Paeds · haematology-oncology-and-transfusion
Sickle cell disease: diagnosis and health maintenance
Also known as Sickle cell disease · SCD · Sickle cell anaemia · HbSS disease · Hemoglobin SC disease · Sickle cell disorder
Fellowship guide to the diagnosis and health maintenance of sickle cell disease in children. Covers the autosomal recessive haemoglobinopathy born of the glutamate-to-valine substitution at beta-globin position six, the newborn-screen diagnosis by haemoglobin electrophoresis or high-performance liquid chromatography, and the four pillars of lifelong care: penicillin V prophylaxis from two months at 125 mg twice daily under three years and 250 mg twice daily at three years and over to five years, encapsulated-organ immunisation, hydroxyurea from nine months at a starting dose of 20 mg per kg per day, and annual transcranial Doppler surveillance from ages two to sixteen with chronic transfusion keeping HbS under 30 percent when the velocity reaches 200 cm per second or more.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A single changed letter in the DNA, a single amino acid swapped in the haemoglobin molecule, can reshape a whole childhood. Sickle cell disease is the inherited haemoglobinopathy in which a glutamate is replaced by a valine at the sixth position of the beta-globin chain, producing an abnormal haemoglobin called haemoglobin S. When haemoglobin S gives up its oxygen, it sticks to itself, forms long rigid fibres, and bends the red cell into the crescent or sickle shape that gives the disease its name. The sickled cell blocks small blood vessels and tears itself apart, so the child lives with pain, chronic anaemia, and organ injury. [11]
The disease is common, and it is global. It is most frequent in children whose families come from sub-Saharan Africa, but it also appears across the Mediterranean, the Middle East, and South Asia, because the sickle gene once protected against falciparum malaria. Most children today are diagnosed within the first weeks of life through newborn screening, before they have been ill at all, and the work of the paediatric team is to keep them well through a package of prophylaxis, immunisation, and surveillance. [5]
Three ideas make this topic central to the paediatric exam. The first is diagnosis, because the haemoglobin electrophoresis or high-performance liquid chromatography read from a heel-prick blood spot settles the genotype in the newborn period. The second is penicillin prophylaxis, because a tablet given twice a day transformed the survival of these children. The third is the surveillance of stroke, because the transcranial Doppler finds the child at risk before the stroke happens, and chronic transfusion stops it. The 2014 National Heart, Lung, and Blood Institute panel report and the 2020 American Society of Hematology cerebrovascular guideline together define modern care and remain the most testable sources. [5][8]
Classification
Sickle cell disease sorts itself by which two beta-globin genes the child carries, and the genotype sets the severity. The most common and the most severe form is sickle cell anaemia, in which the child carries two sickle genes and makes haemoglobin S almost exclusively, written as HbSS. When the sickle gene is inherited alongside a different abnormal beta-globin gene, the result is one of the compound heterozygous diseases: haemoglobin SC disease, written HbSC, and sickle beta-thalassaemia, written HbS beta-thalassaemia. The beta-zero form of sickle beta-thalassaemia makes no normal beta-globin and runs a course close to HbSS, while the beta-plus form makes some and runs milder. [11]
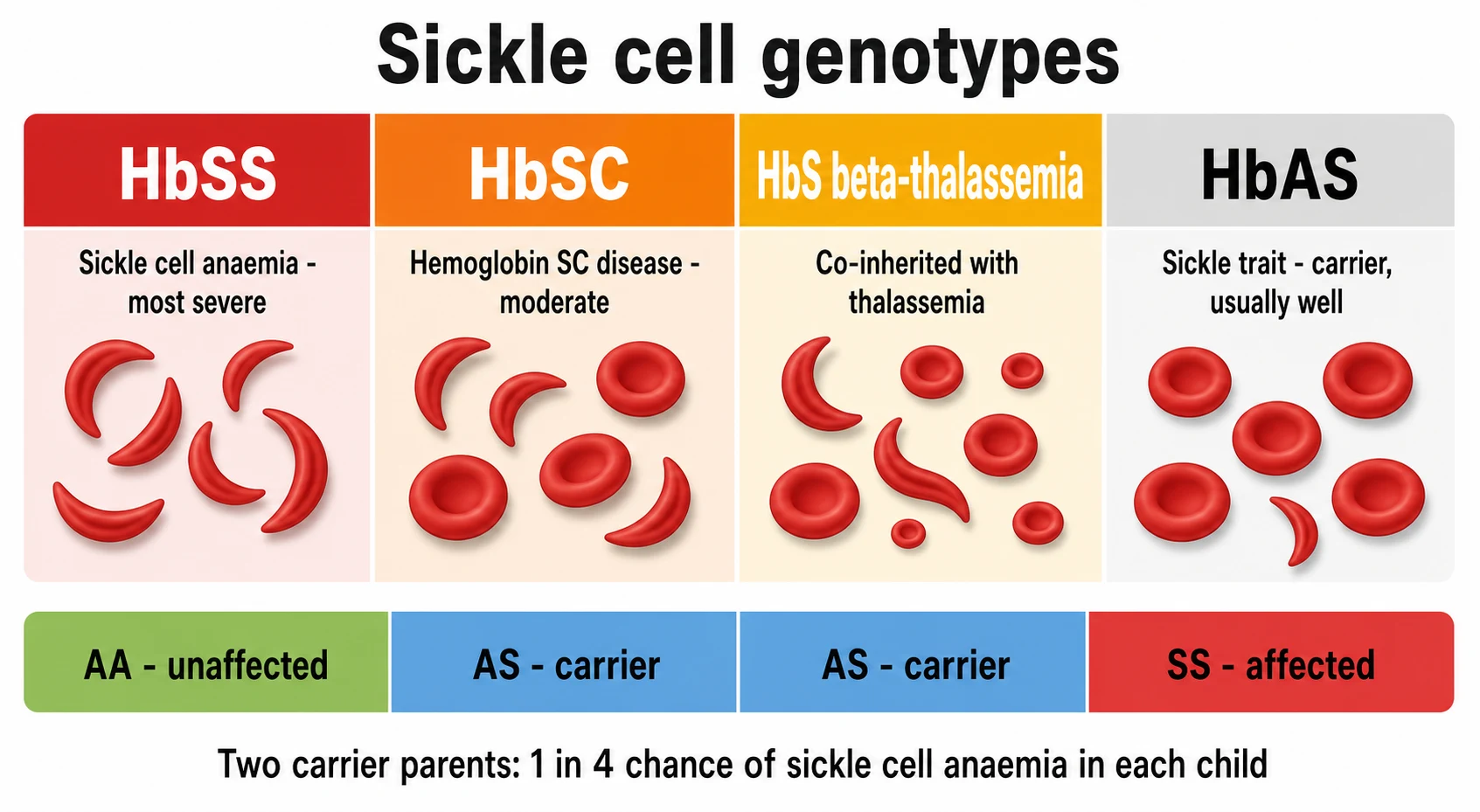
The child who carries only one sickle gene and one normal gene has sickle cell trait, written HbAS, and this is not a disease. The trait carrier makes roughly half normal haemoglobin A and half haemoglobin S, the cells do not sickle under ordinary conditions, and the child is well. The trait matters in two ways for the paediatrician. It matters for the family, because two carrier parents have a one in four chance of a child with sickle cell anaemia in each pregnancy, and it matters for the carrier, because the trait can cause problems under extreme hypoxia, dehydration, or high altitude, and rarely renal concentrating defects. [5]
A second, functional classification runs alongside the genotype and tracks what the spleen is doing, because the spleen is the organ the disease injures first. The infant is born with a working spleen, but the repeated sickling and the slow flow through the splenic cords infarct it, so that by the age of two to four years in HbSS the spleen is functionally absent. This functional asplenia is the reason the child is defenceless against the encapsulated bacteria, the pneumococcus, the meningococcus, and Haemophilus influenzae type b, and it is the reason penicillin and immunisation sit at the heart of health maintenance. [5]
Epidemiology & Risk Factors
Sickle cell disease is one of the most common single-gene disorders in the world, and the most common inherited condition picked up by newborn screening in many high-income countries. It is found wherever the sickle gene is common, which is wherever falciparum malaria was historically intense, because a single sickle gene makes the red cell a poor home for the malaria parasite. The carrier rate reaches one in ten or more in parts of sub-Saharan Africa, and the disease affects roughly one in three hundred to one in five hundred African American or Black African newborns. [11]
The inheritance is autosomal recessive, and the family risk is the risk the fellow traces at the bedside. A carrier mother and a carrier father each pass one sickle gene to a child with a one in two chance, and the chance that a child inherits both and has disease is one in four. This pattern is the reason the brothers, the sisters, and the future pregnancies of an affected child are offered testing, and it is the reason the parents are counselled that each subsequent child carries the same one in four risk. A new mutation is rare, so the carrier state is almost always present in one or both parents. [5]
The single most important determinant of severity, beyond the genotype, is whether the child receives the full health-maintenance package. A child diagnosed at birth, started on penicillin, fully immunised, given hydroxyurea, and screened with transcranial Doppler can expect to reach adulthood. The same child, diagnosed late after a first sepsis or a first stroke, carries the burden the modern package was built to prevent, which is why access to newborn screening and to a sickle cell service, rather than the gene alone, now shapes the outcome. [10]
Pathophysiology
The change at the centre of the disease is a single amino acid. In haemoglobin S, a valine replaces the glutamate that normally sits at the sixth position of the beta-globin chain, a change written as p.Glu6Val and caused by a point mutation in the HBB gene. The substitution matters because valine is hydrophobic, and it sits on the outside of the deoxygenated haemoglobin molecule where the normal glutamate, which is charged and hydrophilic, never caused trouble. When haemoglobin S gives up its oxygen, a sticky hydrophobic patch is exposed, and it fits neatly into a pocket on a neighbouring haemoglobin molecule. [11]
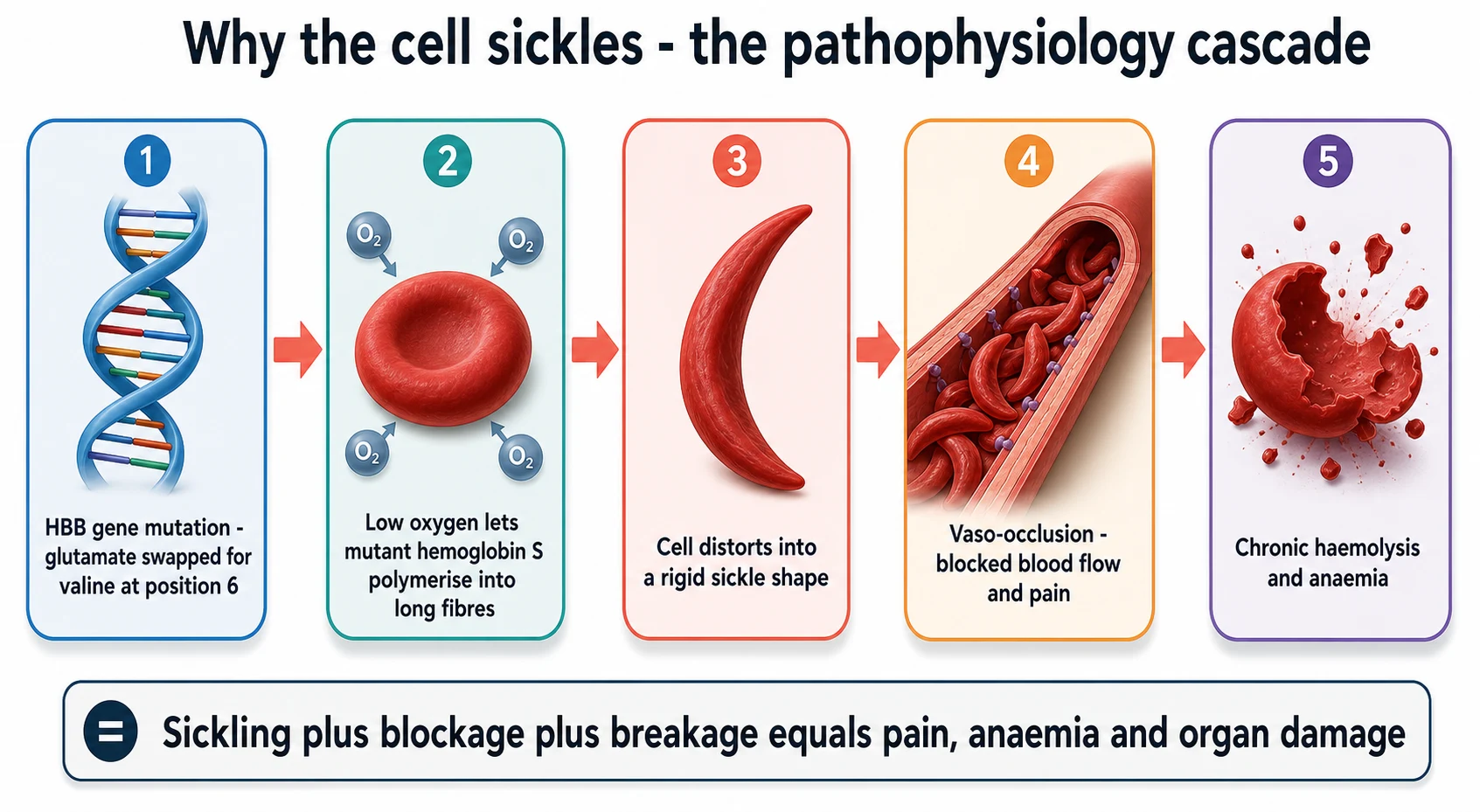
The mechanism of the sickling is polymerisation, and it is reversible only when the cell is well oxygenated. Once enough haemoglobin S molecules have docked together, they form long fibres that stretch the cell into the rigid crescent. A cell that sickles once can recover, but after repeated cycles it becomes permanently deformed, dense, and sticky, and it is this old, damaged cell that blocks vessels and breaks down. The triggers are anything that slows the blood, lowers the oxygen, or makes the blood more acid, and the common ones in childhood are fever, dehydration, hypoxia, acidosis, and cold. [11]
Two consequences flow from the sickling, and they explain almost everything the fellow sees at the bedside. The first is vaso-occlusion, in which the rigid, sticky sickled cells jam in the small vessels and the slow, sticky flow that follows recruits more sickled cells and white cells, producing ischaemia and the pain that is the hallmark of the disease. The second is chronic haemolysis, in which the damaged red cells are destroyed at an accelerated rate, producing the chronic anaemia, the jaundice, and the high reticulocyte count. The spleen, the lungs, the brain, and the kidneys are the organs that suffer first and most. [11]
The protection that fetal haemoglobin provides is the key that unlocks the treatment. Fetal haemoglobin, haemoglobin F, takes over the oxygen carrying in the fetus and falls in the first months of life, which is why the newborn with sickle cell disease is well at birth and becomes ill only as the haemoglobin F falls. Haemoglobin F blocks the polymerisation of haemoglobin S, because it does not carry the sickle mutation and it dilutes the concentration of haemoglobin S in the cell. This is the very reason that hydroxyurea works, because it raises the haemoglobin F level and so slows the sickling, and it is why the timing of treatment follows the fall in fetal haemoglobin. [2]
Clinical Presentation
The newborn with sickle cell disease is well, and this is the central clinical fact that makes newborn screening so powerful. Because fetal haemoglobin protects the cell, the infant is protected in the first months, and the complications begin only as the fetal haemoglobin falls through the second half of the first year. The first sign is often dactylitis, the hand-foot syndrome, a painful swelling of the hands and the feet that appears between six months and two years, and it is often the event that first brings the undiagnosed infant to medical attention in places without screening. [9]
As the fetal haemoglobin falls, the chronic anaemia declares itself. The child is pale, the conjunctivae are washed out, and there is a mild scleral icterus from the chronic haemolysis. The haemoglobin in HbSS typically sits between 60 and 90 g per litre, and the child runs a reticulocyte count of 5 to 15 percent as the marrow works hard to replace what is broken down. The infant cohort of the BABY HUG study showed that even in the first year the severity of the anaemia tracks closely with the burden of clinical events, which is the physiological basis for treating early. [9]
The acute presentations, which are covered in depth in the sister topic on acute complications, are the events that health maintenance exists to prevent. Acute splenic sequestration, in which blood pools suddenly in the spleen, strikes the infant under three years before the spleen has fully infarcted. Overwhelming pneumococcal sepsis strikes the functionally asplenic child and was the chief killer before penicillin prophylaxis. Acute chest syndrome, a combination of sickling, infection, and fat embolism in the lungs, is the leading cause of death after the first decade. Stroke, both overt and silent, struck roughly one in ten children before transcranial Doppler screening. [5]
The chronic picture is one of a child growing up with a multisystem disease. Growth is often delayed, puberty is late, and the child may miss school for pain episodes. The spleen shrinks through infancy until it is no longer palpable, while the heart enlarges to pump the thin anaemic blood. Gallstones from the chronic haemolysis appear from late childhood, the kidneys lose concentrating ability and cause enuresis, and the retina develops sickle retinopathy. Recognising the whole child, not only the crises, is the work of the long-term clinic. [11]
Differential Diagnosis
The child with a low haemoglobin, a high reticulocyte count, and jaundice has a chronic haemolytic anaemia, and the fellow must separate sickle cell disease from the other causes. The blood film and the haemoglobin electrophoresis are the two tests that settle the direction, because the sickled cells and the haemoglobin S peak are diagnostic, and they are present from birth. The other haemolytic anaemias have their own film and their own electrophoresis picture. [11]
Sickle cell disease (HbSS)
sickled cells on film
- Autosomal recessive, p.Glu6Val mutation, HbS over 80 percent
- Chronic haemolytic anaemia, haemoglobin 60 to 90 g per litre
- Sickled cells and target cells on the blood film
- Painful crises, dactylitis, splenic sequestration
Haemoglobin SC disease
milder compound heterozygote
- One sickle gene and one haemoglobin C gene
- Milder anaemia, fewer crises, fewer strokes
- Target cells and rare sickled cells on the film
- Higher risk of retinopathy and splenic sequestration
Sickle beta-thalassaemia
co-inherited thalassaemia
- One sickle gene and one beta-thalassaemia gene
- Beta-zero runs like HbSS, beta-plus is milder
- Low mean corpuscular volume and target cells
- Elevated haemoglobin A2 and haemoglobin F point to thalassaemia
Hereditary spherocytosis
membrane haemolysis
- Autosomal dominant, spherocytes on the film
- Negative sickle solubility and normal electrophoresis
- Osmotic fragility increased, extravascular haemolysis
- No sickled cells, no haemoglobin S
Hereditary spherocytosis is the membrane disorder most often confused with sickle disease, because both produce a chronic haemolytic anaemia with jaundice and gallstones, but the film shows spherocytes rather than sickled cells and the electrophoresis is normal. Glucose-6-phosphate dehydrogenase deficiency produces episodes of haemolysis and anaemia, but it is X-linked, it is episodic and triggered by drugs or fava beans, and the electrophoresis is again normal. The autoimmune haemolytic anaemias give a positive direct antiglobulin test, which sickle disease never does. [11]
The chief pitfall for the fellow is the child whose electrophoresis shows both haemoglobin S and another abnormality, because the compound heterozygous diseases behave differently from HbSS. The mean corpuscular volume is the first clue to co-inherited thalassaemia, because a low mean corpuscular volume in a child with haemoglobin S points to sickle beta-thalassaemia rather than HbSS, and the haemoglobin A2 level confirms it. The message for the exam is that the diagnosis is settled by the combination of the blood film and the haemoglobin electrophoresis, and the genotyping shapes the prognosis and the surveillance. [11]
Clinical & Bedside Assessment
The assessment of the child with sickle cell disease at the clinic visit is a systematic search for the complications the surveillance is built to catch. The visit begins with the history, and the two questions that matter most are whether the child has had any pain, any fever, and any change in colour or energy since the last visit, and whether the prophylaxis is being taken. Adherence to penicillin and to hydroxyurea is asked about directly, because missed doses are the commonest reason for breakthrough infection or escalating disease. [5]
The examination looks for the signs of chronic haemolysis, of organ injury, and of growth. The conjunctivae are checked for pallor and the sclerae for icterus, the abdomen is palpated for the spleen and the liver, and the heart is auscultated for the flow murmur of the chronic anaemia. The spleen is the organ the fellow follows most closely in the infant, because a sudden enlargement is splenic sequestration, and a shrinking then absent spleen is the functional asplenia that drives the prophylaxis. Growth is plotted at every visit, because faltering growth and delayed puberty are common. [5]
The structured clinic visit for a child with sickle cell disease
Ask about pain, fever, pallor, and adherence to penicillin and hydroxyurea since the last visit
Plot the weight, the height, and the body mass index, and check the pubertal stage for the delayed growth of chronic disease
Examine the conjunctivae for pallor and the sclerae for icterus, the abdomen for the spleen and the liver, and the heart for the flow murmur
Teach the parents to palpate the spleen and to recognise the pallor and lethargy of splenic sequestration
Review the full blood count and the reticulocyte count, and book the annual transcranial Doppler between ages two and sixteen
Confirm the immunisations are up to date, and refill the penicillin and the hydroxyurea
The teaching of the parents to recognise the danger signs is a clinical act as important as any examination finding. The parents are taught to palpate the spleen and to bring the child in at once if it enlarges or if the child becomes pale, lethargic, or breathless, because these are the signs of acute splenic sequestration. They are taught that any fever above 38.5 degrees Celsius is an emergency, because the functionally asplenic child can be septicaemic within hours, and they are taught the sick-day rules for keeping the child warm, hydrated, and out of the cold. [5]
Investigations
The diagnosis of sickle cell disease is a laboratory diagnosis, and it is made by separating the haemoglobins. The first test, in the newborn period, is the heel-prick blood spot that is analysed by one of three methods: high-performance liquid chromatography, isoelectric focusing, or haemoglobin electrophoresis. Each method separates the haemoglobins by their physical properties, and each identifies the pattern of haemoglobin S, haemoglobin F, and the absence or near-absence of haemoglobin A that defines the disease. [5]
The confirmatory test is a repeat of the same method on a fresh sample, because the newborn screen is a screening test and the confirmation settles the genotype. The pattern in HbSS shows haemoglobin S and haemoglobin F with little or no haemoglobin A. The pattern in HbSC shows haemoglobin S and haemoglobin C. The pattern in sickle beta-thalassaemia shows haemoglobin S with a raised haemoglobin A2 and little haemoglobin A in the beta-zero form, or some haemoglobin A in the beta-plus form. When the result is ambiguous, deoxyribonucleic acid testing of the HBB gene settles the mutation directly. [5]
The timing and the method of newborn screening vary by region, but the principle is the same everywhere: identify the child before the fall in fetal haemoglobin brings the first complication. Australia, Aotearoa New Zealand, the United Kingdom, the United States, and Canada all include sickle cell disease in their universal newborn screening panels. The confirmatory methods and the reporting of carrier status differ, and the fellow should know the local schedule and the local cut-off ages for each intervention. [5]
The bloods that are tracked at every clinic visit are the full blood count, the reticulocyte count, and the haemoglobin S percentage, and each tells the fellow something different. The full blood count tracks the steady-state haemoglobin, typically 60 to 90 g per litre in HbSS, and it is the baseline against which a drop is judged. The reticulocyte count tracks the marrow response and is typically 5 to 15 percent. The haemoglobin S percentage, measured during transfusion programmes, tracks the effect of the transfusion, and the target during chronic transfusion for stroke prevention is under 30 percent. [5]
Management — Resuscitation
Sickle cell disease is a chronic disease, but it carries a small set of acute, time-critical dangers that the fellow must meet with the same discipline as a resuscitation. The first and the most feared in the young child is overwhelming sepsis. The functionally asplenic child who develops a fever above 38.5 degrees Celsius has overwhelming encapsulated sepsis until proven otherwise, and the response is a prompt parenteral broad-spectrum antibiotic that covers the pneumococcus, such as ceftriaxone, after a blood culture, together with assessment for shock and dehydration. The aim is antibiotic within one hour, because the death from pneumococcal sepsis in the asplenic child can be measured in hours. [5]
The second danger is acute splenic sequestration, which strikes the infant under three years before the spleen has fully infarcted. Blood pools suddenly in the enlarging spleen, the haemoglobin falls by 20 g per litre or more, the platelets fall, and the child becomes hypovolaemic. The response is a prompt transfusion to restore the circulating volume, and the parents are taught to bring the child in at the first sign of pallor, lethargy, or a rapidly enlarging spleen. Recurrent episodes often lead to splenectomy, and a single severe episode warrants a long-term transfusion programme or splenectomy in some centres. [5]
[5]The third danger is the impending stroke, which the transcranial Doppler is built to catch. A child with an abnormal transcranial Doppler, a time-averaged mean velocity of 200 cm per second or more, is in a pre-stroke state, and the response is the urgent commencement of a chronic transfusion programme to bring the haemoglobin S under 30 percent and to keep it there. A child who presents with an overt stroke, a sudden hemiparesis or seizure, is transfused urgently, often by exchange transfusion, and is then committed to a chronic transfusion programme for secondary prevention. [3][8]
Management — Definitive & Stepwise
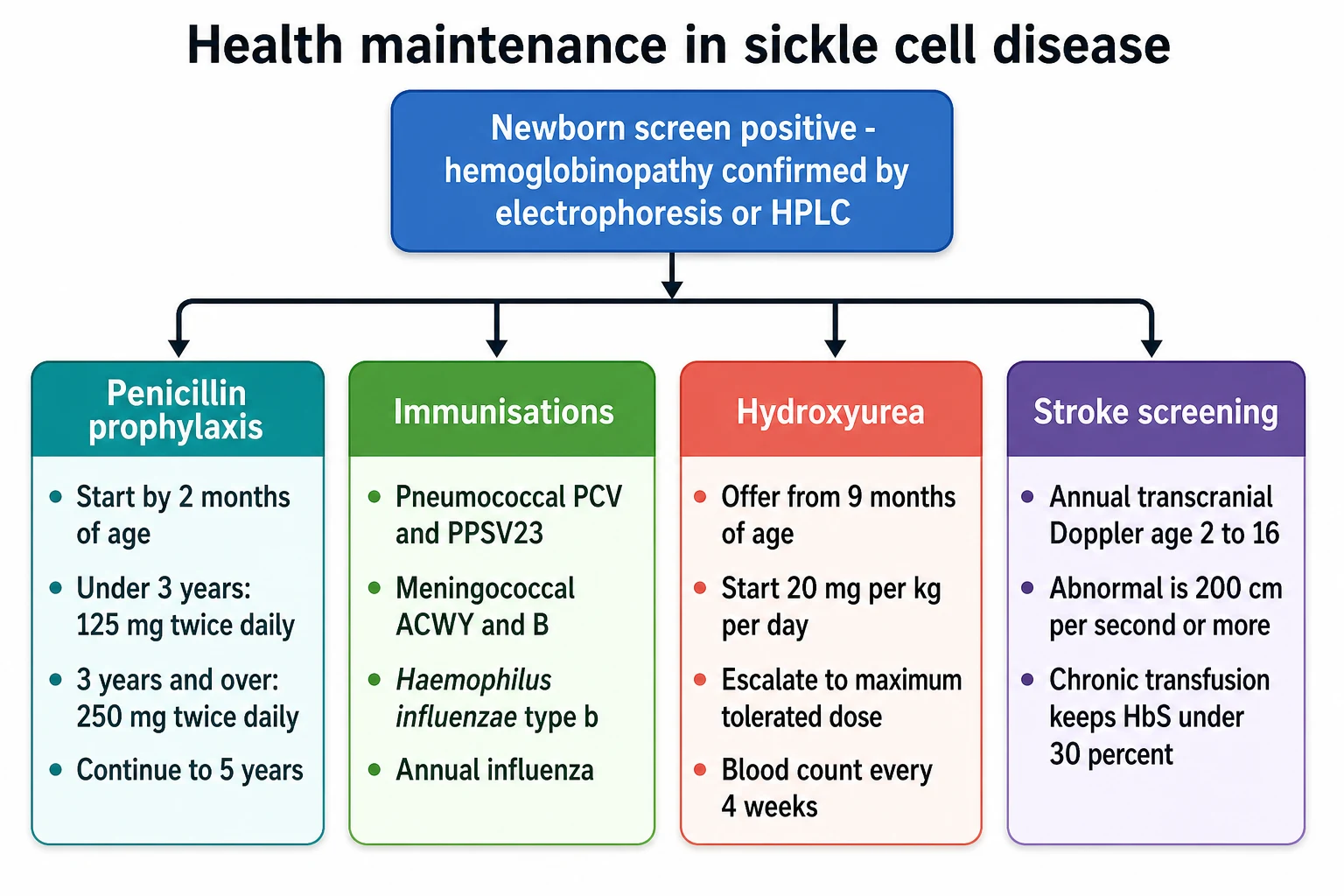
The definitive management of sickle cell disease is health maintenance, and it rests on four pillars that begin at diagnosis and run for life. The first pillar is penicillin prophylaxis, the intervention that more than any other transformed the survival of these children. The randomised PROPS trial of Gaston and colleagues, published in 1986, showed that oral penicillin reduced the incidence of pneumococcal sepsis by 84 percent in children with sickle cell anaemia, and the trial was stopped early because the benefit was so clear. The standard is to begin oral penicillin V by two months of age, and the dose is 125 mg twice daily under three years and 250 mg twice daily at three years and over. [1]
Penicillin V prophylaxis for sickle cell disease
Dose
125 mg orally twice daily for children under three years, and 250 mg orally twice daily for children three years and over, started by two months of age and continued to five years
The question of when to stop penicillin is asked in every exam. The National Heart, Lung, and Blood Institute panel recommends continuing prophylaxis to at least five years of age in HbSS, and the decision to stop at five is made for the child who is fully immunised against the pneumococcus, who has had no invasive pneumococcal infection, and who has reliable access to care. Prophylaxis is continued beyond five years for the child who has had a splenectomy, a prior invasive pneumococcal infection, or who is on immunosuppression, and many centres continue it through childhood as a matter of local policy. [5]
The second pillar is immunisation against the encapsulated organisms, and it runs alongside the routine childhood schedule. The functional asplenia leaves the child defenceless against the pneumococcus, the meningococcus, and Haemophilus influenzae type b, so these are given in addition to the routine vaccines. The pneumococcal cover combines the conjugate vaccine on the routine schedule with the 23-valent polysaccharide vaccine given at two years and again five years later, the meningococcal cover adds the conjugated ACWY vaccine and the serogroup B vaccine, and the child also receives the annual influenza vaccine. [5]
Encapsulated-organ immunisation in sickle cell disease
Dose
Routine pneumococcal conjugate vaccine series, then 23-valent pneumococcal polysaccharide vaccine at two years with a second dose five years later; meningococcal ACWY and serogroup B vaccines; Haemophilus influenzae type b; and annual influenza vaccine
The third pillar is hydroxyurea, the disease-modifying drug that is now offered to nearly every child with HbSS or sickle beta-zero-thalassaemia. The Multicenter Study of Hydroxyurea, published by Charache and colleagues in 1995, showed in adults that hydroxyurea reduced the frequency of painful crises, the acute chest syndrome, and the transfusion requirement, and the long-term follow-up by Hankins and colleagues confirmed sustained benefit and acceptable safety from infancy through adolescence. The current recommendation is to offer hydroxyurea to all children with HbSS or sickle beta-zero-thalassaemia from nine months of age, regardless of severity, because the drug changes the trajectory of the disease. [2][6]
Hydroxyurea for sickle cell disease in children
Dose
Starting dose 20 mg per kg per day orally once daily, escalated by 5 mg per kg per day every eight weeks to the maximum tolerated dose, typically 25 to 35 mg per kg per day, with a full blood count every four weeks
The mechanism of hydroxyurea explains both its power and its monitoring. The drug raises the fetal haemoglobin level, which dilutes the haemoglobin S and blocks its polymerisation, and it lowers the white cell count, which reduces the inflammation that drives vaso-occlusion. The dose is escalated every eight weeks to the maximum tolerated dose, the highest dose that keeps the blood counts safe, and the full blood count is checked every four weeks during the escalation and the maintenance. The goal is a fetal haemoglobin level as high as can be achieved and a steady reduction in the number of crises and the transfusion requirement. [2][6]
The fourth pillar is the surveillance of stroke by transcranial Doppler, and it is the intervention that turned stroke from a common complication into a preventable one. The STOP trial, published by Adams and colleagues in 1998, showed that children with an abnormal transcranial Doppler, a time-averaged mean velocity of 200 cm per second or more, had a 90 percent reduction in first stroke when started on a chronic transfusion programme. The annual screening is done from ages two to sixteen, and the abnormal result triggers the chronic transfusion that keeps the haemoglobin S under 30 percent. [3][8]
[5] [3]The TWiTCH trial extended the transfusion strategy and gave the fellow an exit from lifelong iron overload. Ware and colleagues showed in 2016 that a child who had been on chronic transfusion for an abnormal transcranial Doppler for at least a year, with a normal current Doppler and no silent cerebral infarct, could be switched to hydroxyurea without an increase in stroke risk. The transfusion is continued for long enough to suppress the abnormal flow, the hydroxyurea is escalated to the maximum tolerated dose, and the phlebotomy that follows reduces the iron load. The non-inferiority of hydroxyurea means many children can stop transfusion and the iron chelation that goes with it. [7]
Chronic transfusion programme for stroke prevention
Dose
Simple or exchange transfusion every three to four weeks to keep the haemoglobin S under 30 percent and the haemoglobin around 90 to 100 g per litre
Specific Subtypes & Scenarios
Haemoglobin SC disease runs a milder course than HbSS, and the fellow must know both what it shares and what it changes. The child has a less severe anaemia, a haemoglobin that often sits between 90 and 110 g per litre, fewer painful crises, and a lower risk of stroke. The spleen often remains palpable for longer, so acute splenic sequestration and splenic infarction remain a risk into later childhood. Two complications are more prominent in SC disease than in HbSS: proliferative sickle retinopathy, which needs ophthalmic surveillance, and avascular necrosis of the femoral head. Hydroxyurea is used for the symptomatic child, and transfusion is reserved for the acute complication. [11]
Sickle beta-thalassaemia comes in two forms that differ in severity, and the difference rests on how much normal beta-globin the thalassaemia gene allows. The beta-zero form makes no normal beta-globin, so the disease runs a course close to HbSS, with the same anaemia, the same stroke risk, and the same need for penicillin, hydroxyurea, and transcranial Doppler. The beta-plus form makes some normal beta-globin, so the disease runs milder, with a less severe anaemia and fewer crises. The mean corpuscular volume is low in both, a clue that distinguishes sickle beta-thalassaemia from HbSS, and the haemoglobin A2 is raised. [11]
The first two years of a child diagnosed on newborn screening
The adolescent with sickle cell disease stands at the junction of the paediatric and the adult service, and the fellow is often the one who plans the crossing. Hydroxyurea is discussed for its teratogenicity and the need for reliable contraception, the young woman is counselled on the higher risk of a sickle crisis in pregnancy and the need for joint care, and the transition to the adult haemoglobinopathy service is planned early and run over years rather than handed off overnight. The aim is a young person who knows the diagnosis, carries the prophylaxis, and arrives in adult care connected to a service rather than lost to follow-up. [5]
Complications & Pitfalls
The first pitfall is stopping penicillin too early or letting it lapse. The protection of penicillin prophylaxis depends on the child taking the twice-daily dose, and the family that stops at the first sign of wellness, or that misses doses through poor access or competing demands, loses the protection the PROPS trial established. The fellow asks about adherence at every visit, refills the prescription before it runs out, and frames the prophylaxis as the single most important thing the family does in the early years. [1][5]
The second pitfall is the myelosuppression of hydroxyurea. The drug is escalated to the maximum tolerated dose, and the blood count is checked every four weeks precisely because the dose that helps is close to the dose that suppresses the marrow. A falling neutrophil count under 2.0 times ten to the nine per litre, a falling platelet count, or a falling haemoglobin is a sign to hold or reduce the dose, and the dose is then re-titrated at a lower level. The fellow who escalates without monitoring, or who does not act on the cytopenia, will run the child into a neutropenic sepsis or a bleeding event. [2][5]
[4]The third pitfall is the iron overload of chronic transfusion, which is the price paid for the protection of the transfusion programme. Each unit of blood delivers about 200 mg of iron, and the body has no way to excrete it, so the iron accumulates in the liver, the heart, and the endocrine glands and causes cirrhosis, cardiomyopathy, and endocrine failure. The iron is monitored with a ferritin and a liver iron content, and chelation is started when the ferritin exceeds 1000 micrograms per litre. Deferasirox is the usual first-line chelator in children, with deferiprone and deferoxamine as alternatives. The TWiTCH switch to hydroxyurea, where it is possible, is the best way to stop the iron accumulating. [7][8]
The fourth pitfall is alloimmunisation, the formation of antibodies against the donor red cells, which is common in the transfused child with sickle cell disease because of the different red cell antigen profiles of donor and recipient populations. Alloimmunisation makes future cross-matching harder and can cause haemolytic transfusion reactions, so the child is phenotyped for the major red cell antigens before the first transfusion and is matched for them wherever possible. [5]
Prognosis & Disposition
The modern outlook for a child with sickle cell disease is one of survival into adulthood, and this is the central fact the fellow carries into every counselling. The paper of Quinn and colleagues, published in 2010, showed that the survival of children and adolescents with sickle cell disease in a modern American cohort had risen to over 90 percent to age eighteen, and the improvement was driven by the very package of newborn screening, penicillin prophylaxis, and transcranial Doppler surveillance that this topic describes. The death that was once common in early childhood is now rare, and the chief threats have shifted to the adult years. [10]
The disposition of the child is shared between the primary care team and the sickle cell centre, and the fellow coordinates the two. The primary care team runs the routine immunisations, the growth and development, and the acute presentations, while the sickle cell centre runs the annual review, the transcranial Doppler, the hydroxyurea, and the transfusion programme. The child with HbSS or sickle beta-zero-thalassaemia is followed in the centre from diagnosis, and the child with milder disease is followed more lightly but never lost. [5]
The stroke-free survival, in particular, is the measure of the transcranial Doppler programme. The annual screen, the prompt transfusion of the abnormal Doppler, and the TWiTCH switch where possible mean that the overt stroke, once a common event, is now preventable, and the silent cerebral infarct, once undetected, is now the focus of active research. The long-term outlook for the child who stays on the programme is a childhood without a stroke and an adult life with a brain intact. [3][8]
Special Populations
The migrant, the refugee, and the Indigenous family carry the extra burden of access, and the fellow who works in a region with a mixed population must plan for it. The child born in a country without newborn screening may arrive undiagnosed, and the first presentation may be the first crisis or the first stroke, so the fellow has a low threshold to test the haemoglobins of any child from a high-prevalence population with an unexplained anaemia or pain. The family with limited language, limited transport, or limited trust in the service needs the prophylaxis and the appointments organised around them, not the other way around. [5]
The adolescent girl who is thinking about pregnancy carries a set of risks that the fellow addresses before, not after, the conception. Sickle cell disease raises the risk of a pain crisis, of acute chest syndrome, and of pre-eclampsia in pregnancy, and the pregnancy is managed jointly with the obstetric and the haematology teams. Hydroxyurea is stopped before a planned pregnancy because of its teratogenicity, and the young woman is counselled on reliable contraception while she is on the drug. The partner is offered testing, because the carrier status of the partner sets the risk for the child. [5]
SICKLE
The child in a rural or remote setting, far from a sickle cell centre, is managed by the local team in partnership with the centre through telehealth and shared protocols. The local team holds the emergency plan for fever and for sequestration, keeps the penicillin and the immunisations running, and ships the child to the centre for the annual review and the transcranial Doppler. The fellow who works in such a setting values the local network, because the child who is connected to the centre through it does as well as the child who lives next door to it. [5]
Evidence, Guidelines & Regional Differences
The evidence base for health maintenance in sickle cell disease is one of the strongest in paediatric haematology, and it is built on four landmark trials. The PROPS trial of 1986 established penicillin prophylaxis, the Multicenter Study of Hydroxyurea of 1995 established hydroxyurea in adults, the STOP trial of 1998 established transfusion for the abnormal transcranial Doppler, and the TWiTCH trial of 2016 established the switch to hydroxyurea. Each trial changed practice, and each is the answer the fellow gives when asked for the evidence. [1][2][3][7]
PROPS - penicillin prophylaxis, Gaston 1986
Key finding
Oral penicillin reduced the incidence of pneumococcal sepsis by 84 percent in children with sickle cell anaemia, and the trial was stopped early for benefit.
STOP - transcranial Doppler and transfusion, Adams 1998
Key finding
Chronic transfusion reduced the risk of first stroke by about 90 percent in children with an abnormal transcranial Doppler of 200 cm per second or more.
TWiTCH - transfusion to hydroxyurea, Ware 2016
Key finding
Hydroxyurea was non-inferior to chronic transfusion for maintaining transcranial Doppler velocities in children already transfused for a year with a normal Doppler and no silent infarct.
The guidelines that translate this evidence into practice differ slightly by region, and the fellow names the one being followed. The National Heart, Lung, and Blood Institute panel report of 2014, summarised by Yawn and colleagues, is the most widely used comprehensive guideline, and it sets the penicillin doses, the hydroxyurea timing, and the transcranial Doppler schedule. The American Society of Hematology 2020 guideline on cerebrovascular disease, led by DeBaun, sets the stroke prevention detail. The British Society for Haematology and the United Kingdom Screening Committee set the practice in the United Kingdom, and the local newborn screening programme sets the diagnosis. [5][8]
The chief controversy is the timing of penicillin cessation and the place of universal hydroxyurea. Some centres stop penicillin at five years for the fully immunised child with no prior sepsis, while others continue it through childhood, and the evidence supports either when access to care is good. The move to offer hydroxyurea to every child with HbSS from nine months, regardless of severity, is now the consensus, having shifted from a severity-based to a universal approach, and the long-term safety data of Hankins underpin it. The fellow follows the local guideline and explains the choice to the family. [5][6]
Exam Pearls
The single most testable fact is the amino acid change, so learn it word for word. The mutation substitutes a valine for the glutamate at the sixth position of the beta-globin chain, written as p.Glu6Val, and it is caused by a point mutation in the HBB gene. The fellow who can state this exactly, and who can explain that the valine is hydrophobic and so exposes a sticky patch on deoxygenation, has the molecular heart of the disease. [11]
The penicillin doses are the second most testable set, and they are asked as a pair. Under three years the dose is 125 mg twice daily, and at three years and over it is 250 mg twice daily, started by two months of age and continued to five years. The fellow who gives only the start age without the doses, or who confuses the two age bands, loses the marks. The PROPS trial name and the 84 percent reduction in pneumococcal sepsis are the supporting evidence. [1][5]
[3] [7]The hydroxyurea facts round out the high-yield set. The starting dose is 20 mg per kg per day, it is escalated by 5 mg per kg per day every eight weeks to the maximum tolerated dose, and the full blood count is checked every four weeks. The drug is offered to every child with HbSS or sickle beta-zero-thalassaemia from nine months of age, it works by raising the fetal haemoglobin, and the myelosuppression is the toxicity to watch. The fellow who holds these four numbers, the penicillin doses, the TCD threshold, the transfusion target, and the hydroxyurea dose, carries the whole topic. [2][5]
References
- [1]Gaston MH, Verter JI, Woods G Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med, 1986.PMID 3086721
- [2]Charache S, Terrin ML, Moore RD Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med, 1995.PMID 7715639
- [3]Adams RJ, McKie VC, Hsu L Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med, 1998.PMID 9647873
- [4]Abboud MR, Yim E, Musallam KM Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood, 2011.PMID 21633086
- [5]Yawn BP, Buchanan GR, Afenyi-Annan AN Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA, 2014.PMID 25203083
- [6]Hankins JS, Aygun B, Nottage K From infancy to adolescence: fifteen years of continuous treatment with hydroxyurea in sickle cell anemia. Medicine (Baltimore), 2014.PMID 25526439
- [7]Ware RE, Davis BR, Schultz WH Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open-label, phase 3, non-inferiority trial. Lancet, 2016.PMID 26670617
- [8]DeBaun MR, Jordan LC, King AA American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Adv, 2020.PMID 32298430
- [9]Lebensburger JD, Miller ST, Howard TH Influence of severity of anemia on clinical findings in infants with sickle cell anemia: analyses from the BABY HUG study. Pediatr Blood Cancer, 2012.PMID 22190441
- [10]Quinn CT, Rogers ZR, McCavit TL Improved survival of children and adolescents with sickle cell disease. Blood, 2010.PMID 20194891
- [11]Rees DC, Williams TN, Gladwin MT Sickle-cell disease. Lancet, 2010.PMID 21131035