Paeds · haematology-oncology-and-transfusion
Neutropenia and neutrophil disorders
Also known as Neutropenia · Low neutrophil count · Severe congenital neutropenia · Cyclic neutropenia · Autoimmune neutropenia of infancy · Benign ethnic neutropenia
Fellowship guide to neutropenia and the neutrophil disorders in children. Covers the absolute neutrophil count thresholds that define and grade neutropenia from mild to severe, the neutrophil kinetic model that localises the mechanism to marrow production failure, peripheral destruction, splenic sequestration or marrow retention, and the full differential from common transient post-viral and chemotherapy causes through autoimmune neutropenia of infancy and alloimmune neonatal neutropenia to the inherited syndromes including ELANE severe congenital neutropenia, cyclic neutropenia, Shwachman-Diamond with SBDS, GATA2 deficiency, WHIM with CXCR4, Chediak-Higashi with LYST and Barth syndrome with TAZ. Details the workup of the incidental low count, anti-neutrophil antibody testing, bone marrow and gene panel, the first-hour management of febrile severe neutropenia with empiric anti-pseudomonal beta-lactam, the lifelong granulocyte colony-stimulating factor of severe congenital neutropenia with annual marrow surveillance for myelodysplastic syndrome and acute myeloid leukaemia, the curative role of haematopoietic stem cell transplant, and the reassuring prognosis of benign ethnic neutropenia and autoimmune neutropenia of infancy.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A worried parent brings in a healthy-looking three-year-old for a repeat blood count. The first count, taken during a viral cold a week ago, showed a neutrophil count of 0.8 times ten to the ninth per litre. Today the count is normal. This is the commonest face of paediatric neutropenia: a brief, benign, post-viral dip that resolves on its own. The skill is to recognise that face confidently, and at the same time to know the small minority in whom a low neutrophil count is the first clue to a congenital marrow failure syndrome, a leukaemia, or an immune process. [1]
Neutropenia means an absolute neutrophil count below the age- and population-appropriate lower limit. For practical purposes in older children and adults that lower limit is 1.5 times ten to the ninth per litre. The absolute neutrophil count is not printed by accident; it is calculated from the full blood count as the white cell count multiplied by the percentage of segmented neutrophils plus bands, all divided by one hundred. Severity is graded in three bands, and the band matters, because infection risk climbs sharply only in the severe band. Mild neutropenia is 1.0 to 1.5, moderate is 0.5 to 1.0, and severe is under 0.5 times ten to the ninth per litre. [1]
Neutrophils are the front-line defence against pyogenic bacteria and fungi. They turn over rapidly, surviving only hours in the circulation, so the count is a sensitive read-out of the balance between marrow production and peripheral use. When that balance breaks, the child becomes vulnerable to the very organisms that live on the skin, in the gut and in the mouth, and the first sign can be a single spike of fever that demands same-hour antibiotics. The fellowship candidate must hold three things at once: the thresholds that grade severity, the kinetic model that localises the mechanism, and the gene list that names the congenital syndromes. [1]
Classification
The cleanest way to classify neutropenia is to hold three axes in mind at once: severity, duration, and mechanism. Severity sets the infection risk and is read straight off the absolute neutrophil count. Duration sorts the child into the transient majority and the chronic minority, using three months as the conventional cut-off. Mechanism is the deepest axis, because it localises where along the neutrophil pipeline the problem sits, and it sorts every cause into one of a small number of families. [1]
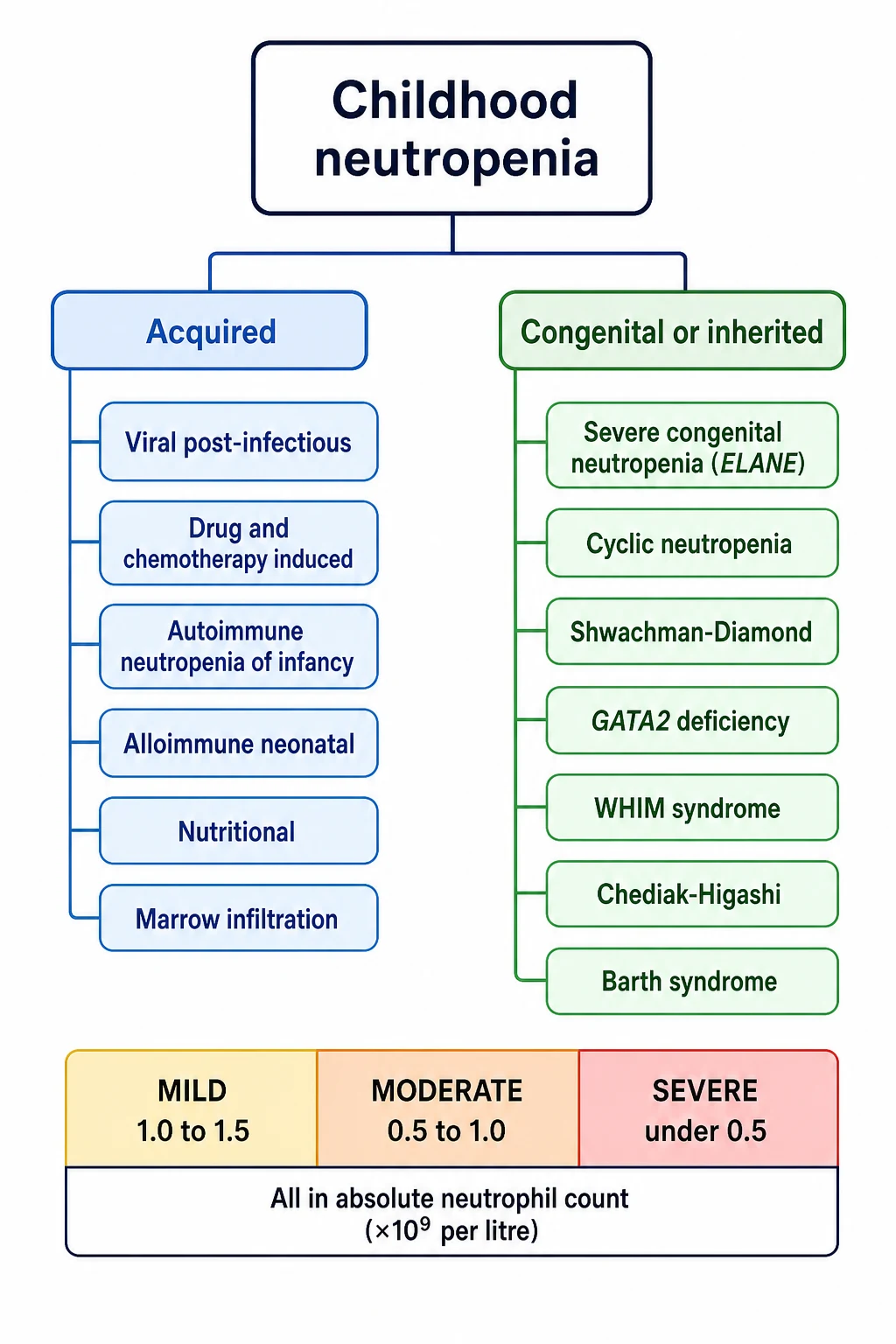
The acquired causes are the common ones. A viral illness in the preceding week or two is the single commonest trigger in childhood, from Epstein-Barr virus, cytomegalovirus, parvovirus B19, human herpesvirus 6, respiratory syncytial virus and influenza through to hepatitis viruses and human immunodeficiency virus. Drugs come next, and the chemotherapy agents are the most predictable offenders, followed by anticonvulsants such as carbamazepine and valproate, antibiotics such as co-trimoxazole, anti-thyroid drugs, non-steroidal anti-inflammatories, and the psychotropic clozapine. The immune causes are autoimmune neutropenia of infancy, the commonest chronic isolated neutropenia in a young child, and secondary autoimmune neutropenia in systemic lupus erythematosus or Evans syndrome. Nutritional deficiency of vitamin B12, folate or copper can present with neutropenia, as can marrow infiltration from acute leukaemia or storage disease and splenic sequestration. [1]
The congenital causes are rare but high-yield, and the candidate who can pair each syndrome with its gene and its patterned phenotype demonstrates command of the topic. The benign variant, benign ethnic neutropenia, sits apart and is defined by ancestry: a child of African, Middle Eastern or Yemenite Jewish descent with a chronically low circulating count but a normal marginated pool, normal neutrophil function and no excess of infections. It is caused by the Duffy-null regulatory variant in the ACKR1 gene, and its importance is negative: it must be recognised so the child is neither over-investigated nor wrongly labelled as immunocompromised. [11][10]
Epidemiology & Risk Factors
Mild transient neutropenia is extremely common in early childhood. Almost any viral infection can drive the neutrophil count down for a few days to weeks, and most of these children are never referred. At the other end of the spectrum, severe congenital neutropenia is rare, with an incidence of around one to two per million live births per year, and ELANE mutations account for roughly half of the autosomal dominant cases. Cyclic neutropenia is rarer still, with no clear ethnic predominance. [2]
The congenital neutropenia syndromes carry recognisable inheritance patterns that the examiner rewards. Severe congenital neutropenia from ELANE is autosomal dominant and the commonest form; the Kostmann form from biallelic HAX1 loss is autosomal recessive and classically described in northern Scandinavian families. Shwachman-Diamond syndrome from SBDS is autosomal recessive. Chediak-Higashi and GATA2 deficiency are autosomal recessive and autosomal dominant respectively. Barth syndrome is X-linked recessive, so it appears in boys with dilated cardiomyopathy and skeletal myopathy. The candidate who names the inheritance alongside the gene has the full picture. [2][6]
The risk factors for an acquired low count are the ones to take a deliberate history for. A recent viral illness is the commonest. Chemotherapy or radiotherapy is the most predictable iatrogenic cause, and the oncology child on treatment is the population at highest absolute risk of febrile neutropenia. Specific drugs carry a reputation: carbamazepine, valproate, clozapine, the anti-thyroid drugs, co-trimoxazole and chloramphenicol. Severe malnutrition with vitamin B12, folate or copper deficiency is a recognised cause, especially in exclusively breast-fed infants of deficient mothers or in children with malabsorption. Marrow infiltration from acute leukaemia is the diagnosis never to miss when a low neutrophil count sits with anaemia and thrombocytopenia. [1]
Pathophysiology
The way to understand neutropenia is to picture the neutrophil as a product on an assembly line with a short shelf life. It is made in the marrow, released into the blood, spends a brief time stuck to the vessel wall, and then leaves for the tissues to fight infection. A drop in the circulating count means the line is broken at exactly one of those points, and naming that point names the mechanism. [1]
The marrow makes neutrophils through the myeloid maturation cascade. A myeloblast becomes a promyelocyte, then a myelocyte, a metamyelocyte, a band, and finally a segmented neutrophil, over roughly ten to fourteen days. The mature cells are released into the circulating pool, which is the fraction a full blood count actually samples. An equal number sit in the marginated pool, adherent to the vessel wall, ready to roll out to a site of infection. Egress to the tissues then completes the journey. Because the circulating pool is only half of the total, a low full-blood-count number does not always mean a low total body neutrophil mass, a fact that explains benign ethnic neutropenia. [1]
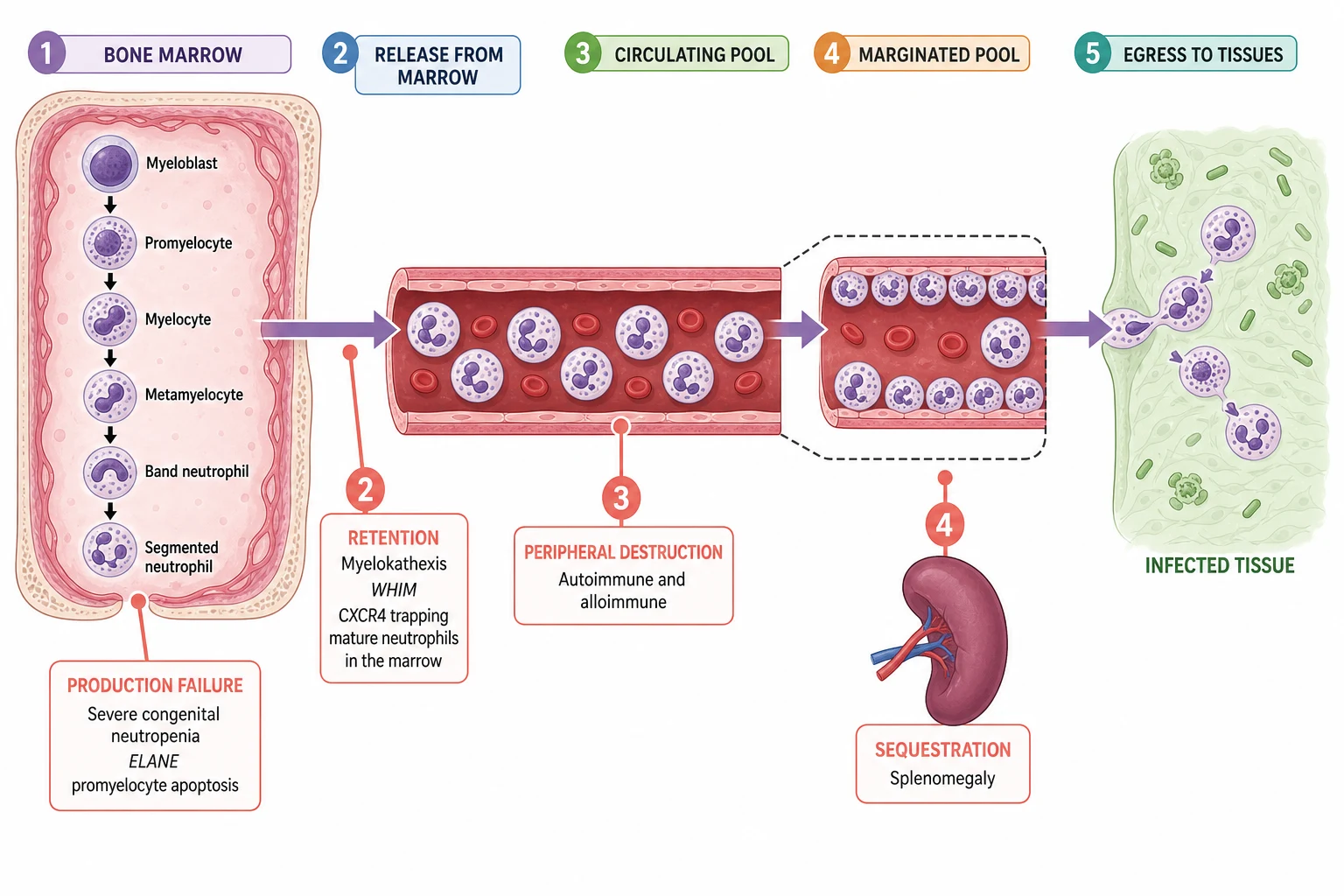
Severe congenital neutropenia is a production failure at the marrow. In the common ELANE form, mutations cause misfolded neutrophil elastase that triggers the unfolded-protein response and apoptosis of the promyelocyte, so granulopoiesis arrests at the promyelocyte stage and almost no mature neutrophils reach the blood. The autosomal recessive HAX1 form (Kostmann disease) causes the same arrest through loss of a mitochondrial anti-apoptotic protein. The marrow in both shows the diagnostic picture of a maturation arrest, and the blood shows a severe persistent neutropenia from infancy. [6]
Cyclic neutropenia shares the ELANE mechanism but with a twist. The same misfolded elastase drives apoptosis, but a feedback loop within the stem-cell compartment makes the production oscillate on a roughly twenty-one day cycle. For three to five days of each cycle, the count crashes, the mouth ulcerates, and the child spikes fevers; then production recovers and the cycle repeats. The regularity of the cycle is the clinical signature. [5]
The peripheral mechanisms are different. Autoimmune and alloimmune neutropenia are destruction: immunoglobulin G antibodies coat the neutrophil, most often against the HNA-1a or HNA-1b antigens, and the spleen clears it. The marrow tries to keep up, shows a maturation arrest from overstimulation, and the count stays low until the antibody clears. WHIM syndrome is the opposite, a retention defect: a gain-of-function mutation in the CXCR4 chemokine receptor traps mature neutrophils inside the marrow in the picture called myelokathexis, so the marrow is full and the blood is empty. Splenic sequestration simply removes circulating neutrophils, and the count corrects if the spleen is removed or shrinks. [14][9]
Infection risk depends on two things together: the depth of the neutropenia and its duration, and the integrity of the mucosal barrier. A count under 0.5 raises the risk of pyogenic sepsis, and a count under 0.1 is profound. Breakdown of the skin and mucosa, common with chemotherapy and with the congenital syndromes, opens a route for the very organisms the neutrophil is meant to clear. That is why oral and perianal hygiene are part of management, not just hygiene. [1]
Clinical Presentation
Most childhood neutropenia is found by chance, on a full blood count taken for another reason, in a child who is entirely well. The second commonest presentation is the child with a pattern of recurrent or unusual infections. The third, and the one that demands same-hour action, is the febrile child who happens to be neutropenic. [1]
The symptomatic child with chronic neutropenia presents with infections of the surfaces the neutrophil defends: the skin and soft tissue, the mouth and oropharynx, the paranasal sinuses, the ears and the lungs. The recurring pictures are cellulitis and furunculosis, aphthous and periodontal ulceration, gingivitis that can progress to loose teeth, perianal cellulitis or fissuring, otitis media, sinusitis, and pneumonia. These infections are often caused by the typical pyogenic organisms, Staphylococcus aureus and the Gram-negatives, and they tend to come back because the neutrophil reservoir is empty. [1]
The danger picture is febrile neutropenia. The definition is a single axillary or oral temperature of 38.5 degrees Celsius or higher, or 38.0 degrees or higher sustained over an hour, in a child with a neutrophil count under 0.5 (or under 1.0 with an expected fall). The child must be treated as presumed bacteraemic, because the signs of sepsis are blunted when there are no neutrophils to generate them, and a wait can be fatal. This is covered in detail in the febrile neutropenia topic; the principle that belongs here is that any severely neutropenic child with fever is an emergency. [1]
The neonate with alloimmune neutropenia presents differently and is easy to miss. The mother, sensitised to paternal human neutrophil antigens during pregnancy, passes immunoglobulin G across the placenta that clears the fetal neutrophils. The neonate presents in the first weeks of life with delayed umbilical-cord separation, omphalitis, skin pustulosis, or a frank neonatal sepsis, and the clue is a persistently low neutrophil count with no other explanation. The maternal autoimmune form behaves similarly when the mother herself has autoimmune neutropenia. [9]
The congenital syndromes carry patterned clues that the candidate must actively seek, because naming the syndrome changes the management and the prognosis. Shwachman-Diamond pairs neutropenia with failure to thrive, fatty or loose stools from exocrine pancreatic insufficiency, and skeletal changes. Chediak-Higashi shows partial oculocutaneous albinism, recurrent pyoderma, and the pathognomonic giant intracytoplasmic granules on the film, with a risk of a fatal accelerated phase. Barth syndrome, in a boy, pairs neutropenia with dilated cardiomyopathy and skeletal myopathy. GATA2 deficiency presents with disseminated non-tuberculous mycobacterial infection, widespread warts, and a progressive loss of monocytes and dendritic cells that culminates in myelodysplasia. Cyclic neutropenia announces itself by its rhythm: the symptoms recur in a regular three-week cycle, with mouth ulcers and fever clustered at the predictable nadir. [12][13]
Differential Diagnosis
The differential is built in three moves. The first move is to confirm the neutropenia is real and not a laboratory artefact. The second is to decide whether the neutropenia is isolated or part of a pancytopenia. The third, within isolated neutropenia, is to weigh the immune and the congenital causes against the common benign ones. [1]
The artefacts come first. Neutrophils can clump in the ethylenediaminetetraacetic-acid tube, giving a falsely low count; a fresh sample, sometimes in citrate or heparin, resolves it. A post-viral dip is not an artefact but it is transient, and a repeat count two to four weeks later is normal. The single most useful habit, and the one the examiner rewards, is to repeat the count before launching a workup. [1]
The isolated-versus-pancytopenia split is decisive. Pancytopenia, with anaemia and thrombocytopenia alongside the neutropenia, points to marrow failure or infiltration: aplastic anaemia, acute leukaemia, myelodysplastic syndrome, storage disease, or overwhelming sepsis. It demands an urgent bone marrow. Isolated neutropenia, with a normal haemoglobin and platelet count, points instead to the immune and congenital causes, and the workup is gentler. [1]
Within the isolated chronic neutropenia of a well infant, the main contenders are autoimmune neutropenia of infancy, benign ethnic neutropenia, and the inherited disorders. The discriminating features are the age and clinical state, the ancestry, the antibody result, the marrow, and the gene panel. The functional neutrophil disorders belong to a related but distinct family: chronic granulomatous disease and leucocyte adhesion deficiency usually have a normal or raised neutrophil count with recurrent or unusual infections, and they are distinguished by functional assays such as the dihydrorhodamine or nitroblue tetrazolium test rather than by the count. They are covered in the primary immunodeficiency topic. [1]
Clinical & Bedside Assessment
Begin with an overall sick-or-well judgement and a paediatric assessment of airway, breathing and circulation. The febrile severely neutropenic child is an emergency, and the assessment there is brief and goal-directed toward empiric antibiotics. In the stable child, the history is where the diagnosis is won. Ask about the age of onset and whether the count was incidental or followed an illness. Take a precise inventory of the infections: the site, the frequency, the severity, the organisms grown, and the response to treatment. [1]
The drug and treatment history is essential and must be exhaustive. Name the high-risk drugs by name: chemotherapy, carbamazepine, valproate, clozapine, anti-thyroid drugs, co-trimoxazole, chloramphenicol, and the non-steroidal anti-inflammatories. Ask about the family origin and the family history, because the Duffy-null variant and the inherited syndromes both travel with ancestry and consanguinity; probe for early infant deaths, siblings with neutropenia, and relatives with cardiomyopathy or immunodeficiency. Ask about growth, stool pattern, and any syndrome clues such as loose or fatty stools, skeletal anomalies, unusual skin or hair colour, and cardiac symptoms in a boy. [1]
The focused examination looks for growth parameters and dysmorphism, the skin and oral cavity (mouth ulcers, gingivitis, periodontal disease, perianal fissures or cellulitis), lymphadenopathy, and hepatosplenomegaly. Seek the syndrome signs deliberately: the partial albinism of Chediak-Higashi, the short stature and skeletal changes of Shwachman-Diamond, the cardiac murmur and myopathy of Barth syndrome in a boy, the warts and skin lesions of GATA2 deficiency. Look for any focus of infection, and in the neonate examine the umbilicus, the skin, and any pustules. [12][7]
[1]Investigations
The core test is a repeat full blood count and differential on a fresh sample, with a peripheral blood film. The repeat confirms a true absolute neutrophil count and checks the other cell lines, and the film is where the syndromic clues live. Look for dysplasia, for blasts that point to leukaemia, for the giant intracytoplasmic granules of Chediak-Higashi, for a left shift or toxic granulation of sepsis, and for the dual neutrophil populations sometimes seen in benign ethnic neutropenia. If the count is unexpectedly low in an asymptomatic person, repeat it on a citrate or heparin sample to exclude ethylenediaminetetraacetic-acid clumping. [1][12]
For the child with chronic or unexplained neutropenia, the targeted tests follow the mechanism. Anti-neutrophil antibody testing confirms autoimmune neutropenia of infancy and the secondary autoimmune causes. Immunoglobulins and a lymphocyte subset screen for an associated immune deficiency, and an antinuclear antibody with double-stranded DNA screens for systemic lupus erythematosus. Vitamin B12, folate and copper levels catch the nutritional causes. A bone marrow aspirate and trephine, with cytogenetics, is obtained when the neutropenia is severe, persistent, part of a pancytopenia, or accompanied by dysplasia or blasts; in severe congenital neutropenia it stages the disease and screens for clonal evolution. [1][4]
Genetic testing is now central, not optional. An inherited neutropenia gene panel covering ELANE, HAX1, SBDS, CXCR4, GATA2, LYST and TAZ identifies the congenital syndromes and guides counselling, surveillance and the transplant decision. In the neonate, anti-neutrophil antibodies and human neutrophil antigen typing of the mother and the baby confirm alloimmune neutropenia and distinguish it from a transient neonatal sepsis dip. [6][9]
[4] [2]Management — Resuscitation
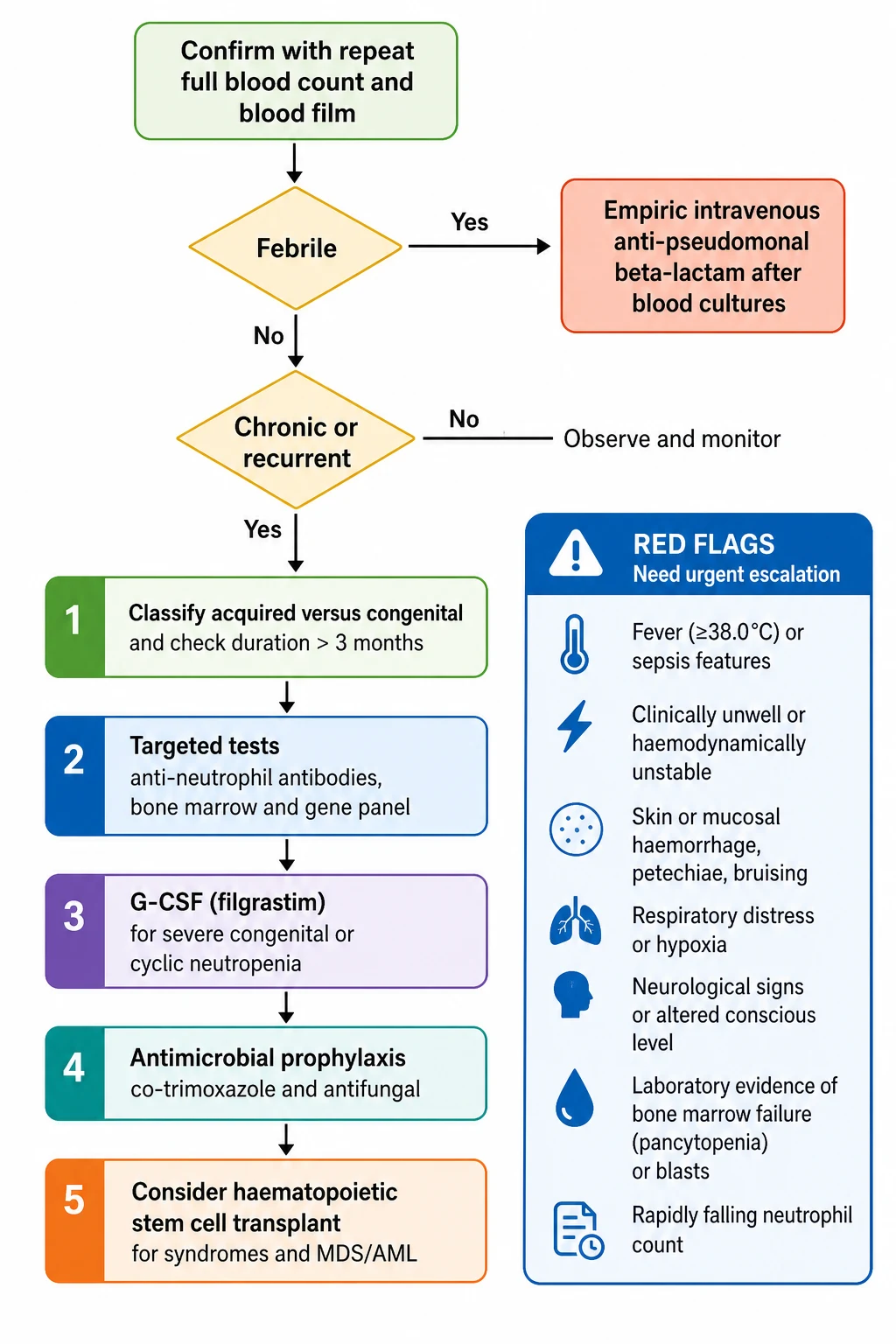
The single time-critical scenario is febrile neutropenia. It is defined as a temperature of 38.5 degrees Celsius or higher on one occasion, or 38.0 degrees or higher sustained over an hour, in a child with a neutrophil count under 0.5 (or under 1.0 with an expected fall). The child is assessed and resuscitated as for sepsis. Blood cultures are drawn from every lumen of any central venous line and peripherally, a urinalysis is sent, and a chest radiograph is taken where indicated. [1]
Empiric broad-spectrum intravenous anti-pseudomonal monotherapy is then started within the first hour. The standard choices are ceftazidime, piperacillin-tazobactam, cefepime or meropenem, weight-dosed by the local oncology or sepsis protocol, with vancomycin added for suspected line or soft-tissue infection or haemodynamic instability. The detailed oncology pathway, including risk-stratification and the duration of therapy, is covered in the febrile neutropenia topic. The principle that belongs here is that any severely neutropenic child with a fever is presumed bacteraemic until proven otherwise, and a delay to antibiotics costs lives. [1]
The mucosal barrier needs protection. Maintain fastidious oral hygiene with regular tooth brushing and antiseptic mouthwash, and perianal hygiene with sitz baths and avoidance of constipation. Avoid rectal examinations, rectal temperatures and suppositories, because the traumatized mucosa seeds infection. Stop any offending drug. In shock, follow paediatric sepsis resuscitation with fluid boluses of 10 mL per kilogram of isotonic crystalloid titrated to perfusion, and escalate to vasoactive support and intensive care. [1]
The first hour of febrile severe neutropenia
Recognise fever of 38.5 degrees Celsius or higher (or 38.0 over an hour) with an absolute neutrophil count under 0.5 as febrile neutropenia, a presumed bacteraemia
Assess airway, breathing and circulation and take a brief focus, weighing the risk of occult shock with blunted signs
Draw blood cultures from every central line lumen and peripherally, send a urinalysis, and image only where indicated
Start empiric intravenous anti-pseudomonal monotherapy within the first hour: ceftazidime, piperacillin-tazobactam, cefepime or meropenem, weight-dosed by protocol, adding vancomycin for suspected line infection or instability
Maintain oral and perianal hygiene, stop any offending drug, and avoid rectal instrumentation
Escalate to intensive care for shock, with fluid boluses of 10 mL per kilogram isotonic crystalloid titrated to perfusion and vasoactive support
Management — Definitive & Stepwise
The definitive management depends on the mechanism, and the cleanest way to hold it is as a ladder from the reassuring to the curative. At the bottom, transient and benign neutropenia needs only observation. In the middle, the immune causes need targeted, often time-limited, therapy. At the top, the congenital syndromes need lifelong granulocyte colony-stimulating factor, prophylaxis, surveillance, and sometimes transplant. [1]
[1] [3]For transient post-viral neutropenia in a well child, the management is observation and a repeat count in two to four weeks; no investigation is needed if the child is well and the count normalises. For autoimmune neutropenia of infancy, the management is equally expectant, because the condition resolves spontaneously in the great majority by age four to five. Granulocyte colony-stimulating factor, intravenous immunoglobulin or corticosteroids are reserved for the child with severe or recurrent infections, not used routinely for a well child with a low count. Over-treatment of a benign condition is the pitfall the examiner probes. [1]
For severe congenital neutropenia and cyclic neutropenia, the cornerstone is lifelong granulocyte colony-stimulating factor (filgrastim or lenograstim) given subcutaneously. The Severe Chronic Neutropenia International Registry documented a sustained response and improved survival on long-term filgrastim, and the standard is to start at a low dose and titrate to an absolute neutrophil count that prevents infection, most often a target in the 1.5 to 2.0 band. The child is taught home injection, monitored for response, and enrolled in annual marrow surveillance. For cyclic neutropenia, the same drug attenuates the oscillation and shortens the cycle. [3][5]
For Shwachman-Diamond syndrome, the management combines pancreatic enzyme replacement and fat-soluble vitamin supplementation for the exocrine pancreatic insufficiency, granulocyte colony-stimulating factor for severe neutropenia, and active surveillance for marrow failure and leukaemic transformation under the consensus guidelines. For alloimmune neonatal neutropenia, the management is supportive, with granulocyte colony-stimulating factor or intravenous immunoglobulin reserved for the severely infected neonate; the condition resolves as the maternal antibody clears over weeks to months. For benign ethnic neutropenia, the management is recognition alone: correct documentation in the record, reassurance of the family, and no treatment. [8][9]
Haematopoietic stem cell transplant is the curative option for the congenital syndromes with marrow failure, immunodeficiency, or leukaemic transformation. The clearest indications are Chediak-Higashi (ideally before the accelerated phase), GATA2 deficiency with myelodysplasia or severe infection, Shwachman-Diamond with myelodysplastic syndrome or leukaemia, and severe congenital neutropenia that has transformed to myelodysplastic syndrome or acute myeloid leukaemia. Antimicrobial prophylaxis with co-trimoxazole for Pneumocystis and an antifungal agent is used in chronic severe neutropenia and after transplant. [12][13]
Specific Subtypes & Scenarios
Severe congenital neutropenia is the archetype of the inherited disorders. It presents in early infancy with severe persistent neutropenia under 0.5, recurrent severe infections of the skin, mouth and lungs, and a marrow maturation arrest at the promyelocyte stage. ELANE is the commonest gene, inherited as an autosomal dominant trait; the autosomal recessive HAX1 variant (Kostmann disease) is classically described in northern Scandinavian families. The standard of care is lifelong granulocyte colony-stimulating factor, started early and titrated to a protective count, with annual marrow surveillance for the cumulative risk of myelodysplastic syndrome and acute myeloid leukaemia. [2][4]
Cyclic neutropenia is distinguished by its rhythm. Driven by ELANE mutations, it produces a regular twenty-one day cycle in which the count crashes for three to five days, the mouth ulcerates, and the child spikes fevers, before recovering. The regularity is the diagnostic clue; serial counts two to three times a week for six weeks will map the cycle. Granulocyte colony-stimulating factor attenuates the oscillation and shortens the cycle, and most children do well on it. [5][6]
Autoimmune neutropenia of infancy is the commonest chronic isolated neutropenia in a child under three. An otherwise well infant, with a median onset around eight months, is found to have a mild to moderate neutropenia and positive anti-neutrophil antibodies, most often against HNA-1a or HNA-1b. The course is benign and self-limiting, with spontaneous resolution in the great majority by age four to five. Granulocyte colony-stimulating factor, intravenous immunoglobulin, or corticosteroids are reserved for the child with severe or recurrent infections, and the family is reassured and given a clear safety-net. [1]
Alloimmune neonatal neutropenia mirrors rhesus haemolytic disease. The mother is sensitised to paternal human neutrophil antigens during pregnancy and passes immunoglobulin G across the placenta, clearing the fetal neutrophils. The neonate presents in the first weeks of life with omphalitis, delayed cord separation, skin pustulosis, or a frank neonatal sepsis, and the count is severely and selectively low. Anti-neutrophil antibodies and human neutrophil antigen typing of mother and baby confirm the diagnosis, and the condition resolves over weeks to months as the antibody clears. [9]
Shwachman-Diamond syndrome is the one to name when neutropenia pairs with gut and bone. Caused by biallelic SBDS mutations, it combines neutropenia with exocrine pancreatic insufficiency (fatty, foul-smelling stools and failure to thrive), skeletal dysplasia, and short stature, and it carries a risk of marrow failure and leukaemia. The management combines pancreatic enzyme replacement and fat-soluble vitamins, granulocyte colony-stimulating factor for severe neutropenia, and surveillance under the consensus guidelines. [7][8]
Chediak-Higashi syndrome is the one with the striking film. Caused by biallelic LYST mutations, it presents with partial oculocutaneous albinism, recurrent pyogenic infections, and the pathognomonic giant intracytoplasmic granules in the neutrophils, and it carries the danger of an accelerated phase, a haemophagocytic syndrome that is often fatal without transplant. The definitive treatment is haematopoietic stem cell transplant, ideally before the accelerated phase. GATA2 deficiency presents with disseminated non-tuberculous mycobacterial infection, widespread warts, and a progressive loss of monocytes and dendritic cells, culminating in myelodysplasia and leukaemia; the curative treatment is transplant. WHIM syndrome, from a CXCR4 gain-of-function mutation, gives warts, hypogammaglobulinaemia, bacterial infections, and the myelokathexis picture of a marrow full of mature neutrophils that cannot exit. [12][13][14]
Complications & Pitfalls
The principal acute complication is overwhelming sepsis. The responsible organisms are the pyogenic bacteria of the skin and gut: the Gram-negatives such as Pseudomonas aeruginosa, Escherichia coli and Klebsiella, and the Gram-positive Staphylococcus aureus. In prolonged severe neutropenia, especially with chemotherapy and mucosal breakdown, invasive fungal infection with Candida, Aspergillus or the mucorales becomes a real and lethal risk. The mucosal complications of chronic neutropenia include periodontal disease and tooth loss from recurrent gingivitis, and perianal cellulitis. [1]
The defining long-term complication of severe congenital neutropenia is transformation to myelodysplastic syndrome and acute myeloid leukaemia. The Severe Chronic Neutropenia International Registry and the work of Rosenberg and colleagues documented a cumulative incidence that rises with the years on granulocyte colony-stimulating factor, to a rough twenty per cent over ten years, and the acquisition of clonal cytogenetic abnormalities such as CSF3R mutations, monosomy 7 or trisomy 21 is the harbinger. Annual marrow surveillance with cytogenetics is the defence. [4]
The pitfalls are the ones the examiner probes. The first is accepting a single low count without repeating it on a fresh sample, and so missing ethylenediaminetetraacetic-acid clumping or a transient post-viral dip. The second is failing to examine the film, and so missing blasts, dysplasia, or the giant granules of Chediak-Higashi. The third is labelling benign ethnic neutropenia as dangerous, over-investigating the child, and excluding the child from school or sport unnecessarily. The fourth is missing autoimmune neutropenia of infancy by not sending anti-neutrophil antibodies, and then over-treating a benign condition. [11][1]
The most dangerous pitfall is failing to treat the febrile severely neutropenic child with prompt empiric antibiotics within the first hour. Two further pitfalls complete the set: forgetting to screen for clonal cytogenetic change on the annual marrow of severe congenital neutropenia, and missing the syndrome clues that point to Shwachman-Diamond, Chediak-Higashi, Barth, or GATA2 deficiency. A candidate who names each pitfall and its defence demonstrates real understanding. [4][12]
Prognosis & Disposition
Prognosis tracks the mechanism. Transient post-viral neutropenia resolves in days to weeks with an excellent outlook. Autoimmune neutropenia of infancy resolves spontaneously in the great majority by age four to five, with a benign prognosis. Benign ethnic neutropenia is a normal variant with a normal life expectancy and no infection excess. These three are the reassuring end of the topic, and recognising them protects the child from harm. [1][11]
Severe congenital neutropenia on long-term granulocyte colony-stimulating factor has a markedly improved survival compared with the pre-treatment era, but it carries the cumulative risk of myelodysplastic syndrome and acute myeloid leukaemia and an ongoing sepsis risk, which is why annual marrow surveillance is mandatory. Cyclic neutropenia on granulocyte colony-stimulating factor has a good prognosis with attenuated cycles. Shwachman-Diamond, GATA2 deficiency, and Chediak-Higashi carry a guarded prognosis dictated by marrow failure, immunodeficiency, and leukaemic transformation, with haematopoietic stem cell transplant as the curative option. [3][4]
Disposition follows the clinical state. A febrile neutropenic child is admitted for intravenous antibiotics under the oncology or sepsis protocol. A well, afebrile child with mild transient neutropenia is observed as an outpatient with a safety-net and a repeat count in two to four weeks. A child with newly diagnosed severe chronic neutropenia is referred to paediatric haematology and started on surveillance and, where indicated, granulocyte colony-stimulating factor and antimicrobial prophylaxis. A child with a congenital syndrome is enrolled in a coordinated multidisciplinary service across haematology, immunology, infectious diseases, dentistry, genetics, and psychology, with a planned transition to adult care. [1]
Special Populations
Neonates are a distinct population because neutrophil kinetics differ. The normal neonatal neutrophil count is higher at birth and falls over the first days of life, and both alloimmune and maternal autoimmune neutropenia present here. A neonate with omphalitis, delayed cord separation, skin pustulosis, or unexplained sepsis may have an isolated severe neutropenia, and the workup includes anti-neutrophil antibodies and human neutrophil antigen typing of mother and baby alongside the sepsis screen. [9]
Infants under three are the population of autoimmune neutropenia of infancy, the commonest chronic isolated neutropenia of this age, where the management is expectant and the family is reassured. Children of African, Middle Eastern, Yemenite Jewish, and some South Asian ancestry are the population of benign ethnic neutropenia, where the priority is correct recognition, documented in the record, to avoid the harm of over-investigation and of wrongly excluding the child from school or sport. The natural history study confirms the benign course. [10][11]
The immunocompromised oncology child on chemotherapy is the population at highest absolute risk, where febrile neutropenia is a separate, protocol-driven topic. Children with congenital neutropenia syndromes are a chronic, technology-dependent population: they learn home granulocyte colony-stimulating factor injection, they need antimicrobial prophylaxis, they attend annual surveillance, and they live with the uncertainty of leukaemic transformation. They need a coordinated multidisciplinary service and a planned transition to adult care, with attention to school, family, and the psychological burden of a chronic condition. [3]
Evidence, Guidelines & Regional Differences
The evidence base rests on the framework review of Newburger and Dale, which sets the thresholds and the workup that the fellowship candidate should be able to reproduce. The Severe Chronic Neutropenia International Registry data of Dale and colleagues document the response to and the outcomes of long-term granulocyte colony-stimulating factor, and the work of Rosenberg and colleagues quantifies the leukaemia and sepsis risk that drives annual marrow surveillance. The pathophysiology and the mutation reviews of Horwitz and of Makaryan explain why ELANE drives both severe congenital and cyclic neutropenia. [1][3][4][6][5]
The congenital-syndrome evidence rests on the SBDS discovery of Boocock and colleagues for Shwachman-Diamond and the consensus guidelines of Dror and colleagues for its management, the Chediak-Higashi review of Kaplan and colleagues, the GATA2 work of Hsu and colleagues, and the WHIM chemokine work of Gulino and colleagues. The benign ethnic neutropenia evidence rests on the Duffy-null regulatory-variant discovery of Reich and colleagues and the natural history study of Lakhotia and colleagues, and the neonatal evidence on the review of Maheshwari. [7][8][12][13][14][10][11][9]
The regional differences are real and examinable. The lower limit of normal for the neutrophil count is genuinely lower in populations of African, Middle Eastern, and Yemenite Jewish ancestry, and several laboratories and guideline bodies now recommend reporting ethnicity-appropriate reference ranges to prevent the over-diagnosis of neutropenia. The oncology febrile-neutropenia pathway, including the choice of empiric agent and the risk-stratification rules, varies between the Australian and New Zealand, United Kingdom, United States, and Canadian protocols, though all share the first-hour antibiotic principle. Gene therapy and base-editing of ELANE for severe congenital neutropenia is an active research frontier that may shift the management of the inherited disorders in the coming decade. [1][11]
In Australia and Aotearoa New Zealand, the workup of the child with an incidental low neutrophil count follows the Newburger and Dale framework, with an emphasis on repeating the count and examining the film before any investigation. Severe congenital neutropenia is managed at a paediatric haematology centre with lifelong filgrastim and annual marrow surveillance under the Severe Chronic Neutropenia International Registry principles. Haematopoietic stem cell transplant for the congenital syndromes and for transformation is coordinated through the national transplant services. Benign ethnic neutropenia is increasingly recognised in the Indigenous, Pacific, and migrant African and Middle Eastern populations, with laboratory reporting moving toward ancestry-appropriate reference ranges. The oncology febrile-neutropenia pathway follows the local paediatric oncology group protocol, with the first-hour empiric anti-pseudomonal beta-lactam as the non-negotiable standard.
Exam Pearls
Neutropenia is an absolute neutrophil count under 1.5 times ten to the ninth per litre, graded as mild 1.0 to 1.5, moderate 0.5 to 1.0, and severe under 0.5; the absolute neutrophil count is the white cell count times the percentage of segmented neutrophils plus bands divided by one hundred. These two facts open the viva. The first move in any low count is to repeat it on a fresh sample and examine the film, because ethylenediaminetetraacetic-acid clumping and post-viral dips are common and spurious. [1]
PEGS-CBG
Severe congenital neutropenia is most often autosomal dominant ELANE, presents in infancy with severe persistent neutropenia and a marrow maturation arrest, and needs lifelong granulocyte colony-stimulating factor with annual marrow surveillance for myelodysplastic syndrome and acute myeloid leukaemia. Cyclic neutropenia is autosomal dominant ELANE with a regular twenty-one day cycle. Shwachman-Diamond is SBDS with neutropenia plus exocrine pancreatic insufficiency and skeletal dysplasia. Chediak-Higashi is LYST with partial albinism and giant granules and a risk of an accelerated phase. WHIM is CXCR4 gain-of-function with myelokathexis. GATA2 deficiency gives monocytopenia with non-tuberculous mycobacterial and wart infection and myelodysplasia. Barth syndrome is X-linked TAZ with cardiomyopathy and neutropenia in a boy. [2][12][13]
Autoimmune neutropenia of infancy is the commonest chronic isolated neutropenia under three, anti-neutrophil-antibody positive, and resolves by age four to five. Alloimmune neonatal neutropenia is maternal immunoglobulin G against paternal human neutrophil antigens and causes omphalitis and neonatal sepsis. Benign ethnic neutropenia is the Duffy-null ACKR1 variant of African ancestry with a normal marginated pool and normal function, and needs no treatment. The single most-tested action point is to treat the febrile severely neutropenic child with prompt empiric anti-pseudomonal beta-lactam within the first hour. [9][11]
[1] [1]References
- [1]Newburger PE, Dale DC Evaluation and management of patients with isolated neutropenia. Semin Hematol, 2013.PMID 23953336
- [2]Welte K, Zeidler C, Dale DC Severe congenital neutropenia. Semin Hematol, 2006.PMID 16822461
- [3]Dale DC, Cottle TE, Fier CJ, Bolyard AA, Bonilla MA, Boxer LA, et al Severe chronic neutropenia: treatment and follow-up of patients in the Severe Chronic Neutropenia International Registry. Am J Hematol, 2003.PMID 12555210
- [4]Rosenberg PS, Alter BP, Bolyard AA, Bonilla MA, Boxer LA, Cham B, et al The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood, 2006.PMID 16497969
- [5]Makaryan V, Zeidler C, Bolyard AA, Skokowa J, Boxer L, Dale DC, et al The diversity of mutations and clinical outcomes for ELANE-associated neutropenia. Curr Opin Hematol, 2015.PMID 25427142
- [6]Horwitz MS, Corey SJ, Grimes HL, Tidwell T, Link DC, Dinauer MC, et al ELANE mutations in cyclic and severe congenital neutropenia: genetics and pathophysiology. Hematol Oncol Clin North Am, 2013.PMID 23351986
- [7]Boocock GR, Morrison JA, Popovic M, Richards N, Ellis L, Durie PR, Rommens JM Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet, 2003.PMID 12496757
- [8]Dror Y, Donadieu J, Koglmeier J, Dodge J, Toiviainen-Salo S, Makitie O, et al Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome. Ann N Y Acad Sci, 2011.PMID 22191555
- [9]Maheshwari A Neutropenia in the newborn. Curr Opin Hematol, 2014.PMID 24322487
- [10]Lakhotia R, Aggarwal A, Link ME, Kelly K, Manchikanti L, Malech HL, Holland SM, et al Natural history of benign ethnic neutropenia in individuals of African ancestry. Blood Cells Mol Dis, 2019.PMID 30909074
- [11]Reich D, Nalls MA, Kao WH, Akylbekova EL, Tandon A, Patterson N, et al Reduced neutrophil count in people of African descent is due to a regulatory variant in the Duffy antigen receptor for chemokines gene. PLoS Genet, 2009.PMID 19180233
- [12]Kaplan J, De Domenico I, Ward DM Chediak-Higashi syndrome. Curr Opin Hematol, 2008.PMID 18043242
- [13]Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux ME, Patel SY, et al Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood, 2011.PMID 21670465
- [14]Gulino AV, Moratto D, Sozzani S, Cavadini P, Otero K, Tassone L, et al Altered leukocyte response to CXCL12 in patients with warts hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome. Blood, 2004.PMID 15026312