Paeds · haematology-oncology-and-transfusion
Neuroblastoma
Also known as Sympathetic nervous system tumour · Neuroblastic tumour · Stage 4S neuroblastoma · Opsoclonus-myoclonus syndrome · Dancing eyes syndrome · MYCN-amplified neuroblastoma
Fellowship guide to neuroblastoma in children. Covers the neural crest origin of the neuroblastic tumours, the International Neuroblastoma Risk Group staging system with the image-defined risk factors, the urinary catecholamine metabolites vanillylmandelic acid and homovanillic acid that confirm ninety percent of tumours, the MYCN amplification that is the single most powerful adverse prognostic marker, the opsoclonus-myoclonus-ataxia syndrome and its poor neurological outcome despite tumour cure, the International Neuroblastoma Pathology Classification of neuroblastoma, ganglioneuroblastoma and ganglioneuroma, the iodine-123 metaiodobenzylguanidine scan for disease extent, the risk-adapted treatment from observation of low-risk disease to the induction, surgery, myeloablative consolidation, radiotherapy and anti-GD2 immunotherapy of high-risk disease, and the spontaneous regression of stage MS disease in infants.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A toddler arrives with a firm mass in the belly that has grown over weeks, and the examination shows a lump that crosses the midline. An infant is brought in with eyes that dance and a body that jerks, and the family is frightened by a child they no longer recognise. Both have neuroblastoma, and both demand a clinician who can move from the bedside pattern to the risk-adapted plan without delay. Neuroblastoma is a tumour of the developing sympathetic nervous system that arises from the primordial neural crest cells, the sympathogonia, that migrate during embryogenesis to form the adrenal medulla and the sympathetic ganglia along the paraspinal chain. It is the commonest extracranial solid tumour of childhood, and it carries a unique biology that spans the range from the tumour that vanishes on its own to the tumour that defies the most intensive treatment in paediatric oncology. [1][8]
The disease earns its place in the examination by the sheer breadth of its behaviour and by the powerful prognostic markers that drive the risk stratification. The tumour sits along the sympathetic chain, most often in the adrenal medulla, and it secretes catecholamines into the urine, where the metabolites vanillylmandelic acid and homovanillic acid confirm over ninety percent of tumours. The MYCN amplification is the single most powerful adverse prognostic marker in the disease, and its presence converts a child into the high-risk group regardless of the stage. The opsoclonus-myoclonus-ataxia syndrome, the dancing eyes and dancing feet, is the paraneoplastic signature that links neuroblastoma to an autoimmune process and that carries a poor neurological outcome even when the tumour is cured. [1][6][7]
The clinical gravity of the disease is concentrated in the risk group, because the modern treatment of neuroblastoma is risk-adapted to a degree that few childhood cancers match. The low-risk and intermediate-risk groups carry a survival above ninety percent with surgery alone or with moderate chemotherapy, while the high-risk group, defined by the MYCN amplification, the metastatic stage in the older child, or both, carries a survival below fifty percent despite the full sequence of induction chemotherapy, surgical resection, myeloablative consolidation with autologous stem cell rescue, radiotherapy and anti-GD2 immunotherapy. The candidate who masters the risk stratification, the staging system and the treatment pathway holds the whole topic, and the one who can hold the biology, the markers and the paraneoplastic syndrome together is the one the boards reward. [1][10][12]
Classification
The classification of neuroblastoma rests on three axes that together define the risk and the treatment, and the examination rewards the candidate who holds all three. The first is the anatomical stage, defined by the International Neuroblastoma Risk Group staging system that replaced the older International Neuroblastoma Staging System and that uses the image-defined risk factors assessed on imaging before any surgery. The second is the histology, defined by the International Neuroblastoma Pathology Classification, which grades the tumour by the degree of differentiation and the mitosis-karyorrhexis index within the context of the age. The third is the biology, defined by the MYCN amplification, the ploidy and the segmental chromosome abnormalities, and it is the biology that carries the most prognostic weight. [2][12]
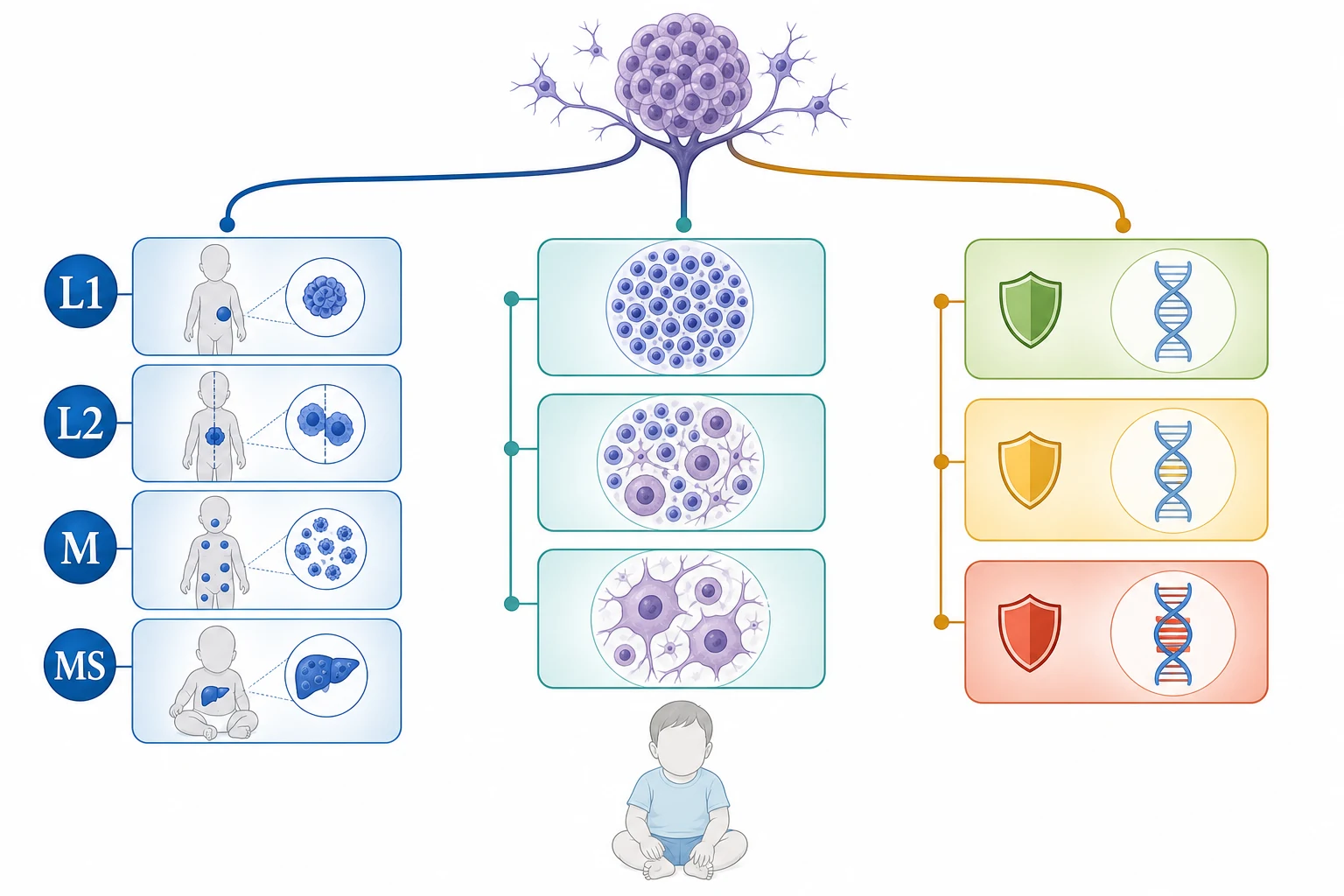
The International Neuroblastoma Risk Group staging system assigns the localised stages L1 and L2, the metastatic stage M, and the special metastatic stage MS. Stage L1 describes a localised tumour that does not involve any vital structure and that is confined to a single body compartment, while stage L2 describes a locoregional tumour that has one or more image-defined risk factors, such as the extension across the midline, the encasement of major vessels or nerves, the infiltration of the porta hepatis or the spinal canal, or the airway compression. Stage M describes the distant metastatic disease in any child, and stage MS describes the metastatic disease confined to the skin, the liver and the bone marrow in a child under eighteen months of age, which is the modern replacement for the old stage four S and which carries the potential for spontaneous regression. [2][3]
The histological spectrum runs from the immature neuroblastoma through the ganglioneuroblastoma to the mature ganglioneuroma, and it reflects the maturation of the neuroblast along the sympathetic line. The neuroblastoma is a Schwannian stroma-poor tumour composed of sheets of small round blue cells, and it is subdivided by the degree of neuroblastic differentiation and the mitosis-karyorrhexis index into the favourable and the unfavourable histology, with the classification set within the age of the child. The ganglioneuroblastoma is a mixed tumour with both the immature neuroblasts and the mature ganglion cells within a Schwannian stroma, and the ganglioneuroma is the fully mature, benign tumour composed entirely of ganglion cells and Schwannian stroma that requires no more than surgical excision. [1][8]
Stage L1
localized, no IDRF
- Tumour confined to one body compartment
- No image-defined risk factors
- Surgical resection is usually curative
- Five-year survival above ninety percent
Stage L2
locoregional with IDRF
- One or more image-defined risk factors
- Crosses midline or encases vessels
- Moderate chemotherapy plus surgery
- Intermediate risk group in most cases
Stage M
distant metastatic
- Metastatic disease beyond the primary site
- Bone, marrow, liver, orbit, skin
- Older children are high risk
- Intensive multimodal therapy required
Stage MS
infant under 18 months
- Skin, liver and marrow only
- Child under eighteen months of age
- Potential for spontaneous regression
- Low or intermediate risk in most cases
Epidemiology & Risk Factors
Neuroblastoma is the commonest extracranial solid tumour of childhood and the commonest tumour of infancy, accounting for roughly eight percent of all childhood cancers and for around fifteen percent of childhood cancer deaths. The median age at diagnosis is about eighteen months, and approximately forty percent of cases are diagnosed in the first year of life. The disease is slightly more common in boys than in girls, and it shows no strong racial or ethnic predilection in the large registries, though the access to the specialist care varies by geography and by socioeconomic status. The incidence is estimated at around one in seven thousand live births, which places neuroblastoma among the commonest solid tumours encountered by the general paediatrician. [1][8]
The age at diagnosis is one of the most powerful prognostic factors in the disease, and it is the fact the boards reward. Children diagnosed under the age of eighteen months carry a substantially better prognosis than those diagnosed over eighteen months, and the age cut-off is built into the staging system itself, because the stage MS designation that confers the potential for spontaneous regression is reserved for the infant under eighteen months. The younger infant may present with the localised or the metastatic disease that still carries a favourable biology, and the spontaneous regression of stage MS disease, observed and marvelled at for decades, is one of the most extraordinary phenomena in oncology. [11]
The familial and the genetic risk factors are present in a small minority, and they matter because they identify the child who warrants genetic counselling. Approximately one to two percent of neuroblastoma cases are familial, with the autosomal dominant inheritance of germline mutations in the paired-like homeobox 2B gene, the PHOX2B, and in the anaplastic lymphoma kinase gene, the ALK. The germline PHOX2B mutation is also associated with the congenital central hypoventilation syndrome and with the Haddad syndrome of neuroblastoma and Hirschsprung disease, and the family with a known germline mutation is offered the surveillance of the at-risk infant. The environmental risk factors are weak and inconsistent, and no strong environmental cause has been identified. [1][8]
Pathophysiology
The pathophysiology of neuroblastoma begins with the neural crest, because the tumour is a developmental disease of the migrating sympathogonia that form the adrenal medulla and the sympathetic chain. During the normal embryogenesis, the neural crest cells delaminate from the dorsal neural tube and migrate ventrally to populate the developing adrenal gland, the sympathetic ganglia and the prevertebral plexuses. The neuroblastoma arises when a population of these migrating sympathogonia fails to differentiate and to mature, and instead proliferates as a clonal tumour that retains the features of the developing neuroblast. The tumour secretes catecholamines, because the sympathogonia are the precursors of the catecholamine-producing chromaffin cells, and the secretion of the catecholamine metabolites into the urine is the biochemical hallmark that confirms the diagnosis. [1][8]
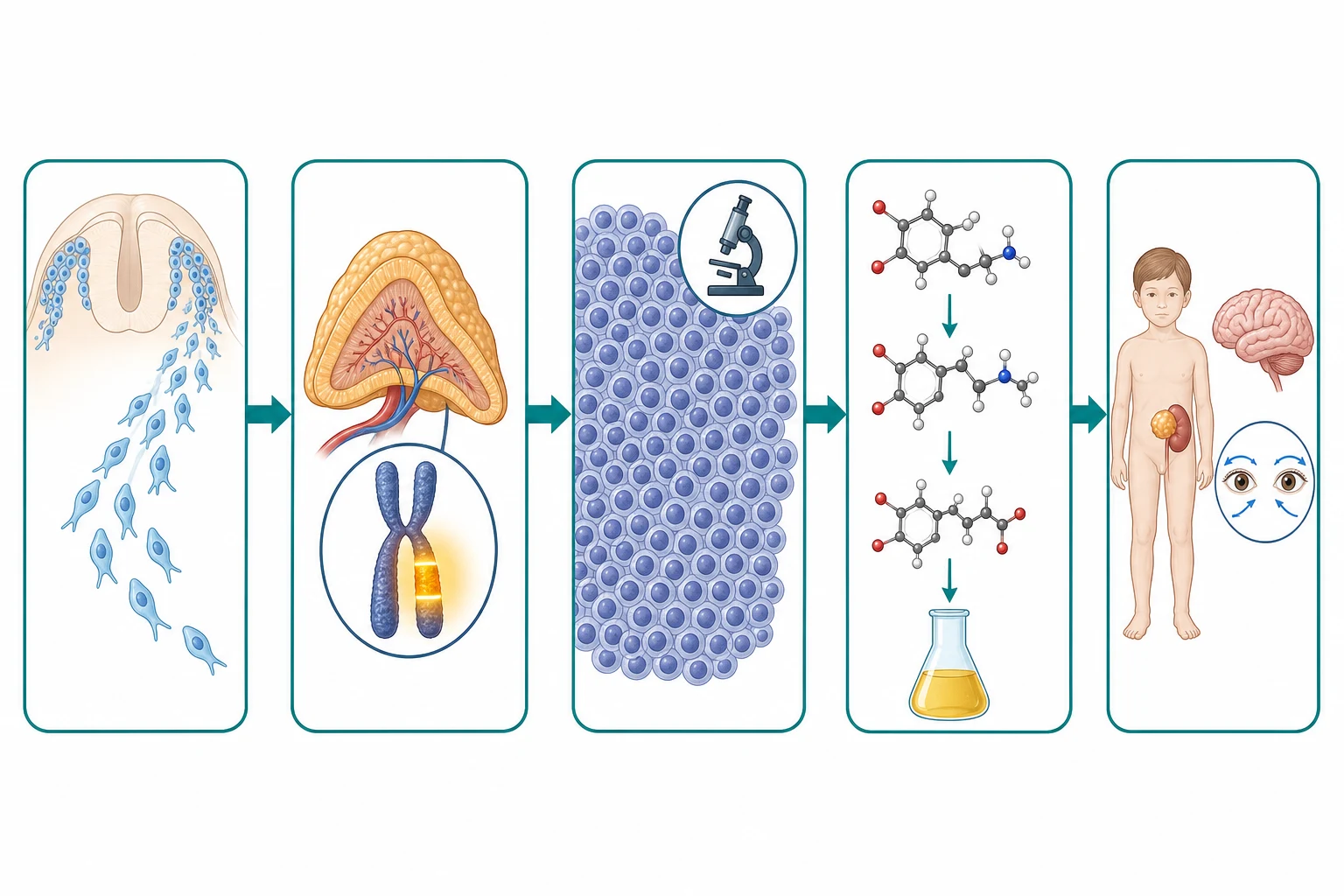
The MYCN amplification is the molecular driver that defines the high-risk tumour, and it is the single most powerful prognostic marker in the disease. The MYCN gene on chromosome two is amplified in roughly twenty percent of neuroblastomas, and the amplification, defined as more than ten copies of the gene per cell, drives the relentless proliferation of the neuroblast and confers the aggressive behaviour. The amplified MYCN tumour grows fast, metastasises early, and resists the conventional chemotherapy, and the presence of the amplification places the child in the high-risk group regardless of the stage or the age. The candidate who can link the MYCN to the prognosis and the risk group is demonstrating the molecular reasoning the boards reward. [5][12]
The catecholamine secretion is the biochemical basis of both the diagnosis and some of the clinical features, and it connects the tumour to the sympathetic lineage. The neuroblastoma cells synthesise dopamine, norepinephrine and epinephrine from the tyrosine precursor, and they metabolise these catecholamines into the urinary metabolites vanillylmandelic acid and homovanillic acid. The measurement of these metabolites in a random urine sample, corrected for the creatinine, confirms the diagnosis in over ninety percent of tumours, and the Eisenhofer group has refined the modern mass spectrometry methods that measure the full panel of catecholamine metabolites with the high sensitivity and the specificity. The catecholamine secretion may cause the hypertension, the flushing, the sweating and the irritability that occasionally accompany the disease. [7]
The opsoclonus-myoclonus-ataxia syndrome is the paraneoplastic neurological manifestation that links neuroblastoma to the autoimmune process, and it is one of the most distinctive syndromes in paediatric medicine. The opsoclonus is the chaotic, multidirectional, involuntary conjugate eye movements that differ from the rhythmic nystagmus, and the myoclonus is the brief shock-like jerks of the limbs and the trunk, and the ataxia is the wide-based unsteady gait. The syndrome occurs in roughly two to three percent of children with neuroblastoma, and it is driven by an autoimmune response against the neural tissue that is cross-reactive with the tumour antigens. The devastating fact is that the neurological outcome is often poor even when the tumour is cured, because the autoimmune cerebellar and brainstem injury persists and causes the cognitive, behavioural and motor sequelae that may last for life. [6][9]
Clinical Presentation
The child with neuroblastoma presents through several doors, and each presentation carries its own story and its own examination favourite. The commonest presentation is the abdominal mass, which is palpable in over half of the cases and which sits in the adrenal or the paraspinal region. The mass of the neuroblastoma characteristically crosses the midline, and this is the bedside sign that distinguishes it from the Wilms tumour of the kidney that stays confined to the flank on one side. The mass is firm, irregular and non-tender, and it may be so large that it distends the abdomen and causes the visible veins. The associated features may include the irritability, the weight loss, the fever and the failure to thrive, and the child may have the pallor and the bruising of the marrow involvement. [1][8]
The metastatic presentation is the one that brings the child through the emergency department with the acute and the alarming signs. The bone and the bone marrow metastases cause the bone pain, the limping and the refusal to walk in the young child, and the marrow involvement causes the pallor, the bruising and the infection of the pancytopenia. The orbital metastasis is the classic presentation that produces the periorbital ecchymosis, the raccoon eyes, and the proptosis, and it is the sign that the boards reward the candidate who recognises. The liver metastasis in the infant may cause the massive hepatomegaly of the stage MS disease, and the skin metastases produce the bluish subcutaneous nodules that have the blueberry-muffin appearance on the skin of the infant. [1]
The paraneoplastic presentations are the syndromes that link neuroblastoma to the distant physiology, and they are the examination favourites that reward the candidate who can connect the bedside sign to the tumour. The opsoclonus-myoclonus-ataxia syndrome brings the child in with the chaotic eye movements, the jerking limbs and the ataxic gait, and it is neuroblastoma until proven otherwise. The vasoactive intestinal peptide secretion by the tumour causes the intractable watery diarrhoea and the hypokalaemia, and it is the secretory diarrhoea that resolves when the tumour is resected. The catecholamine secretion causes the hypertension, the flushing and the sweating, though the florid hypertensive crisis is far less common in the neuroblastoma than in the pheochromocytoma. [6][9]
Dinutuximab beta for high-risk neuroblastoma
Dose
Approximately 10 milligrams per square metre per day for five days of each cycle, with the full course running over approximately twenty weeks after the myeloablative consolidation
Differential Diagnosis
The differential diagnosis of the abdominal mass in the child is the one that the boards probe most directly, and the task is to separate the neuroblastoma from the Wilms tumour and the other masses of the retroperitoneum. The Wilms tumour, the nephroblastoma, presents as a smooth, firm, unilateral flank mass in the young child, typically between one and five years of age, and it does not cross the midline. The neuroblastoma, by contrast, is an irregular, firm mass that often crosses the midline, and it may present in the infant or the toddler. The haematuria favours the Wilms tumour, and the hypertension, the diarrhoea and the opsoclonus favour the neuroblastoma, and the urinary catecholamines and the imaging settle the question before the biopsy. [1][8]
The differential of the orbital mass and the periorbital ecchymosis runs through the trauma, the infection and the malignancy, and the raccoon eyes of the neuroblastoma metastasis are the sign that must not be attributed to the trivial trauma alone. The periorbital cellulitis, the allergic periorbital oedema and the trauma are the common causes of the periorbital swelling, but the bilateral ecchymosis, the proptosis and the limitation of the eye movements in the absence of the fever or the clear trauma demand the imaging and the catecholamine measurement. The leukaemia and the rhabdomyosarcoma may also produce the orbital mass, and the biopsy and the marrow examination distinguish them. [1]
Neuroblastoma
neural crest
- Irregular mass crossing the midline
- Infant or toddler, median 18 months
- Urinary VMA and HVA elevated
- MIBG-avid, calcified on imaging
Wilms tumour
nephroblastoma
- Smooth unilateral flank mass
- One to five years of age
- Does not cross the midline
- Haematuria, displaced but intact kidney
Burkitt lymphoma
rapid growth
- Rapidly growing ileocaecal mass
- Twenty-four-hour doubling
- LDH very high
- Tumour lysis risk
The differential of the opsoclonus and the ataxia is the one that connects neuroblastoma to the wider neurological examination, and it is the point where the paediatric oncology and the paediatric neurology meet. The post-infectious acute cerebellar ataxia is the commonest cause of the acute ataxia in the preschool child, and it is a benign self-limiting condition with the normal conscious state and the negative workup. The opsoclonus, however, is not a feature of the acute cerebellar ataxia, and the child with the opsoclonus and the myoclonus is neuroblastoma until proven otherwise, because the opsoclonus-myoclonus-ataxia syndrome is the paraneoplastic signature of the disease. The candidate who recognises the opsoclonus and sends the urinary catecholamines and the imaging is the one who makes the diagnosis. [6][9]
Clinical & Bedside Assessment
The bedside assessment of the child with a suspected neuroblastoma begins with the general state and the airway, because the child with the large abdominal mass or the mediastinal extension may be in the respiratory distress, and the child with the cervical or the paraspinal mass may have the cord compression that is the neurological emergency. The vital signs include the blood pressure, because the catecholamine secretion may cause the hypertension that demands the management before the biopsy or the surgery. The child is weighed and measured, because the failure to thrive and the weight loss are the common features, and the growth parameters provide the baseline for the treatment. [1][8]
The abdominal examination is the centrepiece of the assessment, and the candidate who can describe the mass precisely is demonstrating the skill the boards reward. The mass is characterised by its site, its size, its consistency, its surface and its mobility across the midline. The neuroblastoma is typically firm, irregular, non-tender and fixed, and it may extend across the midline into the opposite side of the abdomen. The liver is examined for the hepatomegaly of the metastatic disease, and the spleen is sought. The examining hand also palpates the paraspinal region, the neck and the chest wall for the paraspinal, the cervical and the thoracic sympathetic chain tumours that may present as the neck mass, the Horner syndrome or the respiratory compression. [1]
The metastatic sites are examined systematically, because the neuroblastoma seeds to the bone, the marrow, the liver, the skin, the orbit and the lymph nodes. The skin is searched for the blueberry-muffin nodules of the stage MS disease, and the orbits are examined for the periorbital ecchymosis and the proptosis of the metastasis. The bones are examined for the tenderness, the swelling and the pathological fracture, and the child with the refusal to walk or the limping is examined for the bone metastasis. The lymph nodes are mapped, and the nervous system is examined for the opsoclonus, the myoclonus and the ataxia of the paraneoplastic syndrome, and for the weakness or the sensory level of the cord compression. [1][9]
Investigations
The investigation of the child with a suspected neuroblastoma is built around the confirmation of the catecholamine secretion, the imaging of the primary and the metastatic disease, and the biopsy with the histology and the molecular analysis. The urinary catecholamine metabolites are the first-line test, and the measurement of the vanillylmandelic acid and the homovanillic acid in a random urine sample, corrected for the creatinine, confirms the diagnosis in over ninety percent of tumours. The modern mass spectrometry methods measure the full panel of the catecholamine metabolites, and they have the high sensitivity and the specificity that make them the cornerstone of the biochemical diagnosis. The blood tests include the full blood count for the marrow involvement, the renal and hepatic function, and the lactate dehydrogenase as the marker of the tumour burden. [7][1]
The imaging begins with the ultrasound, which confirms the abdominal mass and its relationship to the kidney, and it proceeds to the cross-sectional imaging for the staging. The contrast-enhanced magnetic resonance imaging or the computed tomography of the primary site defines the anatomy, the image-defined risk factors and the resectability, and the magnetic resonance imaging is the preferred modality because it avoids the radiation and it better defines the vascular encasement and the intraspinal extension. The iodine-123 metaiodobenzylguanidine scan is the functional imaging that maps the disease extent, because the metaiodobenzylguanidine is taken up by the catecholamine-producing cells, and it is positive in over ninety percent of neuroblastomas. The iodine-123 MIBG scan is the single most important staging investigation, and it identifies the primary, the regional and the distant disease in one whole-body image. [3][4]
The bone marrow aspirate and trephine biopsy is performed bilaterally, because the neuroblastoma metastasises to the marrow in over half of the metastatic cases, and the marrow involvement is confirmed by the clumps of the small round blue cells in the aspirate or the trephine. The bone marrow is also the site of the staging and the response assessment, and the bilateral sampling reduces the false-negative rate. The biopsy of the primary tumour or the involved node is obtained by the percutaneous core biopsy or the surgical excision, and the tissue is sent for the histology, the immunohistochemistry and the molecular analysis. [1][8]
The molecular analysis of the tumour tissue is the key to the risk stratification, and it is the investigation that the boards reward the candidate who names. The MYCN amplification is assessed by the fluorescence in situ hybridisation or the multiplex ligation-dependent probe amplification, and it is defined as more than ten copies of the gene per cell. The DNA ploidy is measured by the flow cytometry, and the hyperdiploid tumour with the near-triploid DNA content carries a more favourable prognosis than the near-diploid tumour. The segmental chromosome abnormalities, particularly the one-p deletion and the eleven-q deletion, are assessed by the comparative genomic hybridisation or the single-nucleotide polymorphism array, and the segmental aberrations carry a worse prognosis than the whole-chromosome gains. [5][12]
Management — Resuscitation
The resuscitation of the child with a newly diagnosed neuroblastoma is governed by the acute complications and the principle that the dangerous elements are treated before the diagnosis is fully confirmed. The child with the spinal cord compression from the intraspinal extension of the paraspinal tumour is a neurological emergency, and the management begins with the dexamethasone and the urgent magnetic resonance imaging, with the surgical decompression or the chemotherapy as the definitive treatment. The child with the respiratory compromise from the large abdominal mass or the cervical or thoracic tumour is assessed and supported, and the airway is protected before any procedure. [1][8]
The child with the hypertension from the catecholamine secretion is managed with the antihypertensive agents before the biopsy or the surgery, and the anaesthesia team is involved early because the handling of the tumour may release the catecholamine surge. The child with the tumour lysis syndrome, though less common than in the Burkitt lymphoma, may develop the hyperkalaemia, the hyperphosphataemia and the acute kidney injury from the rapid destruction of the bulky tumour, and the prophylaxis with the hyperhydration and the rasburicase is begun before the chemotherapy in the high-risk child. The child with the pancytopenia from the marrow involvement receives the transfusion of the red cells and the platelets, and the febrile neutropenia is managed with the empiric antipseudomonal beta-lactam. [1]
The urgent management of spinal cord compression in neuroblastoma
Recognise the back pain, the weakness, the sensory level and the bladder or bowel dysfunction as the cord compression until proven otherwise
The paraspinal neuroblastoma with the intraspinal extension is the classic cause
Give the intravenous dexamethasone urgently to reduce the cord oedema
Approximately one milligram per kilogram as the loading dose
Obtain the urgent whole-spine magnetic resonance imaging
The MRI defines the level and the degree of the cord compression
Coordinate the neurosurgical and the oncological decompression
Surgical decompression or urgent chemotherapy depending on the case
Management — Definitive & Stepwise
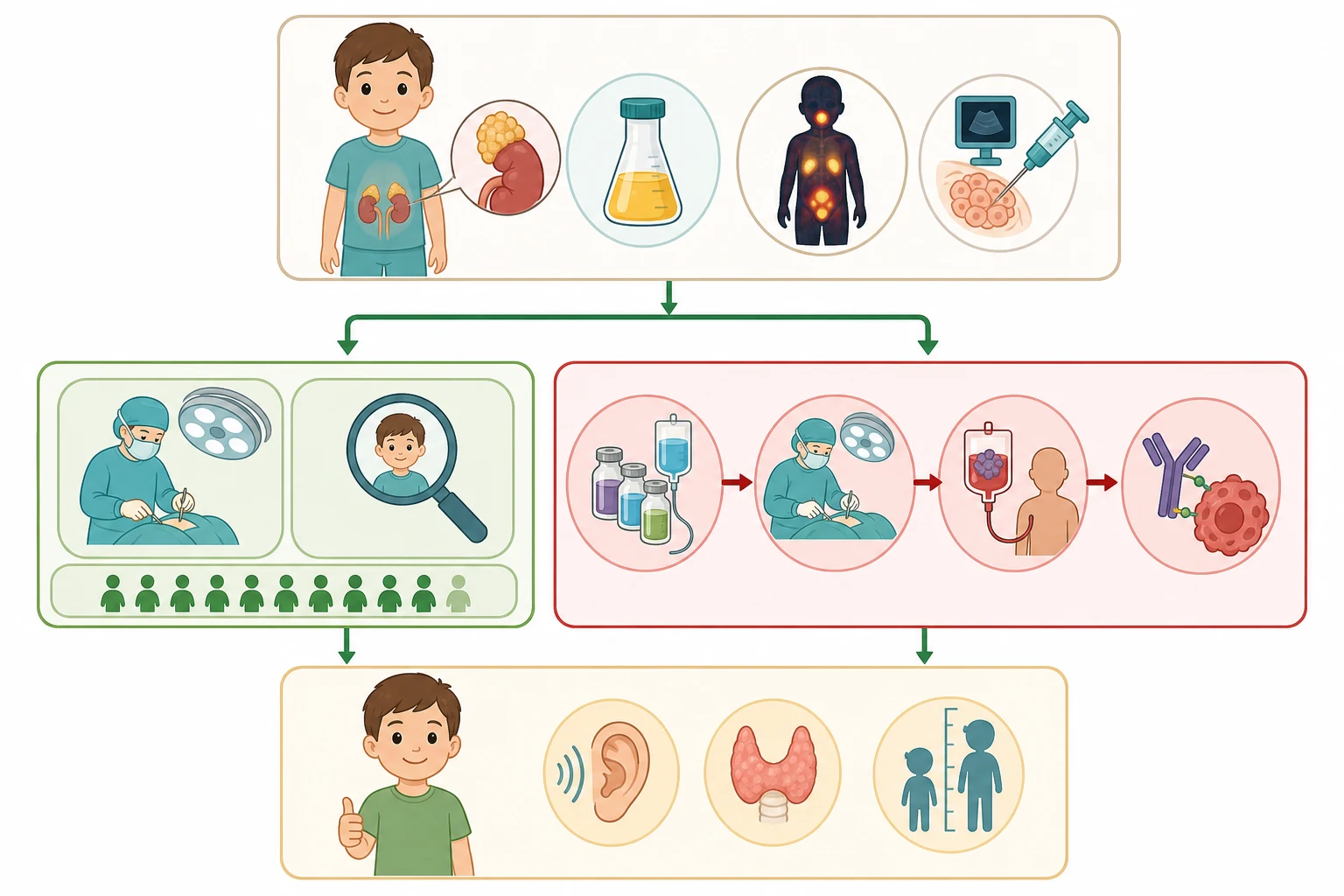
The definitive treatment of neuroblastoma is risk-adapted, and the risk group is assigned from the stage, the age, the histology, the MYCN status, the ploidy and the segmental aberrations through the International Neuroblastoma Risk Group classification. The low-risk group, which includes the localised L1 tumours without the MYCN amplification and the favourable-biology stage MS disease, is managed with the surgical resection or the observation alone, because the localised tumour without the image-defined risk factors is cured by the complete excision, and the stage MS disease in the infant may regress spontaneously. The five-year survival of the low-risk group is above ninety-five percent, and the chemotherapy is reserved for the symptomatic infant or the incompletely resected tumour. [1][12]
The intermediate-risk group, which includes the localised L2 tumours with the image-defined risk factors and the favourable biology, and the infants under eighteen months with the localised or the metastatic disease without the MYCN amplification, is managed with the moderate chemotherapy and the surgery. The chemotherapy uses the combination of the carboplatin, the etoposide, the cyclophosphamide and the vincristine, and the surgery is performed after the initial chemotherapy to reduce the tumour bulk and to facilitate the complete resection. The five-year survival of the intermediate-risk group is above ninety percent, and the treatment is designed to minimise the late effects while maintaining the high cure rate. [1][8]
The high-risk group, which includes the children over eighteen months with the metastatic disease, the children of any age with the MYCN amplification, and the children with the refractory or the relapsed disease, is treated with the most intensive regimen in paediatric oncology. The treatment runs through the induction chemotherapy to reduce the tumour, the surgical resection of the primary, the myeloablative consolidation with the autologous stem cell rescue, the external beam radiotherapy to the primary site, and the anti-GD2 immunotherapy with the dinutuximab beta in combination with the interleukin-2 and the isotretinoin. The dinutuximab beta is the monoclonal antibody that targets the GD2 disialoganglioside on the surface of the neuroblastoma cell, and its addition to the consolidation has improved the event-free survival, as demonstrated by the SIOPEN trial of Ladenstein and colleagues. The five-year survival of the high-risk group remains below fifty percent despite the full sequence, and the treatment carries the significant toxicity of the hearing loss, the cardiotoxicity, the growth impairment and the second malignancy. [10][1]
The treatment journey of high-risk neuroblastoma
Specific Subtypes & Scenarios
The opsoclonus-myoclonus-ataxia syndrome is the scenario that links neuroblastoma to the autoimmune neurology, and it is the one the boards probe most directly. The child with the opsoclonus, the myoclonus and the ataxia is neuroblastoma until proven otherwise, and the search for the occult tumour, with the urinary catecholamines, the iodine-123 metaiodobenzylguanidine scan and the magnetic resonance imaging of the chest, abdomen and pelvis, is mandatory and urgent. The neuroblastoma associated with the opsoclonus-myoclonus-ataxia syndrome is typically the low-stage, favourable-biology tumour, and the tumour itself carries an excellent prognosis with the surgical resection. The devastating fact is that the neurological outcome is often poor despite the tumour cure, because the autoimmune injury persists and causes the cognitive, behavioural and motor sequelae. The management of the neurological syndrome uses the immunomodulation with the corticosteroids, the intravenous immunoglobulin, the rituximab and the cyclophosphamide, and the early and the aggressive treatment may improve the outcome. [6][9]
The stage MS disease is the scenario that demonstrates the spontaneous regression, and it is the one that captures the fascination of the examination. The infant under eighteen months with the metastatic disease confined to the skin, the liver and the bone marrow carries the unique biology that may undergo the spontaneous maturation or the regression, and the histological counterpart is the maturing neuroblastoma with the favourable biology. The management of the asymptomatic stage MS infant is the observation with the supportive care, because the disease regresses without the treatment in the majority. The symptomatic infant with the respiratory compromise from the massive hepatomegaly or the pancytopenia from the marrow involvement receives the low-dose cyclophosphamide or the hepatic radiotherapy to reduce the tumour burden, and the five-year survival is above ninety percent. [11][1]
The four INRG stages and their meaning
The cervical and the thoracic neuroblastomas are the primary sites outside the abdomen, and they carry their own presentations and their own challenges. The cervical neuroblastoma may present as the neck mass with the Horner syndrome of the ptosis, the miosis and the anhidrosis, and it is distinguished from the other neck masses by the catecholamine secretion and the imaging. The thoracic neuroblastoma, arising from the sympathetic ganglia of the posterior mediastinum, may present as the incidental mass on the chest radiograph or with the respiratory compromise, and it may extend into the spinal canal through the neural foramen to cause the cord compression. The pelvic neuroblastoma, arising from the organ of Zuckerkandl or the pelvic sympathetic chain, may present with the bladder or the bowel dysfunction from the local compression. [1][8]
Complications & Pitfalls
The complications of neuroblastoma divide into the disease-related and the treatment-related, and the candidate who holds both together is the one who manages the survivor as well as the acute patient. The disease-related complications are the cord compression, the airway compromise, the hypertension, the pancytopenia and the opsoclonus-myoclonus-ataxia syndrome, and the acute complications of the bulky abdominal mass. The treatment-related complications are the immediate toxicities of the chemotherapy and the immunotherapy, the mucositis, the febrile neutropenia, the hearing loss, the cardiotoxicity and the capillary leak syndrome, and the late effects that dominate the survivorship. [1][10]
The late effects are the burden of the cure, and they are the reason the treatment has been progressively refined. The cisplatin causes the sensorineural hearing loss that may demand the hearing aid or the cochlear implant, and the baseline and the surveillance audiometry are mandatory in the treated child. The anthracycline causes the dose-dependent cardiotoxicity that may declare decades later, and the echocardiogram is the surveillance tool. The myeloablative therapy with the total body irradiation or the busulfan causes the growth impairment, the endocrine dysfunction and the infertility, and the growth hormone and the endocrine replacement are part of the survivorship. The second malignancy, particularly the myelodysplasia and the leukaemia after the alkylating agents and the topoisomerase inhibitors, is the rare but devastating late effect. [1][8]
The classic diagnostic pitfalls are the ones the examiner probes, because they are the points where the diagnosis is missed or the harm is done. The abdominal mass attributed to the constipation or the faecal loading, the periorbital ecchymosis attributed to the trivial trauma, the opsoclonus attributed to the seizure, and the limping attributed to the transient synovitis are the errors that delay the diagnosis. The failure to send the urinary catecholamines in the child with the suspected abdominal mass, the failure to recognise the opsoclonus as the neuroblastoma paraneoplastic syndrome, and the failure to image the child with the suspected cord compression are the mistakes that change the outcome. [1][9]
Prognosis & Disposition
The prognosis of neuroblastoma is the most risk-group-dependent of any childhood cancer, and the survival figures span the range from the near-universal cure of the low-risk localised disease to the forty-percent survival of the high-risk metastatic disease. The low-risk group has a five-year survival above ninety-five percent with the surgery or the observation alone, and the intermediate-risk group has a survival above ninety percent with the moderate chemotherapy. The high-risk group, defined by the MYCN amplification, the metastatic disease in the child over eighteen months, or both, has a five-year survival below fifty percent despite the full multimodal treatment, and it remains the greatest challenge in the paediatric oncology. [1][12]
The prognostic factors are the age, the stage, the MYCN status, the histology, the ploidy and the response to the treatment, and the International Neuroblastoma Risk Group classification integrates them all into the risk group. The age under eighteen months is favourable, and the age over eighteen months is adverse. The localised stage is favourable, and the metastatic stage in the older child is adverse. The MYCN amplification is the single most powerful adverse marker, and its presence overrides the stage and the age. The favourable histology with the low mitosis-karyorrhexis index is favourable, and the unfavourable histology is adverse. The hyperdiploid DNA content is favourable, and the near-diploid with the segmental aberrations is adverse. [5][12]
The disposition of the child is the specialist paediatric oncology centre, because the diagnosis, the staging, the risk stratification and the risk-adapted treatment demand the multidisciplinary team and the protocol-based care. The child with the high-risk disease is managed in the centre that delivers the myeloablative consolidation and the anti-GD2 immunotherapy, and the child with the opsoclonus-myoclonus-ataxia syndrome is managed jointly with the paediatric neurology. The survivor returns to the primary care and the long-term follow-up clinic, where the late effects are watched and the transition to the adult care is planned. [1][10]
Special Populations
The infant with the neuroblastoma is the patient in whom the biology and the prognosis are uniquely favourable, and the approach is shaped by the potential for the spontaneous regression. The infant under eighteen months with the localised or the stage MS disease carries the favourable biology, and the management is more conservative, with the observation, the low-dose chemotherapy or the surgery. The infant with the massive hepatomegaly of the stage MS disease may require the supportive care for the respiratory compromise, and the low-dose cyclophosphamide or the hepatic radiotherapy is reserved for the symptomatic infant. The neonate with the prenatally detected adrenal mass may undergo the observation with the serial imaging, and many of these neonatal adrenal masses are the benign neuroblastoma or the adrenal haemorrhage that resolves spontaneously. [11][1]
The child with the opsoclonus-myoclonus-ataxia syndrome is the patient in whom the neurological outcome is the primary concern, because the tumour itself carries the excellent prognosis but the autoimmune injury persists. The family is counselled honestly about the tumour prognosis and the neurological prognosis, and the early and the aggressive immunomodulation is begun. The child may require the long-term support for the cognitive, the behavioural and the motor sequelae, and the educational and the developmental support is integrated into the follow-up. The child with the syndromic neuroblastoma, such as the congenital central hypoventilation syndrome with the PHOX2B mutation and the Hirschsprung disease, is managed jointly with the respiratory, the gastrointestinal and the genetic teams. [6][9]
The socioeconomic disadvantage, the remoteness and the migrant or the refugee status affect the access to the diagnosis and the treatment, and the indigenous and the remote populations carry a later presentation and a worse outcome in some regions. The family from the remote area may face the prolonged separation from the community during the months of the high-risk treatment, and the social work and the educational liaison are part of the multidisciplinary care. The access to the specialist centre, the protocol-based treatment and the clinical trial enrolment is the equity issue that the candidate who acknowledges the social determinants demonstrates the breadth the boards reward. [1][8]
Evidence, Guidelines & Regional Differences
The landmark evidence for the neuroblastoma is the body of the International Neuroblastoma Risk Group studies and the cooperative group trials that have refined the risk stratification and the treatment. The International Neuroblastoma Risk Group staging system, published by Monclair and the INRG Task Force in two-thousand-nine, replaced the older surgical staging system with the pre-treatment image-based staging using the image-defined risk factors, and it has become the international standard. The International Neuroblastoma Risk Group classification, published by Cohn and the INRG Task Force in two-thousand-nine, integrated the stage, the age, the histology, the MYCN status and the ploidy into the risk group, and it has harmonised the risk assignment across the cooperative groups. [2][12]
The SIOPEN trial of Ladenstein and colleagues, published in two-thousand-eighteen in the Lancet Oncology, demonstrated that the addition of the interleukin-2 to the anti-GD2 antibody ch14.18 slash CHO, the dinutuximab beta, in the consolidation of the high-risk neuroblastoma improved the event-free survival. The dinutuximab beta has become the standard of care for the high-risk neuroblastoma in the immunotherapy phase, and it is given in combination with the interleukin-2 and the isotretinoin. The Cooperative Group trials across the SIOP Europe, the Children's Oncology Group and the Japanese Study Group have refined the induction, the surgery, the myeloablative consolidation and the radiotherapy, and the outcomes are converging across the groups. [10][1]
The International Neuroblastoma Risk Group staging system is the international standard, and the consensus report of Brisse and the INRG Task Force, published in two-thousand-eleven in Radiology, established the imaging guidelines for the image-defined risk factors that separate the L1 from the L2 stage. The magnetic resonance imaging is the preferred modality for the primary-site imaging in most European and Australasian centres, because it avoids the radiation and it better defines the vascular and the intraspinal extension. The iodine-123 metaiodobenzylguanidine scan is the standard functional imaging across all regions, and the fluorodeoxyglucose positron emission tomography is used for the MIBG-negative tumours that occur in roughly ten percent of cases.
[3][4]The controversies are the ones that the candidate can discuss without pretending to resolve them. The role of the maintenance therapy with the isotretinoin and the immunotherapy in the intermediate-risk disease, the optimal number of the myeloablative cycles, the role of the radiotherapy in the complete responders, and the targeted therapy for the relapsed disease are the open questions. The evidence is weakest in the rare subtypes and in the resource-limited setting, where the access to the MIBG scan, the myeloablative therapy and the anti-GD2 immunotherapy is limited. The precision medicine approaches, targeting the ALK mutation and the alternative lengthening of telomeres pathway, are the emerging therapies that the candidate may name as the future direction. [8][5]
Exam Pearls
The high-yield facts for the examination are the ones that the examiner probes and that the candidate must carry without hesitation. The neural crest origin, the International Neuroblastoma Risk Group staging of L1, L2, M and MS, the urinary catecholamine metabolites vanillylmandelic acid and homovanillic acid, the MYCN amplification as the single most powerful adverse marker, and the opsoclonus-myoclonus-ataxia syndrome are the core of the topic. The risk-adapted treatment from the observation to the intensive multimodal therapy, the spontaneous regression of stage MS, and the survival figures complete the picture. [1][2]
The opsoclonus-myoclonus-ataxia syndrome is the single most testable paraneoplastic syndrome in the topic, and the principle is simple and absolute. The child with the opsoclonus, the myoclonus and the ataxia is neuroblastoma until the urinary catecholamines and the imaging prove otherwise, because the opsoclonus is the chaotic multidirectional dancing-eye movement that is not the rhythmic nystagmus. The neuroblastoma associated with the syndrome is typically the low-stage, favourable-biology tumour, and the tumour itself carries the excellent prognosis with the surgical resection. The devastating fact is the neurological outcome, because the autoimmune injury persists and causes the cognitive, behavioural and motor sequelae, and the early and aggressive immunomodulation may improve the outcome. [6][9]
References
- [1]Matthay KK, Maris JM, Schleiermacher G, et al Neuroblastoma Nat Rev Dis Primers, 2016.PMID 27830764
- [2]Monclair T, Brodeur GM, Ambros PF, et al The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report J Clin Oncol, 2009.PMID 19047290
- [3]Brisse HJ, McCarville MB, Granata C, et al Guidelines for imaging and staging of neuroblastic tumors: consensus report from the International Neuroblastoma Risk Group Task Force Radiology, 2011.PMID 21586679
- [4]Matthay KK, Shulkin B, Ladenstein R, et al Criteria for evaluation of disease extent by (123)I-metaiodobenzylguanidine scans in neuroblastoma: a report for the International Neuroblastoma Risk Group (INRG) Task Force Br J Cancer, 2010.PMID 20424613
- [5]Huang M, Weiss WA Neuroblastoma and MYCN Cold Spring Harb Perspect Med, 2013.PMID 24086065
- [6]Du H, Cai W Opsoclonus-myoclonus syndrome associated with neuroblastoma: Insights into antitumor immunity Pediatr Blood Cancer, 2022.PMID 36094353
- [7]Eisenhofer G, Peitzsch M, Bechmann N, et al Biochemical diagnosis of catecholamine-producing tumors of childhood: neuroblastoma, pheochromocytoma and paraganglioma Front Endocrinol, 2022.PMID 35957826
- [8]Sainero-Alcolado L, Sjoberg Bexelius T, Santopolo G, et al Defining neuroblastoma: From origin to precision medicine Neuro-Oncology, 2024.PMID 39101440
- [9]Rossor T, Yeh EA, Khakoo Y, et al Diagnosis and management of opsoclonus-myoclonus-ataxia syndrome in children: An international perspective Neurol Neuroimmunol Neuroinflamm, 2022.PMID 35260471
- [10]Ladenstein R, Potschger U, Valteau-Couanet D, et al Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): an open-label, randomised, phase 3 trial Lancet Oncol, 2018.PMID 30442501
- [11]Brodeur GM Spontaneous regression of neuroblastoma Cell Tissue Res, 2018.PMID 29305654
- [12]Cohn SL, Pearson AD, London WB, et al The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report J Clin Oncol, 2009.PMID 19047291