Paeds · haematology-oncology-and-transfusion
Lymphoma in children
Also known as Hodgkin lymphoma · Non-Hodgkin lymphoma · Burkitt lymphoma · Lymphoblastic lymphoma · Mediastinal mass syndrome · Superior vena cava syndrome
Fellowship guide to lymphoma in children. Covers the separation of Hodgkin from non-Hodgkin lymphoma, the four high-grade paediatric non-Hodgkin subtypes of Burkitt, lymphoblastic, diffuse large B-cell and anaplastic large cell, the Reed-Sternberg cell and Ann Arbor staging of Hodgkin lymphoma, the Murphy St Jude staging for the non-Hodgkin group, the pathophysiology of c-MYC and t(8;14) in Burkitt and the mediastinal mass of T-lymphoblastic disease, the urgent diagnostic pathway from excision biopsy with flow cytometry and cytogenetics, the anterior mediastinal mass as an anaesthetic catastrophe that forbids sedation before the airway is secured, the stabilisation with tumour lysis prophylaxis using hyperhydration and rasburicase, and the risk-adapted multi-agent chemotherapy that delivers survival above ninety percent.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A teenager arrives with a rubbery lump above the clavicle that has been growing for a month, and a younger child is rushed in breathless and blue in the face with a chest that is full of mass and empty of airway. Both have lymphoma, and both demand that the clinician move fast for very different reasons. Lymphoma is the malignant proliferation of lymphoid cells that begins in the lymph nodes or in extranodal lymphoid tissue, and it is the third commonest cancer of childhood after leukaemia and the brain tumours. The word itself carries less weight than the single distinction that splits it in two at the bedside, because Hodgkin and non-Hodgkin lymphoma behave so differently that the first question asked of any child with a suspicious node is which side of that line the disease falls on. [1][5]
Hodgkin lymphoma is defined by the presence of the Reed-Sternberg cell, a large binucleate B-cell derivative with the owl-eye nucleolus that sits within a background of reactive inflammatory cells. It spreads in an orderly, contiguous fashion from one lymph node group to the next, it tends to stay nodal, and it is one of the most curable cancers in all of medicine. Non-Hodgkin lymphoma, in the child, is a different creature entirely, because the paediatric form is almost always high-grade, extranodal, and fast. The childhood non-Hodgkin lymphomas double in size every twenty-four to forty-eight hours, they seed marrow and the central nervous system, and a delay of days can move a child from curable to catastrophic. [1][3]
The clinical gravity of the disease is concentrated in two emergencies that frame this whole topic, and a candidate who masters them masters the examination question. The first is the anterior mediastinal mass, most often the T-lymphoblastic lymphoma, which compresses the airway and the great vessels and turns the act of going to sleep for a biopsy into a gamble with death. The second is the tumour lysis syndrome, the metabolic collapse that follows the rapid destruction of a bulky lymphoma, which is prevented before the first dose of chemotherapy or not at all. Around these two emergencies sits a curable disease, because the modern risk-adapted chemotherapy of childhood lymphoma delivers survival that exceeds ninety percent across the group. [8][10]
Classification
The first classification the clinician makes is the single most important one, and it is the split between Hodgkin and non-Hodgkin lymphoma. The distinction rests on the morphology and the immunophenotype of the malignant cell, and it drives everything that follows, from the staging system to the chemotherapy backbone to the prognosis. Hodgkin lymphoma carries the Reed-Sternberg cell within a rich reactive background, spreads contiguously, and is staged with the Ann Arbor system. The non-Hodgkin lymphomas of childhood are high-grade and extranodal, grow in sheets without the reactive background, and are staged with the Murphy St Jude system. [1][5]
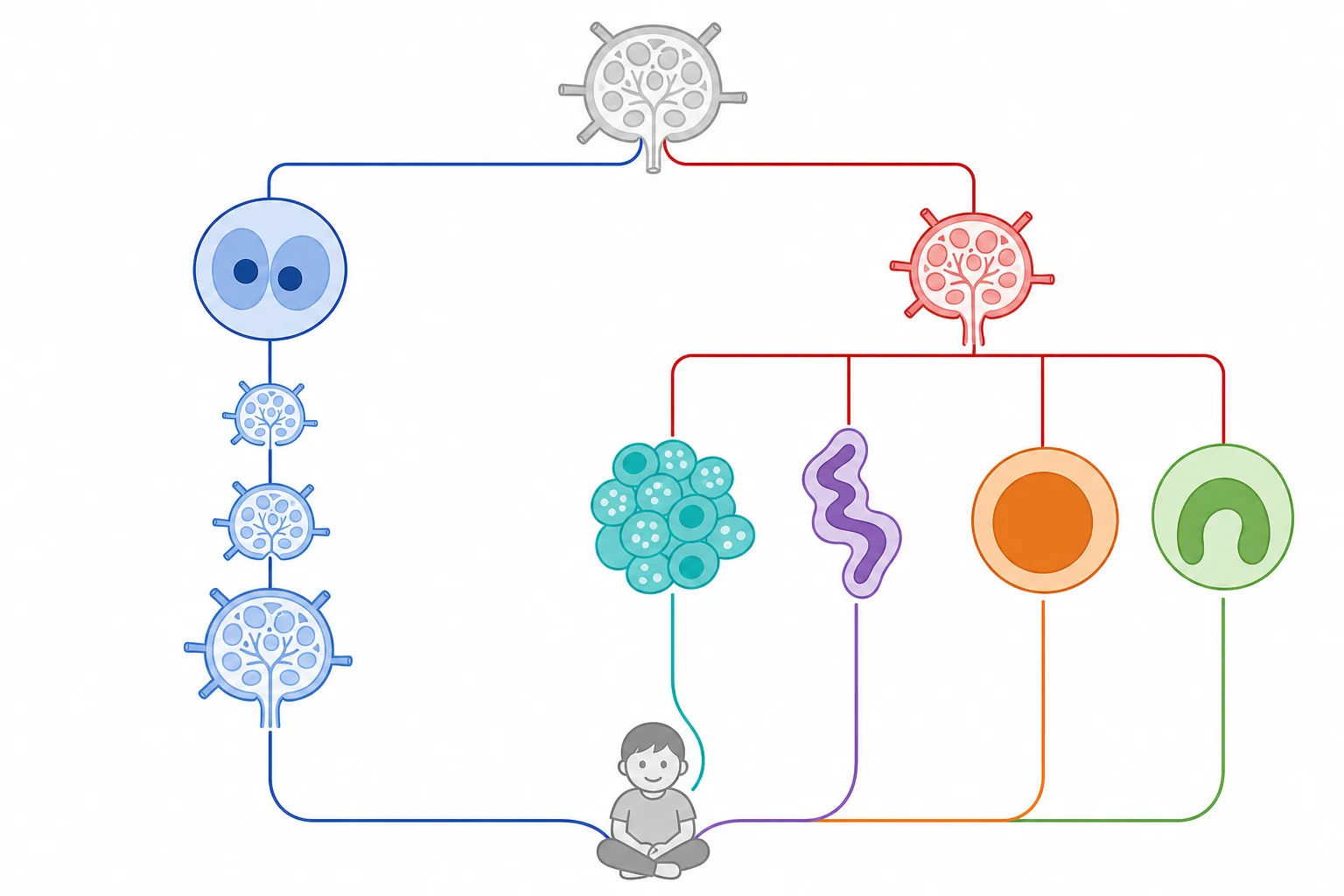
The paediatric non-Hodgkin lymphomas are themselves divided into four major subtypes, and each has a signature presentation, a molecular driver, and a treatment backbone that the boards reward. Burkitt lymphoma is the commonest, driven by the c-MYC translocation t(8;14), and it presents as a rapidly growing mass in the abdomen or the jaw. Lymphoblastic lymphoma is overwhelmingly of T-cell origin and presents as the anterior mediastinal mass with airway compromise. Diffuse large B-cell lymphoma favours older children and adolescents and tends to present in nodal or extranodal sites. Anaplastic large cell lymphoma is CD30-positive and ALK-driven, and it may present in skin, nodes or bone. [3][6][7]
Hodgkin lymphoma carries its own internal classification into the classical form, which holds the great majority, and the nodular lymphocyte-predominant form, which is rarer and behaves almost as an indolent B-cell lymphoma. The classical type is further divided into the nodular sclerosis subtype that dominates the adolescent presentation, the mixed cellularity, the lymphocyte-rich and the lymphocyte-depleted. The nodular sclerosis subtype is the one that sits in the anterior mediastinum and the supraclavicular fossa of the teenager, and it is the form most often encountered in the examination. [1][2]
Hodgkin lymphoma
Reed-Sternberg cell
- Orderly contiguous nodal spread
- Ann Arbor staging with A or B suffix
- Painless cervical or supraclavicular node
- Peak in adolescents; highly curable above ninety percent
Non-Hodgkin lymphoma
high-grade extranodal
- Burkitt with c-MYC t(8;14) and twenty-four-hour doubling
- Lymphoblastic T-cell with mediastinal mass
- Diffuse large B-cell and anaplastic large cell
- Murphy St Jude staging; seeds marrow and CNS
Nodular lymphocyte-predominant
indolent variant
- Rare variant of Hodgkin disease
- CD20-positive popcorn cell
- Localised nodal disease
- Treated more like an indolent B-cell lymphoma
Epidemiology & Risk Factors
Lymphoma accounts for roughly ten to fifteen percent of childhood cancers, which makes it the third commonest malignancy of the paediatric age group after leukaemia and the central nervous system tumours. The two halves of the disease separate sharply by age. Hodgkin lymphoma is rare under five years and rises through adolescence to become the commonest cancer of the teenager, while the non-Hodgkin lymphomas dominate the younger child and peak in the under-ten age band. Boys are affected more often than girls across both groups, and the male predominance is most striking in the younger child with Burkitt lymphoma. [1][5]
Within the non-Hodgkin group, the epidemiology is shaped by geography and by infection in a way that is unique in paediatric oncology and that the boards test directly. Burkitt lymphoma exists in an endemic form and a sporadic form. The endemic form is the equatorial African jaw mass of the young child, almost always associated with the Epstein-Barr virus and with falciparum malaria, while the sporadic form of the high-income country presents as the abdominal and ileocaecal mass and is linked to the Epstein-Barr virus in only a minority of cases. The link between the virus, the malaria-driven polyclonal B-cell proliferation, and the c-MYC translocation is one of the classic stories of oncogenesis. [3][4]
The risk factors for Hodgkin lymphoma cluster around the Epstein-Barr virus, the family history, and the immune state. A history of infectious mononucleosis modestly raises the risk, the virus is found within the Reed-Sternberg cells of a proportion of cases, and the immunodeficiency states, whether inherited, drug-induced or HIV-related, all carry a raised risk of Hodgkin and of the non-Hodgkin lymphomas. The iatrogenic risk after solid organ transplant, the post-transplant lymphoproliferative disorder, is an Epstein-Barr-driven B-cell proliferation that spans the range from a reactive polyclonal expansion to a frank monoclonal lymphoma, and it is a defining example of virus-driven lymphomagenesis. [2][11]
Pathophysiology
The pathophysiology of childhood lymphoma is the pathophysiology of a single cell that has escaped the normal controls of proliferation, maturation and death, and the two halves of the disease escape those controls by different routes. In the non-Hodgkin lymphomas the malignant cell is a lymphoid precursor arrested at a stage of maturation, and it proliferates without the brakes that would normally stop it. The result is a sheet of uniform malignant cells that effaces the normal architecture, grows without the reactive background, and doubles in volume at a terrifying pace. The Burkitt cell, arrested at the germinal-centre stage, carries the c-MYC translocation that drives the relentless proliferation, and its doubling time of twenty-four to forty-eight hours is the fastest of any human tumour. [3][4]
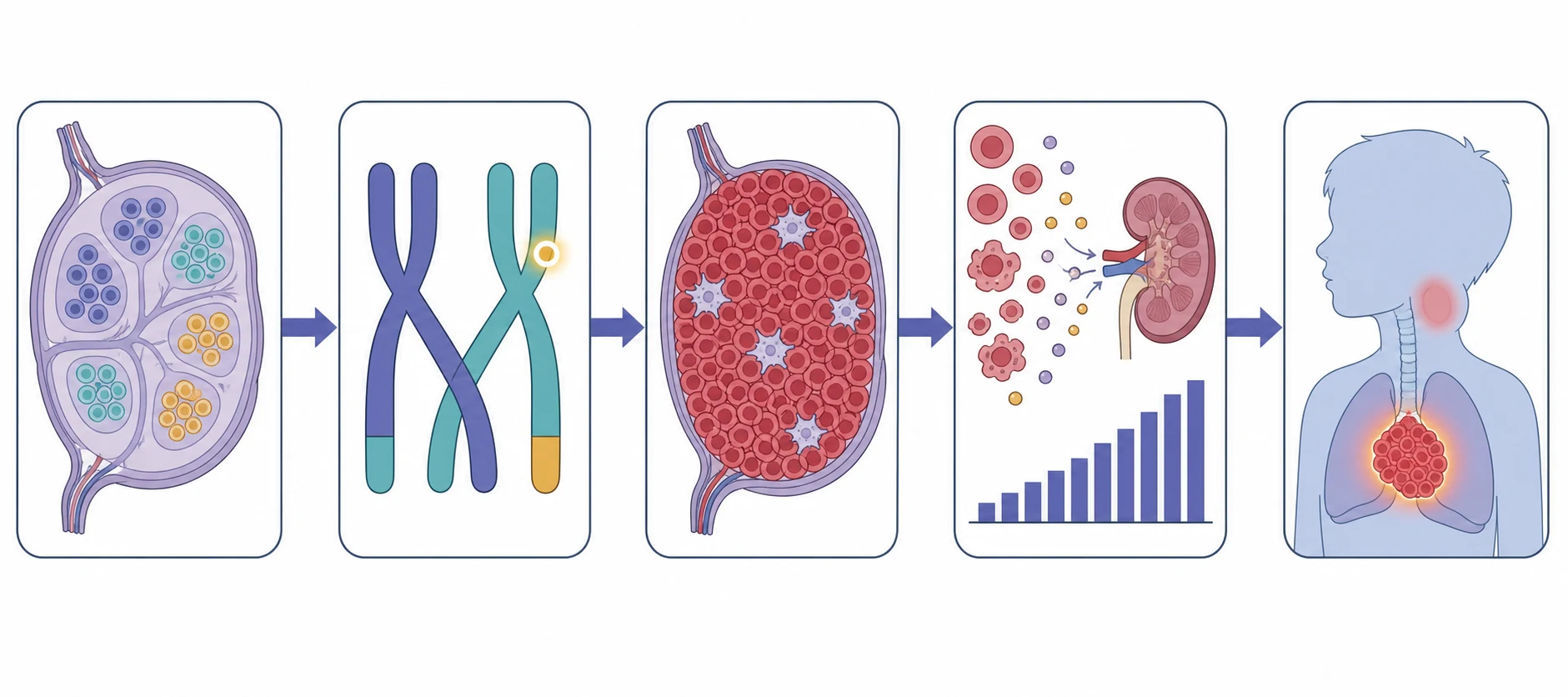
The molecular drivers are the targets of the modern classification, and they are the facts the examination rewards. The c-MYC translocation t(8;14) places the MYC oncogene under the immunoglobulin heavy-chain promoter and drives the Burkitt proliferation. The anaplastic large cell lymphoma carries the translocation that fuses the ALK gene, most often the NPM-ALK t(2;5), and the presence of ALK is a favourable prognostic marker in the child. The lymphoblastic lymphoma shares the biology of T-cell acute lymphoblastic leukaemia, and the distinction between lymphoma and leukaemia rests on the marrow blast burden rather than on the biology. [6][7]
The pathophysiology of the two emergencies is the reason the first hours matter. The mediastinal mass of T-lymphoblastic lymphoma grows in the anterior mediastinum and the thymus, and as it enlarges it compresses the trachea, the main bronchi, the superior vena cava, the pulmonary artery and the heart. The airway narrows, and because the mass and the great vessels surround the trachea, the narrowing is worse in the supine position and under anaesthesia, when the loss of thoracic muscle tone lets the trachea collapse against the mass. The tumour lysis syndrome is the metabolic consequence of the rapid destruction of the bulky tumour, which releases potassium, phosphate and urate faster than the kidneys can clear them, and the hyperphosphataemia drives a precipitate hypocalcaemia. [8][10]
Hodgkin lymphoma has a different and gentler pathophysiology. The malignant Reed-Sternberg cell is a crippled germinal-centre B cell that has lost its antibody machinery, and it survives by hijacking the surrounding inflammatory cells into feeding it with growth signals. The abundant reactive background of lymphocytes, eosinophils, plasma cells and fibrosis is not the tumour but the support tissue the malignant cell has recruited, and the fibrosis is what gives the nodular sclerosis subtype its hard, layered, capsule-like texture. This biology explains the orderly spread, the favourable prognosis, and the way the disease presents as a single slowly growing node rather than a diffuse aggressive mass. [1][2]
Clinical Presentation
The child with lymphoma walks in carrying the story of the disease in the site of the mass, and the history and the examination usually point to the subtype before any test is sent. The persistent, painless, enlarging lymph node is the commonest presentation of Hodgkin lymphoma and of the nodal non-Hodgkin subtypes, and the cervical and supraclavicular chains dominate. The node is typically rubbery rather than hard, it is not tethered or acutely inflamed, and it grows steadily over weeks. A node that has not settled after two weeks of antibiotics, or that is supraclavicular at its first appearance, carries a high enough risk of malignancy to demand referral for biopsy. [1][5]
The B symptoms are the constitutional features that change the staging and the prognosis of Hodgkin lymphoma, and they are named for the suffix they confer. The unexplained fever above thirty-eight degrees Celsius, the drenching night sweats that soak the bedclothes, and the weight loss of more than ten percent of the body weight over six months together make the B category, and a child with any one of them is upstaged to the B suffix. Pruritus, fatigue and the rare alcohol-induced pain in the involved nodes are the less famous but classic associations, and the examiner rewards the candidate who names them. [1]
The non-Hodgkin subtypes declare themselves by their favourite sites. Burkitt lymphoma presents as the rapidly growing abdominal mass with pain, distension and the signs of intussusception or bowel obstruction, most often from an ileocaecal tumour in the sporadic form, and as the jaw or facial mass in the endemic form. Lymphoblastic lymphoma presents as the anterior mediastinal mass with the cough, the dyspnoea, the stridor and the orthopnoea that mark the airway and great-vessel compression, and with the superior vena cava syndrome of the facial plethora, the distended neck and chest veins, and the raised jugular venous pressure. Anaplastic large cell lymphoma may present in the skin, with single or multiple lesions, or with systemic symptoms and a fever that mimics infection. [3][6][7]
Differential Diagnosis
The differential diagnosis of a persistent lymph node in a child is wide, because the vast majority of childhood lymphadenopathy is reactive, but the task is to find the few that are not. The reactive nodes follow the viral upper respiratory infections, the bacterial throat and skin infections, and the atypical infections such as Epstein-Barr virus, cytomegalovirus and toxoplasmosis. The nodes of infection are typically tender, mobile, associated with the signs of the source, and they settle over two to three weeks. The malignant node is painless, rubbery, relentlessly enlarging, and it sits in the supraclavicular fossa or grows despite the antibiotic course. [5]
The differential of the mediastinal mass runs through the four Ts of the anterior mediastinum, and it matters because the approach to each is the same airway-protective one regardless of the tissue of origin. The thymoma, the teratoma and other germ-cell tumours, the thyroid, and the lymphoma all sit in the anterior compartment, and in the child the lymphoma, above all the T-lymphoblastic type, is the commonest. The differential of the rapidly growing abdominal mass runs through the Wilms tumour, the neuroblastoma, the Burkitt lymphoma and the rhabdomyosarcoma, and the site, the age and the imaging narrow it before the biopsy settles it. [9]
Reactive node
self-limiting
- Tender, mobile, with a source infection
- Settles over two to three weeks
- Cervical or submandibular chain
- Normal full blood count and film
Lymphoma node
relentless
- Painless, rubbery, enlarging
- Supraclavicular site or persistent after antibiotics
- Accompanied by B symptoms or organomegaly
- Excision biopsy with flow cytometry
Other malignancy
solid tumour
- Neuroblastoma or rhabdomyosarcoma of node or mass
- Leukaemia with marrow disease
- Hard, fixed or rapidly growing mass
- Biopsy and staging distinguish them
The child with the skin lesions of anaplastic large cell lymphoma is often misdiagnosed at first, because the lesions mimic an infection or an inflammatory disorder and they may partially respond to antibiotics or steroids before revealing their nature. The febrile, irritable child with the systemic form of the disease may be investigated for weeks as a fever of unknown origin before the biopsy of a node or a skin lesion delivers the diagnosis. The candidate who keeps lymphoma in the differential of the persistent unexplained node, the persistent unexplained abdominal mass and the persistent unexplained mediastinal mass is the one who makes the diagnosis in time. [7]
Clinical & Bedside Assessment
The bedside assessment of the child with a suspected lymphoma begins with the airway, because the child with the mediastinal mass is the one who can die during the examination. The child who is stridulous, orthopnoeic or unable to lie flat is assessed upright, kept sitting forward, and moved to a setting where the airway can be secured. The child who is comfortable and lying flat is examined fully. The node is characterised by its site, size, consistency, mobility and tenderness, and the clinician maps every node group, because the distribution drives the staging. The supraclavicular node, the epitrochlear node and the axillary node are sought deliberately, because they are easily missed and they change the picture. [1][5]
The abdominal examination looks for the hepatosplenomegaly and the abdominal mass of the Burkitt or the bulky nodal disease, and it assesses for the ascites and the bowel obstruction. The chest is examined for the signs of the mediastinal mass, the pleural effusion and the pericardial effusion. The skin is searched for the lesions of the anaplastic large cell lymphoma, and the oropharynx is examined for the Waldeyer ring involvement that may signal the head and neck lymphoma. The nervous system is examined for the cord compression that occasionally accompanies the paraspinal or the Burkitt lymphoma, and the bony tenderness is sought for the marrow disease. [3][6]
The severity and the stability are judged at the bedside and they govern the pace of the workup. The child with the airway compromise, the superior vena cava syndrome, or the rapidly compromising abdominal mass is an emergency who is moved to the intensive care and the operating theatre rather than the outpatient clinic. The child with the stable node and no B symptoms is referred urgently but electively for the imaging and the biopsy. The candidate who can look at the child, decide whether the airway is safe, and then describe the focused systematic examination that maps the disease is demonstrating the reasoning the boards reward. [8]
Dexamethasone for the critical mediastinal mass
Dose
Approximately 6 to 10 mg per square metre per day, in divided doses, begun only when the airway is critical and the team accepts that the biopsy will be confounded
Investigations
The investigation of the child with a suspected lymphoma is built around the tissue biopsy, because no lymphoma is diagnosed or treated without the histology, the immunophenotype and the cytogenetics. The excision biopsy of the whole node is the gold standard, because it preserves the architecture that the pathologist needs, and it provides enough tissue for the flow cytometry, the immunohistochemistry, the cytogenetics and the molecular studies. A core biopsy is accepted when the node is inaccessible or the child is too unstable for an excision, and a fine-needle aspirate alone is insufficient because it loses the architecture. The biopsy is sent fresh and in the right medium, and the pathologist is warned in advance. [5][6]
The mediastinal mass forces a modified approach, and the principle is that the airway governs the route. The child with the compromised airway is biopsied awake, under local anaesthesia, with the mass approached through the least dangerous route, because the alternative of a general anaesthetic risks the fatal airway collapse. A mediastinal node is approached by the cervical or supraclavicular route under local anaesthetic where possible, or by an image-guided core biopsy in the upright position. The general anaesthetic is reserved for the child whose airway has been assessed as safe by the anaesthetic and the ENT and the cardiothoracic teams together, with the rigid bronchoscope and the ECMO on standby. [8][9]
The staging imaging follows the biopsy and defines the extent that drives the risk group. The contrast-enhanced computed tomography of the chest, abdomen and pelvis maps the nodal and extranodal disease and measures the bulk. The whole-body imaging, whether the fluorodeoxyglucose positron emission tomography for Hodgkin and the aggressive non-Hodgkin lymphomas or the magnetic resonance imaging in centres that avoid the radiation, completes the staging and provides the metabolic information that guides the response assessment. The bone marrow aspirate and trephine biopsy is performed for the non-Hodgkin lymphomas and for the advanced Hodgkin, and the cerebrospinal fluid is examined for the lymphoblastic and Burkitt subtypes that seed the central nervous system. [1][3]
The laboratory panel supports the diagnosis and the safe delivery of the chemotherapy. The full blood count and film look for the marrow involvement and the circulating malignant cells, the lactate dehydrogenase measures the tumour burden and the prognosis, and the urate, the electrolytes, the calcium, the phosphate and the renal function prepare the child for the tumour lysis prophylaxis. The hepatitis and the HIV and the Epstein-Barr virus serology are sent, the echocardiogram and the pulmonary function tests are baselined before the cardiotoxic and the pulmonary-toxic drugs, and the fertility preservation is discussed with the adolescent before the gonadotoxic chemotherapy. [5][10]
Management — Resuscitation
The resuscitation of the child with a newly diagnosed lymphoma is governed by the two emergencies, and the first principle is that the dangerous elements are treated before the diagnosis is fully confirmed. The child with the airway compromise from a mediastinal mass is kept upright, given no sedation, and moved to the setting where the airway can be secured, and the steroids are reserved for the life-threatening obstruction because they confound the biopsy. The child with the tumour lysis syndrome, or the high risk of it, is begun on the prophylaxis before the first dose of chemotherapy, because the syndrome is prevented or it is managed in crisis. [8][10]
The tumour lysis prophylaxis rests on three legs, and the candidate who reproduces them at the viva passes. The first is the hyperhydration with an isotonic fluid without potassium, run early to maintain a high urine output and to flush the urate, the phosphate and the potassium through the kidneys. The second is the urate lowering, with rasburicase for the high-risk child and allopurinol for the standard-risk, and the rasburicase is given only after the glucose-6-phosphate dehydrogenase screen is clear because it causes haemolysis and methaemoglobinaemia in the deficiency. The third is the close biochemical monitoring, with the potassium, the phosphate, the calcium, the creatinine and the urate measured every four to six hours in the high-risk child, and the renal function watched for the acute kidney injury that the phosphate and the urate crystals cause. [10]
The established tumour lysis syndrome is a metabolic emergency that is treated in the intensive care with the continuous cardiac monitoring, the aggressive fluid and the renal replacement therapy for the refractory hyperkalaemia and the acute kidney injury. The hyperkalaemia is the immediate threat to the rhythm, and it is treated with the cardiac membrane stabilisation, the intracellular shift and the removal, while the hypocalcaemia is corrected only if it is symptomatic, because the calcium supplementation in the face of the hyperphosphataemia precipitates the tissue deposition. The febrile neutropenia, when it accompanies the treated child, is managed with the empiric antipseudomonal beta-lactam within the hour after the blood cultures. [10]
Tumour lysis prophylaxis for the high-risk lymphoma
Recognise the high-risk child: bulky Burkitt, high lactate dehydrogenase, large tumour burden, renal impairment or established biochemical tumour lysis
Risk stratify before the first chemotherapy dose
Begin hyperhydration with an isotonic fluid without potassium to maintain a high urine output
Run early; do not add potassium to the fluid
Give rasburicase for the high-risk child after the glucose-6-phosphate dehydrogenase screen is clear
Allopurinol is the alternative for the standard-risk or the deficient child
Monitor potassium, phosphate, calcium, creatinine and urate every four to six hours
Watch for the acute kidney injury from urate and phosphate crystals
Treat the established syndrome in intensive care with cardiac monitoring and renal replacement therapy
Correct symptomatic hypocalcaemia only; avoid routine calcium with hyperphosphataemia
Management — Definitive & Stepwise
The definitive treatment of childhood lymphoma is risk-adapted multi-agent chemotherapy, and it is delivered in a specialist paediatric oncology centre over months to years, with the backbone shaped by the histology and the stage. Hodgkin lymphoma is treated with the risk-adapted combination chemotherapy that has been progressively de-escalated to spare the child the late effects of the radiotherapy, with the modern protocols such as the EuroNet and the COG regimens reserving the involved-site radiotherapy for the slow responders and the bulky disease. The contemporary survival of childhood Hodgkin lymphoma is among the best in oncology, above ninety-five percent in the early stage and above ninety percent overall. [1][2]
The non-Hodgkin lymphomas are treated with the histology-specific chemotherapy that matches the biology, and the backbone differs by subtype. The Burkitt lymphoma responds to the short, intensive, fractionated regimens that exploit the rapid cycling of the tumour, such as the LMB and the BFM groups, with the central nervous system-directed therapy and the complete removal of the abdominal mass where feasible, and the survival is above ninety percent in the standard-risk. The lymphoblastic lymphoma is treated with the acute lymphoblastic leukaemia-like regimen over two years, because the biology is shared, and the anaplastic large cell lymphoma is treated with the ALCL-specific multi-agent chemotherapy that delivers a survival above seventy-five percent. [3][5][6]
The treatment journey of childhood Hodgkin lymphoma
The surgery has a defined and a limited role in the childhood lymphomas, because the disease is systemic and the chemotherapy is the curative modality, but it matters in two settings. The complete resection of the localised Burkitt abdominal mass may improve the outcome and reduce the chemotherapy intensity, and it is performed when the mass is safely resectable at the presentation or after the initial chemotherapy. The biopsy, whether the excision or the core, is the surgical act that delivers the diagnosis, and it is performed with the airway-protective approach in the mediastinal mass. The radiation has a shrinking role in the paediatric lymphomas, reserved for the selected Hodgkin disease and for the refractory or the relapsed disease, because the late effects in the growing child are substantial. [2][9]
Specific Subtypes & Scenarios
The Burkitt lymphoma is the prototype of the fast-growing paediatric non-Hodgkin lymphoma, and it is the subtype that tests the clinician on the tumour lysis and the rapid response. The endemic form presents as the jaw or the facial mass of the equatorial African child, the sporadic form as the ileocaecal abdominal mass of the high-income country, and the immunodeficiency-associated form in the transplant and the HIV patient. The c-MYC translocation t(8;14) drives the proliferation, the starry-sky histology reflects the high turnover with the scattered macrophages, and the treatment is the short intensive chemotherapy that exploits the rapid cycling. The endemic form in the resource-limited setting is treated with the less intensive regimens that balance the efficacy against the toxicity and the supportive care. [3][4]
The lymphoblastic lymphoma is the subtype that tests the clinician on the mediastinal mass, and it is overwhelmingly of the T-cell origin. It shares the biology and the treatment with the T-cell acute lymphoblastic leukaemia, and the distinction rests on the marrow blast burden, with the lymphoma defined by a marrow involvement under twenty-five percent. The anterior mediastinal mass is the signature presentation, and the airway and the great-vessel compression is the emergency that governs the diagnosis and the early care. The treatment is the acute lymphoblastic leukaemia-like regimen over two years, with the central nervous system-directed therapy, and the survival is comparable to the leukaemia. [6]
The anaplastic large cell lymphoma is the subtype with the distinctive cell and the molecular marker, and it is CD30-positive and driven by the ALK translocation. The horseshoe or the kidney-shaped nucleus is the morphological signature, and the ALK positivity, most often the NPM-ALK t(2;5), is a favourable prognostic marker in the child. It may present in the skin, in the nodes, in the bone or as the systemic disease with the fever and the weight loss, and the cutaneous form in the very young child may regress spontaneously in the rare ALK-positive primary cutaneous variant. The treatment is the ALCL-specific multi-agent chemotherapy, and the survival is above seventy-five percent with the contemporary regimens. [7]
The diffuse large B-cell lymphoma is the subtype of the older child and the adolescent, and it presents in the nodal or the extranodal sites, including the head and neck, the abdomen and the bone. It shares the germinal-centre origin with the Burkitt, and the molecular distinction between the two can be difficult, with the grey-zone lymphoma sitting between them, but the treatment is the B-cell lymphoma regimen and the survival is high. The nodular lymphocyte-predominant Hodgkin lymphoma is the rare indolent variant that behaves more like a B-cell lymphoma, with the CD20-positive popcorn cell, and it is treated with the surgery or the local therapy for the localised disease and the anti-CD20 immunotherapy for the relapsed. [2][5]
Complications & Pitfalls
The complications of childhood lymphoma divide into the disease-related and the treatment-related, and the candidate who holds both together is the one who manages the survivor as well as the acute patient. The disease-related complications are the two emergencies, the mediastinal mass and the tumour lysis syndrome, and the acute complications of the bulky disease such as the cord compression, the bowel obstruction and the airway compromise. The treatment-related complications are the immediate toxicities of the chemotherapy, the mucositis, the febrile neutropenia, the anthracycline cardiotoxicity and the peripheral neuropathy, and the late effects that dominate the survivorship. [2][8]
The late effects are the burden of the cure, and they are the reason the treatment of the childhood lymphoma has been progressively de-escalated. The radiotherapy carries the risk of the second malignancy, the breast cancer in the irradiated adolescent girl, the thyroid dysfunction and the cardiac disease, and the modern Hodgkin protocols aim to omit the radiotherapy in the good responders. The anthracycline carries the dose-dependent cardiotoxicity that may declare decades later, and the gonadotoxic chemotherapy carries the infertility risk that demands the fertility preservation counselling in the adolescent. The endocrine late effects, the growth and the thyroid, and the psychosocial late effects complete the survivorship agenda. [1][2]
The classic diagnostic pitfalls are the ones the examiner probes, because they are the points where the diagnosis is missed or the harm is done. The persistent painless node treated as reactive, the rapidly growing abdominal mass treated as an intussusception, the skin lesions of the anaplastic large cell lymphoma treated as an infection, and the mediastinal mass child sedated for a biopsy are the errors that change the outcome. The mediastinal mass and the anaesthetic is the pitfall above all, because the fatal airway collapse on induction is the event that the airway-protective protocol exists to prevent, and the candidate who can recite the principle and the steps is the one who is safe. [8][9]
The four high-grade paediatric non-Hodgkin subtypes
Prognosis & Disposition
The prognosis of childhood lymphoma is one of the success stories of the paediatric oncology, and the survival figures are the facts the candidate carries into the counselling. Hodgkin lymphoma has a five-year survival above ninety-five percent in the early stage and above ninety percent overall, making it one of the most curable cancers in medicine. The non-Hodgkin lymphomas have a survival above ninety percent in the standard-risk Burkitt and the diffuse large B-cell, comparable to the leukaemia in the lymphoblastic, and above seventy-five percent in the anaplastic large cell. The prognostic factors are the stage, the bulk, the lactate dehydrogenase, the marrow and the central nervous system involvement, and the early response to the chemotherapy. [1][3]
The disposition of the child is the specialist paediatric oncology centre, because the diagnosis, the staging and the treatment demand the multidisciplinary team and the protocol-based care that the specialist centre provides. The child with the mediastinal mass is managed with the anaesthesia and the intensive care and the cardiothoracic teams in the same centre, and the child with the tumour lysis syndrome is managed in the oncology or the intensive care unit with the renal team. The survivor returns to the primary care and the long-term follow-up clinic, where the late effects are watched and the transition to the adult care is planned. [5][9]
The relapsed lymphoma is treated with the salvage chemotherapy and the autologous or the allogeneic haematopoietic stem cell transplant, depending on the histology and the response, and the relapse carries a worse but still meaningful prognosis in the contemporary era. The candidate who can quote the survival, name the prognostic factors, describe the disposition, and plan the survivorship is demonstrating the breadth the boards reward, and the one who can hold the cure and the late effects together is the one who serves the child across the whole journey. [2][7]
Special Populations
The immunocompromised child carries a raised risk of lymphoma that the clinician must hold in mind, because the Epstein-Barr-driven B-cell proliferation spans the range from the reactive to the malignant. The post-transplant lymphoproliferative disorder follows the solid organ or the stem cell transplant, and it is driven by the Epstein-Barr virus under the immunosuppression, and the management rests on the reduction of the immunosuppression, the anti-CD20 immunotherapy, and the cytotoxic chemotherapy for the frank lymphoma. The HIV-positive child carries a raised risk of the Hodgkin and the non-Hodgkin lymphoma, and the inherited immune disorders carry their own lymphoma risk. [11]
The endemic Burkitt lymphoma of the equatorial African child is the disease of the resource-limited setting, and the management differs from the high-income country because the supportive care, the intensive chemotherapy and the tumour lysis management are harder to deliver. The malaria and the Epstein-Barr virus together drive the polyclonal B-cell proliferation that culminates in the c-MYC translocation, and the jaw mass of the young child is the classic presentation. The less intensive regimens, the single-agent cyclophosphamide in the most resource-limited setting, and the improved supportive care have improved the survival in the endemic regions, and the gap with the high-income outcome is narrowing. [3][4]
The adolescent with the Hodgkin lymphoma is the patient in whom the survivorship and the late effects matter most, because the cure is near-certain and the lifespan ahead is long. The fertility preservation, the counselling of the gonadotoxic and the cardiotoxic chemotherapy, the radiotherapy de-escalation to spare the breast and the thyroid, and the transition to the adult care are the elements that the adolescent-centred service delivers. The socioeconomic disadvantage, the remoteness, and the migrant or the refugee status affect the access to the diagnosis and the treatment, and the indigenous and the remote populations carry a later presentation and a worse outcome in some regions. [1][2]
Evidence, Guidelines & Regional Differences
The landmark evidence for the childhood lymphomas is the body of the cooperative group trials that have progressively refined the risk-adapted chemotherapy, and the candidate who knows the shape of the evidence is the one who can defend the treatment. The EuroNet-PHL trials in Hodgkin lymphoma have established the chemotherapy de-escalation with the response-adapted radiotherapy, reserving the involved-node radiotherapy for the slow responders and the bulky disease, and the COG trials have delivered the comparable regimens. The LMB and the BFM trials in Burkitt lymphoma have established the short intensive chemotherapy that delivers the high survival, and the ALCL trials have refined the anaplastic large cell lymphoma regimen. [1][3]
The regional differences are mainly in the approach to the radiotherapy and in the protocol selection, and they matter because the late effects of the radiotherapy are the burden of the cure. The European centres have driven the radiotherapy de-escalation more aggressively, with the PET-guided omission of the radiotherapy in the good responders, while the North American centres have used a different chemotherapy backbone with a similar aim. The choice of the Burkitt and the ALCL regimen differs by the cooperative group, but the outcomes converge. The ANZ and the UK practice follows the European protocols in the main, with the radiotherapy reserved for the selected cases. [1][2]
The European EuroNet-PHL trials and the modern COG trials share the aim of de-escalating the chemotherapy and the radiotherapy to spare the child the late effects, but they differ in the backbone regimen and the radiotherapy field. The European practice uses the OEPA or the OEPA-COPDAC and the response-adapted involved-node radiotherapy, while the North American practice uses the ABVE-PC and the involved-site radiotherapy for the slow responders. The contemporary survival is comparable across the protocols, and the trend is towards the radiotherapy omission in the complete metabolic responders.
[1][2]The controversies are the ones that the candidate can discuss without pretending to resolve them. The role of the radiotherapy in the good responder, the optimal management of the grey-zone lymphoma between the Burkitt and the diffuse large B-cell, the duration of the maintenance in the lymphoblastic lymphoma, and the role of the targeted and the immunotherapy agents in the relapsed disease are the open questions. The evidence is weakest in the rare subtypes and in the resource-limited setting, where the supportive care and the intensive regimens are harder to deliver. [5][7]
Exam Pearls
The high-yield facts for the examination are the ones that the examiner probes and that the candidate must carry without hesitation. The separation of Hodgkin from non-Hodgkin by the Reed-Sternberg cell and the contiguous spread, the four high-grade non-Hodgkin subtypes with their signatures, the Ann Arbor and the Murphy St Jude staging, and the two emergencies of the mediastinal mass and the tumour lysis syndrome are the core of the topic. The survival figures, the prognostic factors, and the late effects complete the picture. [1][3]
The mediastinal mass is the single most testable emergency in the topic, and the principle is simple and absolute. The child with the anterior mediastinal mass and the airway or the great-vessel compromise is never sedated or anaesthetised before the airway is secured, the biopsy is taken under the local anaesthesia where possible, the child is kept upright, and the steroids are reserved for the life-threatening obstruction. The fatal cannot-intubate cannot-ventilate situation on the induction is the event that the airway-protective protocol exists to prevent, and the candidate who can recite the principle and the steps is the one who is safe and who passes. [8][9]
References
- [1]Mauz-Korholz C, Metzger ML, Kelly KM Pediatric Hodgkin Lymphoma J Clin Oncol, 2015.PMID 26304892
- [2]Munir F, Hardit V, Sheikh IN Classical Hodgkin Lymphoma: From Past to Future-A Comprehensive Review of Pathophysiology and Therapeutic Advances Int J Mol Sci, 2023.PMID 37373245
- [3]Lopez C, Burkhardt B, Chan JKC Burkitt lymphoma Nat Rev Dis Primers, 2022.PMID 36522349
- [4]Molyneux EM, Rochford R, Griffin B Burkitt's lymphoma Lancet, 2012.PMID 22333947
- [5]Galardy PJ, Bedekovics T, Hermiston ML Targeting childhood, adolescent and young adult non-Hodgkin lymphoma: therapeutic horizons Br J Haematol, 2016.PMID 27019108
- [6]Temple WC, Mueller S, Hermiston ML Diagnosis and management of lymphoblastic lymphoma in children, adolescents and young adults Best Pract Res Clin Haematol, 2023.PMID 36907639
- [7]Lowe EJ, Woessmann W Anaplastic large cell lymphoma in children and adolescents Br J Haematol, 2025.PMID 40351161
- [8]Pearson JK, Tan GM Pediatric Anterior Mediastinal Mass: A Review Article Semin Cardiothorac Vasc Anesth, 2015.PMID 25814524
- [9]Garey CL, Laituri CA, Valusek PA Management of anterior mediastinal masses in children Eur J Pediatr Surg, 2011.PMID 21751123
- [10]Perissinotti AJ, Bishop MR, Bubalo J Expert consensus guidelines for the prophylaxis and management of tumor lysis syndrome in the United States: Results of a modified Delphi panel Cancer Treat Rev, 2023.PMID 37579533
- [11]Quintanilla-Martinez L, Swerdlow SH, Tousseyn T New concepts in EBV-associated B, T, and NK cell lymphoproliferative disorders Virchows Archiv, 2023.PMID 36216980