia is immunoglobulin G-mediated, reacts at body temperature, and produces spherocytes with extravascular destruction in the spleen. Cold agglutinin disease is immunoglobulin M-mediated, fixes complement in cooler peripheral vessels, and produces red cell agglutination. A mixed picture and the rarer drug-induced and alloimmune causes complete the immune side. [1]
Within the non-immune group, the blood film usually gives the lead. Spherocytes point to a membrane defect, most often hereditary spherocytosis. Bite cells and Heinz bodies point to an enzyme defect, most often glucose-6-phosphate dehydrogenase deficiency. Sickle cells and target cells point to a haemoglobinopathy. Fragmented schistocytes point to mechanical or microangiopathic injury. Each morphological clue carries a confirmatory test, and chasing that clue is far more efficient than ordering every assay at once. [8]
Epidemiology & Risk Factors
Autoimmune haemolytic anaemia is uncommon in children but over-represented in exam material because its diagnosis turns on tests a general paediatrician must interpret. The reported incidence is roughly 1 to 3 per million children per year, with a peak in preschool age and a second rise in adolescence. Around half of paediatric cases follow an infection, most often an upper respiratory or gastrointestinal virus, and a substantial minority are secondary to an autoimmune disease or immunodeficiency. [6]
Warm antibody disease dominates the paediatric immune group and accounts for roughly two-thirds of childhood autoimmune haemolytic anaemia. Cold agglutinin disease is less common in children than in older adults but appears characteristically after Mycoplasma pneumoniae or Epstein-Barr virus infection. Post-infectious immune haemolysis tends to be self-limited over weeks, whereas idiopathic and secondary forms run a more chronic, relapsing course. [5]
Among the inherited causes, hereditary spherocytosis is the most common inherited haemolytic anaemia in people of northern European descent, with a prevalence of around 1 in 2000. Glucose-6-phosphate dehydrogenase deficiency is the most common enzymatic red cell defect worldwide, affecting hundreds of millions and reaching high prevalence in Mediterranean, African, Middle Eastern and Southeast Asian populations. Sickle cell disease and the thalassaemias are concentrated in the same malaria-belt populations and are increasingly encountered across Australia and New Zealand through migration. [7]
Acquired non-immune causes are rarer but carry disproportionate danger. Thrombotic thrombocytopenic purpura, haemolytic uraemic syndrome and disseminated intravascular coagulation share microangiopathic fragmentation on the film and demand urgent action. Paroxysmal nocturnal haemoglobinuria is rare in children but enters the differential of direct-antiglobulin-negative haemolysis with pancytopenia or unexplained thrombosis. [2]
Pathophysiology
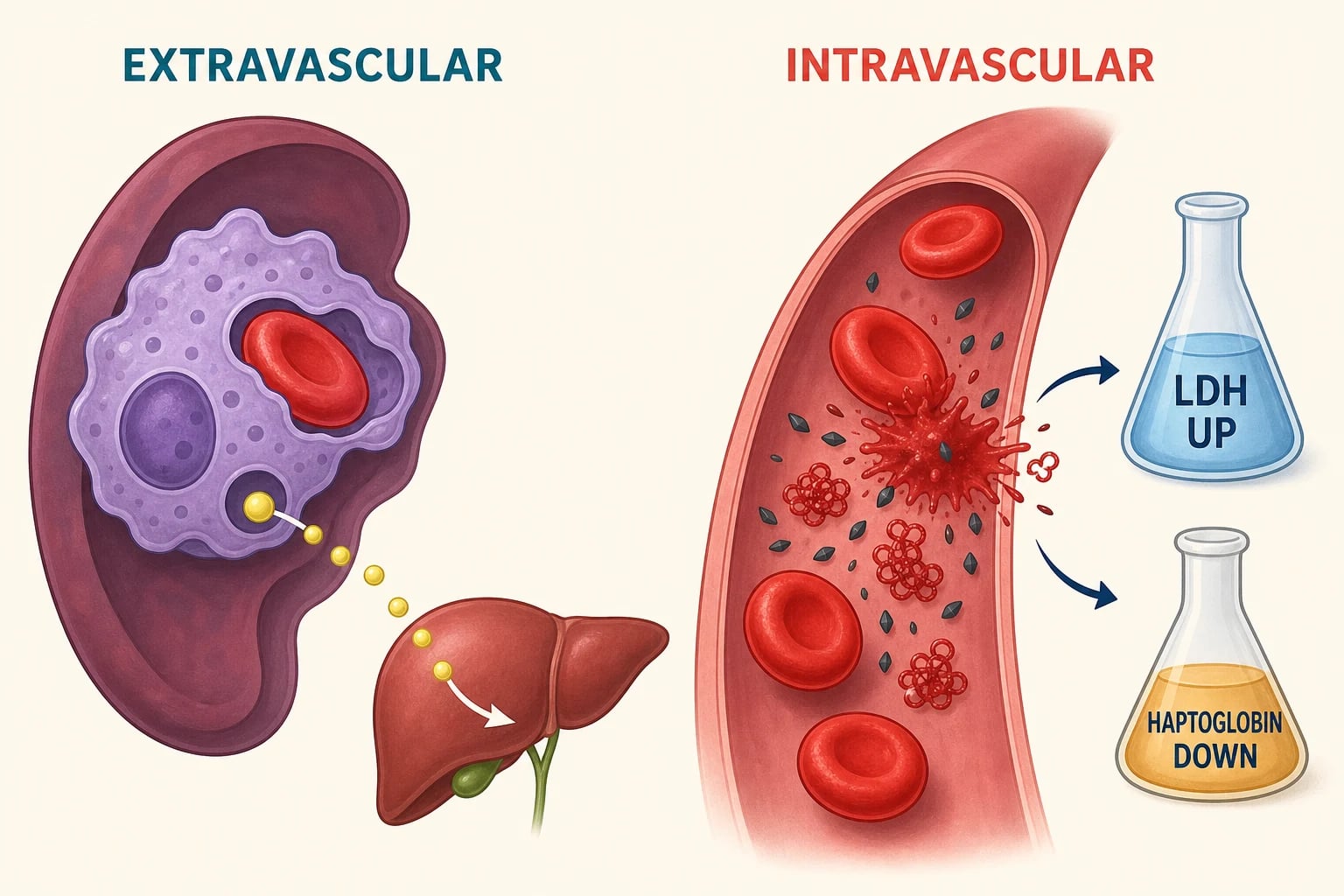
Haemolysis damages red cells either outside or inside the circulation, and the site of destruction shapes the laboratory pattern. Extravascular haemolysis dominates in the spleen and liver, where macrophages clear antibody-coated, membrane-damaged or poorly deformable cells. The released haemoglobin is processed to unconjugated bilirubin, so indirect bilirubin rises and jaundice appears. This pathway is typical of warm autoimmune haemolytic anaemia and hereditary spherocytosis. [1]
Intravascular haemolysis releases free haemoglobin directly into plasma. Haptoglobin binds the free haemoglobin and the complex is cleared, so haptoglobin falls sharply and is often the most sensitive single marker of intravascular destruction. Once haptoglobin is saturated, free haemoglobin circulates, is filtered by the kidney, and produces haemoglobinuria with cola-coloured urine. Lactate dehydrogenase rises as red cells lyse, and these three findings together are the biochemical fingerprint of intravascular haemolysis. [2]
The bone marrow responds to destruction by ramping up erythropoiesis, which is why the reticulocyte count rises. Reticulocytes are large, polychromatic cells, and a brisk reticulocytosis itself raises the mean corpuscular volume. A high mean corpuscular volume with a high reticulocyte count in a jaundiced child is a strong clue that the marrow is responding to haemolysis rather than failing. The exception is the aplastic crisis, where the marrow response collapses and reticulocytes fall. [2]
In warm autoimmune haemolytic anaemia, immunoglobulin G coats the red cell and splenic macrophages either phagocytose the cell or nibble away its membrane, producing the small, dense spherocyte. In cold agglutinin disease, immunoglobulin M binds in cool peripheral skin and fixes complement; the immunoglobulin M then detaches, but complement remains, and the direct antiglobulin test reads positive for complement (C3d) rather than immunoglobulin G. [4]
In the inherited membrane and enzyme disorders, the red cell is destroyed because it is structurally or metabolically abnormal. Hereditary spherocytosis destabilises the vertical membrane protein interactions, losing surface area and producing a rigid spherocyte that the spleen traps. Glucose-6-phosphate dehydrogenase deficiency starves the cell of reduced glutathione, so oxidative stress denatures haemoglobin into Heinz bodies that damage the membrane. The spleen again removes the damaged cells, leaving bite cells on the film. [10]
Clinical Presentation
Children with haemolytic anaemia present along a spectrum from incidental anaemia to life-threatening collapse. The chronic forms most often bring a pale, mildly jaundiced child with fatigue and exercise intolerance, sometimes discovered on a routine blood count. Splenomegaly is a valuable clue because the spleen is both the workplace of extravascular destruction and the site of compensatory extramedullary haematopoiesis. [7]
The pace of onset is itself diagnostic information. A child with glucose-6-phosphate dehydrogenase deficiency typically describes a trigger, eating fava beans or starting a new drug, followed within one to three days by dark urine and increasing pallor. A child with autoimmune haemolytic anaemia more often describes days of progressive tiredness after a viral illness, with jaundice and sometimes fever. Sudden severe pallor with cardiovascular compromise points to a fulminant immune process or an aplastic crisis. [5]
Jaundice in haemolysis is unconjugated, so the stools stay normally coloured and the urine may be dark from urobilinogen or, in intravascular forms, frankly red-brown from haemoglobin. Pigment gallstones are a late complication of any chronic haemolysis and may bring a teenager in with biliary colic before the underlying anaemia is recognised. A family history of gallbladder disease, splenectomy or neonatal jaundice should always be sought. [7]
Certain presentations flag a dangerous cause. Acrocyanosis or Raynaud-type change on cold exposure suggests cold agglutinin disease. Bleeding or bruising alongside haemolysis raises Evans syndrome, where immune haemolytic anaemia accompanies immune thrombocytopenia, or a thrombotic microangiopathy. Neurological change with haemolysis and thrombocytopenia raises thrombotic thrombocytopenic purpura, a true emergency. [4]
Presenting patterns by mechanism
Chronic extravascular: pallor, mild jaundice, splenomegaly, pigment gallstones (hereditary spherocytosis, warm AIHA)
Episodic after trigger: dark urine, sudden pallor, back or abdominal pain (G6PD deficiency)
Post-infectious: jaundice and fatigue days after a virus (warm or cold AIHA)
Intravascular emergency: cola urine, falling haemoglobin, shock (severe AIHA, PNH, mismatched transfusion)
Microangiopathic: fragmentation, thrombocytopenia, organ dysfunction (HUS, TTP, DIC)
Differential Diagnosis
The differential diagnosis begins by confirming that the anaemia is truly haemolytic, because several non-haemolytic causes mimic the picture. Acute blood loss produces pallor and a high reticulocyte count but normal haptoglobin and bilirubin. Marrow recovery from a nutritional deficiency or suppression shows a rising reticulocyte count without destruction. A megaloblastic crisis raises the mean corpuscular volume for a different reason, and ineffective erythropoiesis in B12 or folate deficiency can itself raise bilirubin. [2]
Once haemolysis is confirmed, the direct antiglobulin test divides the differential. A positive test brings autoimmune haemolytic anaemia to the top, and the clinician must then separate primary from secondary disease by screening for systemic lupus erythematosus, common variable immunodeficiency, lymphoproliferative disorder, and recent drugs or transfusion. Cold agglutinins are suggested by the film and confirmed with a cold agglutinin titre and a Mycoplasma or Epstein-Barr serology. [4]
A negative test redirects the workup to intrinsic red cell disorders and mechanical causes. The blood film is now the guide. Spherocytes with a positive family history point to hereditary spherocytosis. Bite cells in a boy after a trigger point to glucose-6-phosphate dehydrogenase deficiency. Sickle cells or target cells point to a haemoglobinopathy. Schistocytes with thrombocytopenia point to a thrombotic microangiopathy or disseminated intravascular coagulation. [8]
Paroxysmal nocturnal haemoglobinuria deserves a deliberate place on the direct-antiglobulin-negative list. It is an acquired clonal loss of glycosylphosphatidylinositol-anchored complement regulators, producing complement-mediated intravascular haemolysis, often with pancytopenia and an unusual thrombosis. Flow cytometry with fluorescent aerolysin, which binds the missing anchor, is the modern diagnostic standard. Wilson disease and oxidant exposure are rarer intravascular causes that should not be forgotten. [2]
Clinical & Bedside Assessment
The bedside assessment serves two purposes: to gauge how sick the child is, and to harvest the historical clues that direct the laboratory workup. Begin with the paediatric assessment triangle and a full set of observations. Tachycardia, a flow murmur, prolonged capillary refill and pallor quantify the cardiovascular strain, and their severity decides whether the child can wait for results or needs urgent transfusion. [5]
Examine the skin and conjunctivae for pallor and icterus, paying attention to whether the jaundice is most visible in the sclera or the skin. Palpate the abdomen for hepatosplenomegaly. Splenomegaly is expected in hereditary spherocytosis and autoimmune haemolytic anaemia, and hepatosplenomegaly with lymphadenopathy points to a secondary cause such as lymphoproliferative disease. Look for acrocyanosis, which suggests cold agglutinin disease, and for petechiae or bruising, which suggest Evans syndrome or a microangiopathy. [7]
The history is where the specific diagnosis usually declares itself. Ask about recent infections, new medications, fava beans, mothballs, and occupational or traditional remedies. Establish the timeline from the first symptom to presentation, because the pace distinguishes an acute crisis from chronic compensated haemolysis. Take a careful family history of anaemia, splenectomy, cholecystectomy, neonatal jaundice, and consanguinity, and record the ethnic background, which raises the prior probability of glucose-6-phosphate dehydrogenase deficiency, sickle cell disease and thalassaemia. [10]
A few focused bedside manoeuvres refine the picture. The cold agglutinin test at the bedside is to warm a blood sample to 37 degrees Celsius and watch agglutination resolve; its persistence at body temperature argues against a simple cold antibody. Inspect a fresh urine sample directly, because haemoglobinuria is easily missed if the sample is old or diluted. A finger-prick blood film viewed at the bedside can be unrevealing, but knowing whether to prioritise the laboratory film changes the order of the day. [4]
Investigations
The investigation strategy runs in three tiers: confirm haemolysis, split immune from non-immune, then confirm the specific cause. The first tier is the same for every child and should be sent at the first venepuncture so that no sample is wasted. The full blood count shows the haemoglobin, mean corpuscular volume, and a reticulocyte count. A high reticulocyte count, typically above 2 per cent or an absolute count above 100 times 10 to the 9 per litre, confirms that the marrow is responding. [2]
The confirmatory biochemistry is reticulocyte count, lactate dehydrogenase, haptoglobin and unconjugated bilirubin. Lactate dehydrogenase rises as cells lyse. Haptoglobin falls or disappears because it binds free haemoglobin and is cleared; a low haptoglobin is among the most sensitive markers of haemolysis, though it is also an acute phase reactant and can be falsely reassuring during inflammation. Unconjugated bilirubin rises from extravascular and intravascular destruction alike. [2]
The second tier is the direct antiglobulin test, the pivotal investigation of the whole algorithm. Performed on ethylenediaminetetraacetic acid anticoagulated blood, it uses antihuman globulin reagent to cross-link antibody or complement on the red cell surface, producing visible agglutination. A polyspecific reagent screens for both immunoglobulin G and complement; if positive, monospecific reagents separate immunoglobulin G from C3d coating and so distinguish warm from cold disease. [3]
The third tier is morphology and confirmatory testing, guided by the film and the direct antiglobulin test result. The peripheral blood film must be examined by someone who knows what to look for, because each morphology carries a specific confirmatory test. A smear read in haste is the most common source of diagnostic delay. [8]
[8]For the membrane disorders, the International Council for Standardization in Haematology recommends flow cytometric eosin-5-maleimide binding as the preferred screening test, with cryohaemolysis and acidified glycerol lysis as alternatives. Sodium dodecyl sulfate polyacrylamide gel electrophoresis of membrane proteins confirms the diagnosis and identifies the defective protein. The old osmotic fragility test is less specific because it is also raised after any recent transfusion. [8]
For glucose-6-phosphate dehydrogenase deficiency, the enzyme assay must be interpreted with care. During an acute haemolytic episode the most severely deficient cells have already been destroyed, leaving relatively normal cells behind, so the assay can return falsely normal. Repeat the assay during remission, several weeks after the episode resolves, and remember that the deficiency is X-linked so affected males and homozygous or compound heterozygous females are symptomatic while heterozygous females are mosaic. [10]
For the haemoglobinopathies, haemoglobin electrophoresis or high-performance liquid chromatography defines the variant. Sickle cell disease is suggested by a positive sickle solubility test, but solubility testing alone cannot distinguish sickle cell trait from disease and does not detect other variants, so it must always be followed by quantification. In the neonate, newborn screening using high-performance liquid chromatography identifies the major haemoglobinopathies early. [11]
Management — Resuscitation
A child who arrives in, or progresses to, cardiovascular compromise from acute haemolysis needs resuscitation alongside investigation. Follow the airway, breathing, circulation framework, establish reliable intravenous access, and take the diagnostic samples before any blood is given. A bedside glucose and blood gas help to exclude hypoglycaemia and acidosis, and a group-and-save or crossmatch must be sent early because transfusion in autoimmune disease is technically difficult. [5]
The decision to transfuse rests on the haemodynamic state, not on a single haemoglobin number. A child who is tachycardic with poor perfusion and a rapidly falling haemoglobin needs urgent red cell support while the workup proceeds. In autoimmune haemolytic anaemia the autoantibody reacts with all donor cells, so the blood bank may be unable to find a fully compatible unit; the least incompatible unit is released and transfused under close observation. [1]
When cold agglutinin disease is suspected, every infused product and the giving set should be warmed, and the child kept in a warm environment, because cold exposure drives further agglutination and haemolysis. In any child receiving urgent transfusion for haemolysis, monitor for a haemolytic transfusion reaction and recheck the haemoglobin after the unit, because ongoing destruction may blunt the expected rise. [4]
Emergency red cell transfusion
Dose
10 to 20 mL per kg
Management — Definitive & Stepwise
The definitive management of haemolytic anaemia is cause-specific, and the diagnostic algorithm above exists to deliver the clinician to the right cause quickly. While the specific diagnosis is pending, supportive care holds the child safely: folic acid supplementation supports the brisk erythropoiesis of chronic haemolysis, hydration protects renal perfusion during intravascular episodes, and a clear plan for rechecking the haemoglobin prevents surprise. [7]
For warm autoimmune haemolytic anaemia, corticosteroids are first-line. They reduce both antibody production and the macrophage clearance of coated cells, and most children respond within the first week. The dose is weaned slowly over months to reduce relapse, and relapsing or steroid-dependent disease is treated with rituximab or splenectomy in selected cases. Intravenous immunoglobulin has a role in severe or refractory disease and in the acutely unstable child. [6]
Prednisolone (warm AIHA)
Dose
1 to 2 mg per kg per day
Cold agglutinin disease responds poorly to corticosteroids and splenectomy because the destruction is complement-mediated and largely hepatic. The priorities are cold avoidance and supportive care, with rituximab the mainstay for persistent disease. Transfusion, when needed, must use warmed products. For the inherited causes, definitive management belongs to the disease-specific topics, but splenectomy is curative for hereditary spherocytosis and is undertaken only after vaccination and with lifelong penicillin prophylaxis planning. [1]
The three diagnostic tiers of haemolysis
Two principles govern the transition from investigation to long-term care. First, every child with chronic haemolysis needs folic acid, surveillance for pigment gallstones and splenic size, and a plan for febrile illness in the asplenic or hyposplenic state. Second, every child with autoimmune haemolytic anaemia needs a search for an underlying driver, because identifying systemic lupus erythematosus or a primary immunodeficiency changes the long-term management as much as treating the haemolysis itself. [5]
Specific Subtypes & Scenarios
Warm autoimmune haemolytic anaemia is the prototype immune cause and the one most likely to appear in a written stem. The autoantibody is immunoglobulin G with a thermal optimum near 37 degrees Celsius, the direct antiglobulin test is positive for immunoglobulin G with or without complement, and the film shows spherocytes. Around half of paediatric cases follow a viral infection, and a substantial minority are secondary to autoimmune disease or immunodeficiency, so a focused secondary screen is part of the workup. [5]
Cold agglutinin disease is the second immune subtype and behaves differently. The immunoglobulin M antibody fixes complement in cool peripheral skin, the red cells agglutinate, and the direct antiglobulin test is positive for C3d only. Mycoplasma pneumoniae and Epstein-Barr virus are the classic triggers in children, and acrocyanosis with cold exposure is the bedside clue. Corticosteroids and splenectomy are unhelpful, and warm products plus cold avoidance are the practical priorities. [4]
Hereditary spherocytosis is the most common inherited membrane disorder and a frequent long-case subject. Autosomal dominant inheritance is found in about three-quarters of families, but new mutations and recessive forms exist, so a negative family history does not exclude it. The classic picture is chronic compensated haemolysis with splenomegaly, a raised mean corpuscular haemoglobin concentration, and spherocytes on the film. The eosin-5-maleimide binding test is the recommended screen, and gel electrophoresis of membrane proteins confirms the defective protein. [7]
Glucose-6-phosphate dehydrogenase deficiency is the most common enzyme defect and the classic episodic cause. Affected boys develop acute intravascular haemolysis after fava beans, sulphonamides, primaquine, rasburicase, naphthalene, or an infection. The film shows bite cells and Heinz bodies on supravital staining, and the enzyme assay must be repeated in remission. Trigger avoidance and parental education are the cornerstones of long-term care. [10]
[10]Sickle cell disease presents a distinctive film with sickled cells, target cells and, in the older child, Howell-Jolly bodies from autoinfarction of the spleen. Haemoglobin electrophoresis or high-performance liquid chromatography confirms the diagnosis and quantifies the haemoglobins. Newborn screening programmes identify most cases early, allowing penicillin prophylaxis and pneumococcal vaccination to prevent overwhelming sepsis in the hyposplenic infant. [11]
Complications & Pitfalls
The principal acute complication is haemolytic crisis with cardiovascular collapse, driven by fulminant immune haemolysis or an aplastic crisis. Pigment gallstones complicate any chronic haemolysis and may bring a teenager to surgery before the underlying cause is recognised. Folate depletion follows the sustained erythropoietic drive of chronic haemolysis, which is why folic acid supplementation is part of long-term care. [7]
A transfusion in autoimmune haemolytic anaemia carries its own risks. The autoantibody reacts with all donor cells, so a seemingly compatible unit may be cleared rapidly, and a delayed haemolytic reaction can deepen the anaemia days after the transfusion. The expected haemoglobin rise may not appear, and rechecking the count after the unit is essential. Underestimating this possibility is a common pitfall. [1]
The diagnostic pitfalls cluster around two errors. The first is ordering a glucose-6-phosphate dehydrogenase assay during the acute attack and accepting a normal result, falsely excluding the diagnosis. The second is interpreting a positive direct antiglobulin test on a recently transfused child as autoimmune disease, when the cause is an alloantibody against the transfused cells. Both errors are avoided by attention to timing and to the transfusion history. [3]
Missed dangerous mimics are the final pitfall. Thrombotic thrombocytopenic purpura presents with haemolysis and thrombocytopenia and needs urgent plasma exchange, so it must not be mistaken for autoimmune haemolytic anaemia with Evans syndrome. Paroxysmal nocturnal haemoglobinuria is rare in children but enters the differential of direct-antiglobulin-negative haemolysis with pancytopenia or unexplained thrombosis, and a low threshold for fluorescent aerolysin flow cytometry shortens the diagnostic delay. [2]
Prognosis & Disposition
The prognosis of haemolytic anaemia depends almost entirely on the underlying cause and the speed of its recognition. Post-infectious autoimmune haemolytic anaemia in a young child usually resolves over weeks and carries an excellent outlook. Idiopathic and secondary autoimmune forms run a more relapsing course and need long-term immunological follow-up, but the overall paediatric mortality is low with modern supportive and transfusion care. [6]
The inherited causes carry a chronic but manageable prognosis when recognised early. Hereditary spherocytosis runs a stable, compensated course in most children, with crises triggered by infection, and splenectomy is reserved for those with severe, transfusion-dependent disease. Glucose-6-phosphate dehydrogenase deficiency has an excellent prognosis once triggers are identified and avoided, and education of the child and family is the most important intervention. [7]
Disposition decisions turn on the haemodynamic stability and the rate of haemoglobin change. A stable child with a clear, confirmed diagnosis can be managed as an outpatient with close review. A child with active haemolysis, a falling haemoglobin, or an unstable observation set belongs in hospital for monitoring and possible transfusion. Every child with autoimmune haemolytic anaemia needs haematology referral, and those with a secondary cause need the relevant subspecialty engaged. [5]
Special Populations
Children from populations with a high prevalence of glucose-6-phosphate dehydrogenase deficiency and the haemoglobinopathies form a distinct group. In Mediterranean, Middle Eastern, Southeast Asian, Pacific Islander, West African and some Indigenous Australian communities, these conditions are common enough that an unexplained anaemia or jaundice should prompt early targeted testing rather than a broad infectious workup. Cultural sensitivity around traditional remedies and foods is important, because some traditional preparations are oxidant triggers. [10]
Migrant and refugee children may present with undiagnosed chronic haemolysis because they have never been screened. A full blood count, film and haemoglobin electrophoresis or high-performance liquid chromatography are reasonable screening tests in a newly arrived child with microcytosis, target cells or an unexplained raised bilirubin. Interpreting results across ethnicities requires knowing the local variant spectrum, because haemoglobin E, alpha-thalassaemia traits and sickle variants each carry their own diagnostic pitfalls. [11]
Children with immune dysregulation form a second special group. Autoimmune haemolytic anaemia may be the presenting feature of common variable immunodeficiency, autoimmune lymphoproliferative syndrome, or systemic lupus erythematosus, and a child with recurrent or relapsing haemolysis deserves an immunological and autoimmune evaluation rather than repeated courses of steroids alone. Recognising the underlying driver changes the long-term plan. [6]
Children with chronic haemolysis and an enlarged spleen need clear guidance about splenic safety. The risk of overwhelming post-splenectomy infection applies to the surgical patient, but functional hyposplenism from sickle autoinfarction carries the same danger and requires the same pneumococcal, meningococcal and Haemophilus vaccination and penicillin prophylaxis planning. Families should be taught to recognise the features of severe infection and to present early. [11]
Evidence, Guidelines & Regional Differences
The diagnostic framework rests on internationally accepted laboratory standards. The International Council for Standardization in Haematology guidance on the non-immune hereditary red cell membrane disorders established eosin-5-maleimide binding flow cytometry as the preferred screen and moved practice away from the less specific osmotic fragility test. The British Committee for Standards in Haematology guideline on hereditary spherocytosis codified the clinical, family and laboratory criteria that most centres now use. [8]
The autoimmune haemolytic anaemia literature has consolidated the warm-versus-cold framework around the direct antiglobulin test pattern, the antibody class and the thermal optimum. Recent paediatric scoping reviews emphasise the importance of an underlying-cause search in every child, because secondary disease is commoner than historically supposed and changes management. Standardisation of the direct antiglobulin test, including monospecific reagents to separate immunoglobulin G from C3d, has reduced misclassification. [6]
[8]Regional differences reflect both population genetics and access. In high-prevalence regions, newborn screening for sickle cell disease and targeted glucose-6-phosphate dehydrogenase education are standard public-health measures that reduce early mortality. In Australia and New Zealand, universal newborn screening does not currently include sickle cell disease everywhere, so clinicians serving migrant communities must maintain a low threshold for haemoglobinopathy testing. Access to specialist flow cytometry and membrane-protein analysis is uneven, and referral pathways vary between metropolitan and remote services. [11]
Ongoing controversies include the optimal use of the direct antiglobulin test in the recently transfused child, the interpretation of weakly positive tests in ill children without haemolysis, and the role of extended molecular testing in undiagnosed hereditary haemolytic anaemia. Next-generation sequencing panels for the inherited haemolytic anaemias are increasingly available and are reshaping the workup of the direct-antiglobulin-negative child whose film and first-line assays are unrevealing. [9]
Exam Pearls
SLICE the haemolysis film
References
- [1]Scheckel CJ, Go RS Autoimmune Hemolytic Anemia: Diagnosis and Differential Diagnosis. Hematol Oncol Clin North Am, 2022.PMID 35282951
- [2]Siddon AJ, Tormey CA The chemical and laboratory investigation of hemolysis. Adv Clin Chem, 2019.PMID 30797470
- [3]Zantek ND, Koepsell SA, Tharp DR Jr, Cramer JP The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol, 2012.PMID 22566278
- [4]Bass GF, Tuscano ET, Tuscano JM Diagnosis and classification of autoimmune hemolytic anemia. Autoimmun Rev, 2014.PMID 24418298
- [5]Blackall D, Dolatshahi L Autoimmune Hemolytic Anemia in Children: Laboratory Investigation, Disease Associations, and Treatment Strategies. J Pediatr Hematol Oncol, 2022.PMID 35235549
- [6]Zhang C, Charland D, O'Hearn K, et al Childhood autoimmune hemolytic anemia: A scoping review. Eur J Haematol, 2024.PMID 38894537
- [7]Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ Guidelines for the diagnosis and management of hereditary spherocytosis--2011 update. Br J Haematol, 2012.PMID 22055020
- [8]King MJ, Garçon L, Hoyer JD, et al ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol, 2015.PMID 25790109
- [9]Wu Y, Liao L, Lin F The diagnostic protocol for hereditary spherocytosis-2021 update. J Clin Lab Anal, 2021.PMID 34689357
- [10]Luzzatto L, Nannelli C, Notaro R Glucose-6-Phosphate Dehydrogenase Deficiency. Hematol Oncol Clin North Am, 2016.PMID 27040960
- [11]Brandow AM, Liem RI Advances in the diagnosis and treatment of sickle cell disease. J Hematol Oncol, 2022.PMID 35241123