Paeds · haematology-oncology-and-transfusion
Thalassaemia syndromes
Also known as Beta-thalassaemia major · Cooley anaemia · Thalassaemia major · Alpha-thalassaemia · Haemoglobin H disease · Thalassaemia intermedia
Fellowship guide to the thalassaemia syndromes in children, the inherited disorders of globin-chain synthesis that range from the transfusion-dependent beta-thalassaemia major of early childhood to the lethal Hb Bart hydrops fetalis of four-gene alpha-thalassaemia. Covers the pathophysiology of unbalanced globin production, ineffective erythropoiesis and chronic haemolysis, the diagnosis by haemoglobin electrophoresis or high-performance liquid chromatography with a raised haemoglobin F, and the three pillars of major-disease care: regular leucodepleted transfusion keeping the pre-transfusion haemoglobin at 90 to 100 g per litre every two to five weeks, iron chelation with deferasirox started at 20 mg per kg per day once the ferritin exceeds 1000 micrograms per litre, and curative haematopoietic stem cell transplant or betibeglogene autotemcel gene therapy. Includes the alpha-thalassaemia gene-deletion ladder, transfusional iron overload monitored by cardiac T2 star magnetic resonance imaging, the luspatercept BELIEVE trial, and the alpha-thalassaemia, HbE beta-thalassaemia and thalassaemia intermedia subtypes.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A child born with thalassaemia makes haemoglobin that is structurally normal but quantitatively wrong, because one of the two globin protein chains is produced too slowly or not at all. The name covers a family of inherited anaemias in which the balance between alpha and beta globin is lost, the spare chains clump and destroy the red cell from within, and the child is left with severe, chronic, microcytic anaemia. The commonest and most severe form is beta-thalassaemia major, once called Cooley anaemia, in which the child makes little or no beta-globin and becomes dependent on lifelong blood transfusion from infancy. [12]
The thalassaemias are among the most common single-gene disorders in the world, and they cluster wherever malaria was historically intense, across the Mediterranean, the Middle East, South Asia and South-East Asia, because carrying one thalassaemia gene once protected a child against falciparum malaria. The severity spans a wide range, from the silent carrier who is never unwell, through the transfusion-dependent major, to the four-gene alpha-thalassaemia that kills the fetus in utero as Hb Bart hydrops fetalis. The paediatric fellow meets the whole spectrum, but the centre of gravity is the child with major disease whose life depends on a transfusion and chelation programme that begins in the first year. [12]
Three ideas make this topic central to the exam. The first is the molecular imbalance between the globin chains, because the unpaired alpha chains explain the ineffective erythropoiesis, the chronic haemolysis and the bone expansion that together shape the disease. The second is the transfusion programme, because a regular transfusion that keeps the pre-transfusion haemoglobin at 90 to 100 g per litre suppresses the abnormal marrow and lets the child grow. The third is iron chelation, because every transfusion loads the body with iron it cannot excrete, and the heart, the liver and the endocrine glands fail if that iron is not removed. The 2026 primer of Piel and colleagues and the landmark chelation trials together define modern care. [12][4]
Classification
The thalassaemias sort themselves by which globin gene is affected and by how many genes are lost, and the two questions together set the severity. The beta-thalassaemias arise from mutations in the HBB gene on chromosome 11, and because each child carries two beta genes the disease is either present or absent in its major form. A child who inherits two severe alleles makes little or no beta-globin and has beta-thalassaemia major, transfusion-dependent from infancy. A child who inherits milder alleles, or one severe and one mild allele, makes enough beta-globin to survive without regular transfusion and has beta-thalassaemia intermedia. The carrier who carries one abnormal allele and one normal allele has beta-thalassaemia trait, microcytic but well. [12]
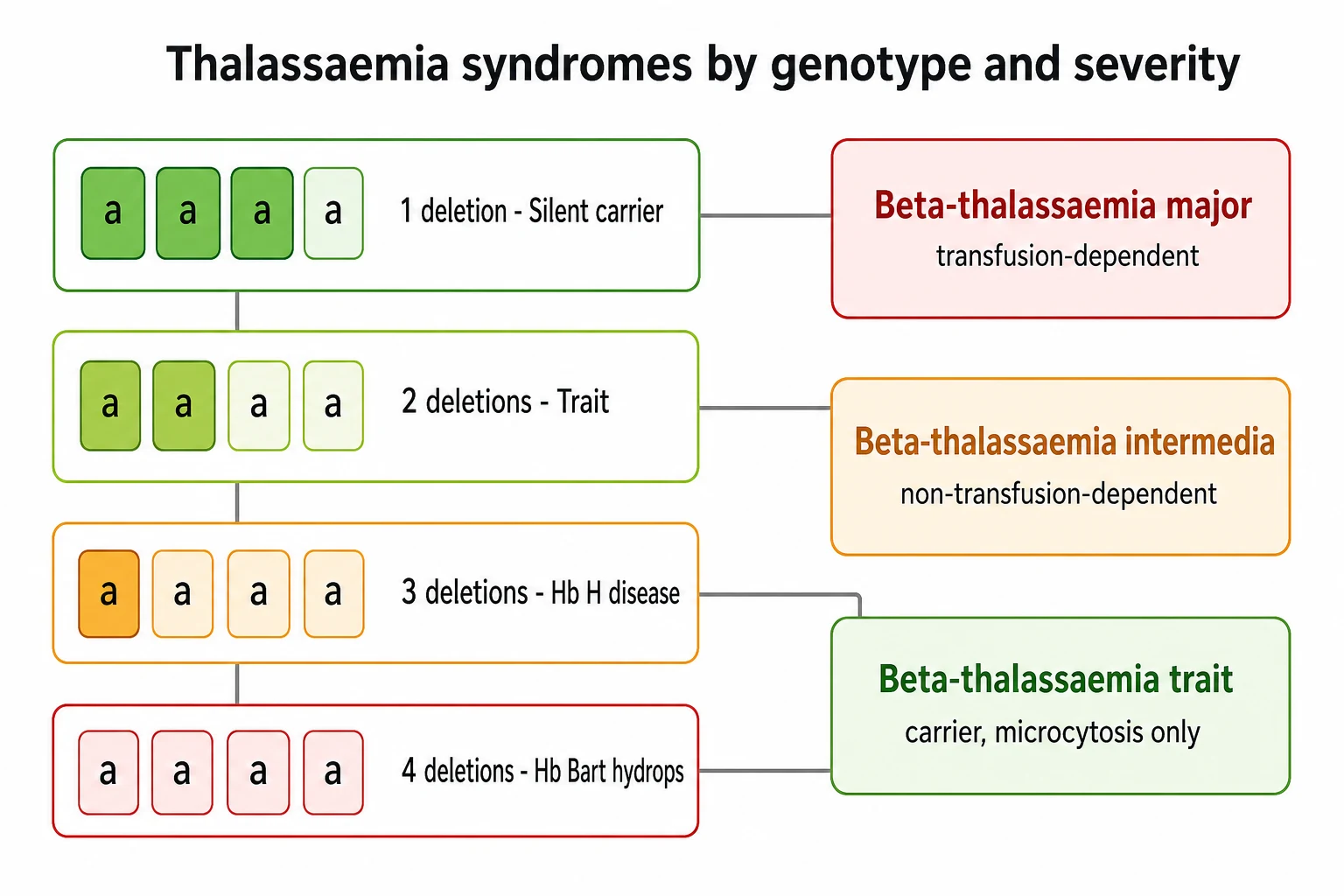
The alpha-thalassaemias run on a different logic, because there are four alpha genes arranged in pairs on chromosome 16, and the severity rises stepwise with the number deleted. The loss of one gene is the silent carrier state, microcytosis so mild it is never noticed. The loss of two genes is alpha-thalassaemia trait, a mild microcytic anaemia that is the form most often found on a routine blood count. The loss of three genes is haemoglobin H disease, a moderate chronic haemolytic anaemia in which the unpaired beta chains join in fours to form an unstable haemoglobin that precipitates in the red cell. The loss of all four genes is Hb Bart hydrops fetalis, in which the fetus cannot make functional haemoglobin and dies in utero or survives only with intrauterine transfusion. [9]
A third, clinically important genotype sits across the boundary of beta-thalassaemia and a structural variant, and it is the commonest form of severe thalassaemia in much of South-East Asia. Haemoglobin E is a beta-chain variant that is itself synthesised poorly, and when it is inherited alongside a beta-thalassaemia allele the result is HbE beta-thalassaemia, a disease of widely varying severity that is often transfusion-dependent. The fellow who works in a region with migrants from South-East Asia meets this genotype often, and the message for the exam is that the clinical severity is set by the net output of beta-globin, whether the cause is two thalassaemia alleles or a thalassaemia allele paired with haemoglobin E. [12]
Epidemiology & Risk Factors
The thalassaemias are the most common monogenic disorders in humans, and the carrier frequencies are highest where malaria was once endemic, because a single thalassaemia gene reduced the severity of falciparum infection in early childhood. The beta-thalassaemia carrier rate reaches several percent across the Mediterranean, the Middle East and South Asia, and the alpha-thalassaemia carrier rate is even higher in parts of South-East Asia and southern China, where the four-gene deletion that causes Hb Bart hydrops fetalis remains a leading cause of perinatal mortality. The global burden falls hardest on the regions least able to afford the transfusion and chelation programmes that keep a child alive. [12]
The inheritance is autosomal recessive in both the alpha and the beta forms, and the family risk is the risk the fellow traces at the bedside. A carrier mother and a carrier father each pass one abnormal beta gene to a child with a one in two chance, and the chance that a child inherits both and has major disease is one in four. The alpha disorders inherit on the same principle but on four genes, so the risk of Hb Bart hydrops fetalis is one in four only when both parents carry the deletion in cis, the so-called cis-deletion that is common in South-East Asia. Consanguinity raises the chance that both parents carry the same allele, and it is the reason a family history of anaemia, splenomegaly or early death is asked about directly. [12]
The single most important determinant of outcome, beyond the genotype, is access to safe blood and to chelation. A child born into a health system with screened, leucodepleted blood, viral-tested donors and affordable oral chelation can expect to reach adulthood, while a child born where these are scarce dies of untreated anaemia, of transfusion-transmitted infection, or of the cardiac failure of unchelated iron overload. The gap in outcome is the reason carrier screening, prenatal diagnosis and counselling sit at the public-health heart of thalassaemia care, and the reason the fellow frames the disease as much as a matter of access as of genes. [6]
Pathophysiology
The defect at the centre of every thalassaemia is an imbalance between the globin chains, and the consequences of that imbalance explain the whole clinical picture. In beta-thalassaemia the beta-globin chain is made too slowly or not at all, while the alpha chain continues to be produced at the normal rate, so the red cell precursors fill with unpaired alpha chains. These spare alpha chains are unstable, they precipitate inside the developing red cell, and they trigger the death of the precursor before it ever leaves the bone marrow. This death of red cell precursors within the marrow is called ineffective erythropoiesis, and it is the dominant cause of the anaemia. [12]
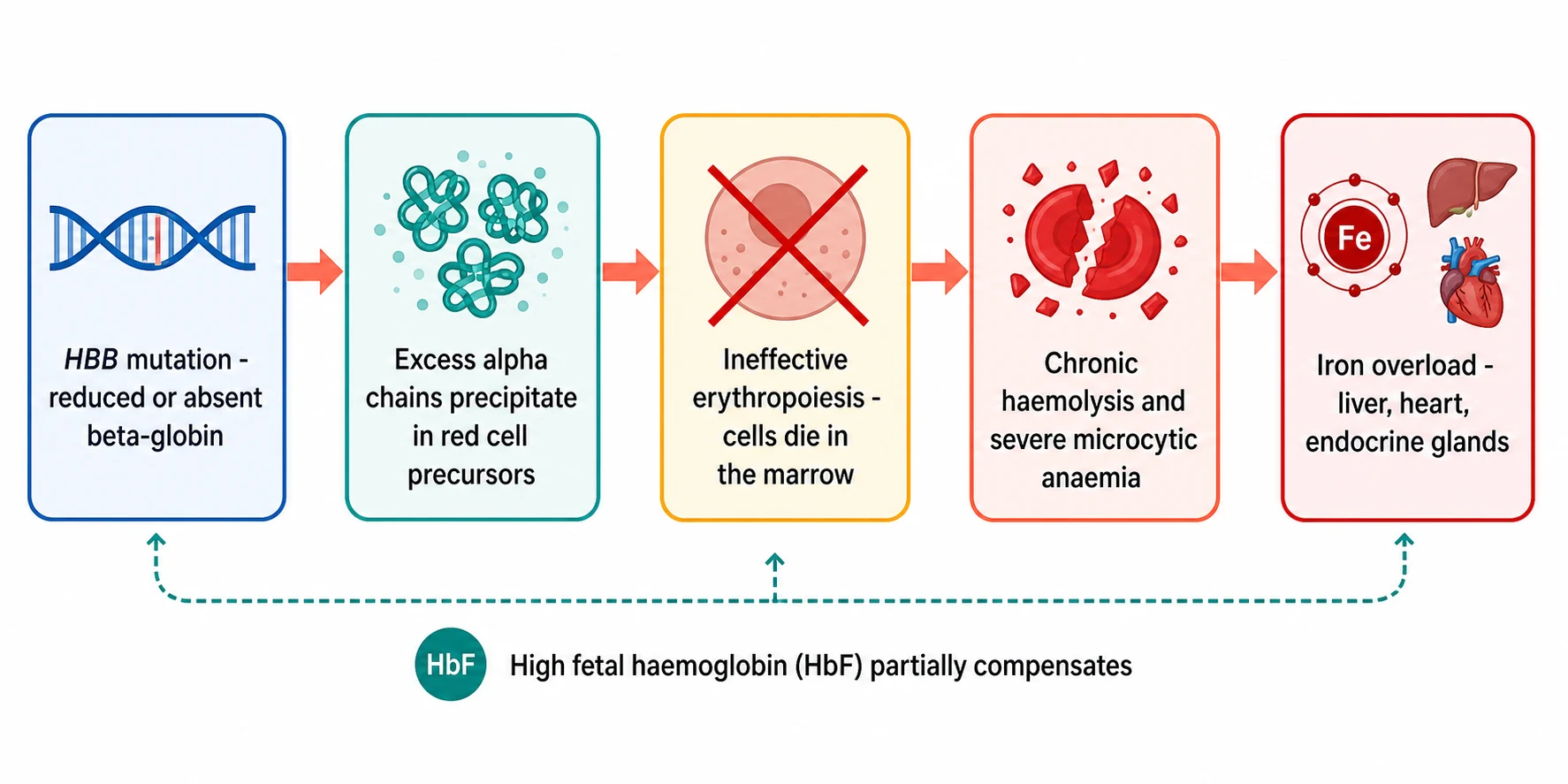
The red cells that do reach the circulation are abnormal and fragile, and they are destroyed prematurely in the spleen, a process of chronic haemolysis that adds to the anaemia and drives the hepatosplenomegaly. The marrow, starved of adequate oxygen delivery, expands to try to make more red cells, and the expansion thins the bones, distorts the face and shifts the blood picture towards the classic skeletal changes of untreated disease. Fetal haemoglobin, made of two alpha and two gamma chains, takes over some of the oxygen carrying because the gamma chain is produced normally, which is why the newborn is well at birth and becomes ill only as the fetal haemoglobin falls through the first year, and why drugs that raise fetal haemoglobin are part of modern treatment. [12]
Two further consequences complete the pathophysiology and both are iatrogenic. The first is transfusional iron overload, because each unit of red cells delivers about 200 mg of iron and the body has no physiological route to excrete it, so the iron accumulates in the liver, the heart and the endocrine glands and causes cirrhosis, cardiomyopathy and endocrine failure. The second is increased gastrointestinal iron absorption, because the ineffective erythropoiesis signals the gut to take up more iron, so that even the child who is not transfused, such as one with thalassaemia intermedia, slowly loads iron over the years. The iron, more than the anaemia, is what kills the adult with thalassaemia. [11][7]
Clinical Presentation
The newborn with beta-thalassaemia major is well, and this is the clinical fact that mirrors sickle cell disease and that explains the delayed presentation. Fetal haemoglobin carries the oxygen in the first months, so the infant looks healthy at birth, and the anaemia declares itself only as the fetal haemoglobin falls through the second half of the first year. The typical presentation is therefore between six and twelve months, with a pale, irritable infant who feeds poorly, fails to thrive, and is found to have a severe microcytic anaemia, often below 70 g per litre, with a large liver and spleen. In a region without newborn screening this is the event that brings the child to diagnosis. [6]
The untreated child who presents late carries the skeletal stigmata of a marrow fighting to expand, and these are the signs the exam still tests even though they are now rare wherever transfusion is available. The skull develops frontal bossing and a crew-cut appearance on the radiograph as the marrow pushes the bone outward, the maxilla enlarges to give the chipmunk facies with dental malocclusion, and the long bones thin and fracture easily. These changes are the visible record of ineffective erythropoiesis left unchecked, and they recede once a regular transfusion programme suppresses the marrow drive, which is why the fellow aims to start transfusion before the skeleton is reshaped. [6]
The alpha-thalassaemias present on a different timeline that follows the number of genes lost. The silent carrier and the trait carrier are never clinically unwell, and the finding is a microcytosis on a routine count that does not respond to iron. Haemoglobin H disease presents in childhood with a moderate chronic haemolytic anaemia, a haemoglobin typically between 70 and 100 g per litre, intermittent jaundice and splenomegaly, and episodes of worsening anaemia with infection, oxidant drugs or pregnancy. Hb Bart hydrops fetalis presents before birth, as a hydropic fetus with massive oedema, a hugely enlarged placenta and a haemoglobin too low to sustain extrauterine life, and it is detected on obstetric ultrasound in a pregnancy at risk. [9][10]
The chronic, transfused picture is the one the fellow meets in the clinic, and it is the picture of a child living with a multisystem disease. Growth is often delayed and puberty is late because iron loads the anterior pituitary, the child may be glucose intolerant or frankly diabetic as the pancreas loads, and the heart enlarges as the myocardium loads with iron. The spleen enlarges from the haemolysis and the extramedullary haematopoiesis, and over years it may accelerate the transfusion need through hypersplenism. Recognising the whole child, across the transfusion, the iron, the endocrine and the cardiac axes, is the daily work of the thalassaemia clinic. [11]
Differential Diagnosis
The infant who presents with a severe microcytic anaemia has a short list of causes, and the fellow must separate beta-thalassaemia major from the iron deficiency that is far commoner, and from the other microcytic disorders. The first clue is the red cell indices and the red cell count, because thalassaemia produces a marked microcytosis with a high or normal red cell count, while iron deficiency produces a microcytosis with a low red cell count. The ferritin settles iron deficiency, and the haemoglobin electrophoresis settles the thalassaemia, but the two are distinguished most quickly at the index visit by the indices and the clinical context. [12]
Beta-thalassaemia major
HbF raised on electrophoresis
- Autosomal recessive, HBB mutation, absent or reduced beta-globin
- Severe microcytic anaemia under 70 g per litre, presents at 6 to 12 months
- Raised haemoglobin F on electrophoresis, hepatosplenomegaly
- Transfusion-dependent from infancy, iron overload without chelation
Beta-thalassaemia intermedia
later, milder onset
- Two milder alleles or one severe one mild
- Anaemia 70 to 100 g per litre without regular transfusion
- Later presentation, splenomegaly, extramedullary haematopoiesis
- Iron overload from absorption, thrombosis and pulmonary hypertension
Haemoglobin H disease
three alpha-gene deletions
- Three of four alpha genes deleted
- Moderate haemolytic anaemia 70 to 100 g per litre
- Hb H inclusions on supravital staining, splenomegaly
- Worsens with infection, oxidants and pregnancy
Iron deficiency anaemia
low ferritin, low red cell count
- Dietary or blood loss, commonest microcytosis of childhood
- Low ferritin, low transferrin saturation, low red cell count
- Normal electrophoresis, responds to iron therapy
- No hepatosplenomegaly unless severe and chronic
The other inherited microcytic anaemias each have a distinguishing feature. Alpha-thalassaemia trait shares the microcytosis but runs a milder course and a milder anaemia, and the electrophoresis is often normal, so the diagnosis rests on the family history, the persistent microcytosis and, where needed, genetic testing. Lead poisoning produces a microcytic anaemia with basophilic stippling and a history of exposure. Sideroblastic anaemia produces a microcytic or dimorphic picture with ringed sideroblasts on the marrow. The chief pitfall is treating a thalassaemic child with iron, because the iron worsens the overload without helping the anaemia, so the ferritin and the electrophoresis are checked before any iron is given. [12]
Clinical & Bedside Assessment
The clinic visit for a child with thalassaemia major is a systematic search for the adequacy of the transfusion, the burden of the iron, and the injury to the organs that the iron targets. The visit begins with the history, and the questions that matter most are whether the child feels well between transfusions, whether the spleen is enlarging, and whether the growth and the puberty are on track. The interval between transfusions is asked about directly, because a shortening interval that demands more frequent or larger transfusions is the sign of rising hypersplenism. [6]
The examination looks for the signs of chronic anaemia, of iron injury and of marrow expansion. The conjunctivae are checked for pallor, the sclerae for the icterus of haemolysis, and the abdomen for the size of the liver and the spleen, because a spleen that grows across the transfusion years drives the transfusion need upward. Growth is plotted at every visit, because faltering growth and delayed puberty are the earliest signs of anterior pituitary iron loading. The heart is auscultated for the gallop or the murmur that signals early iron cardiomyopathy, and the skin is checked for the grey-brown bronzing of chronic iron excess. [11]
The structured clinic visit for a child with thalassaemia major
Ask about energy between transfusions, the transfusion interval, and any fevers or abdominal fullness since the last visit
Plot the weight, the height and the body mass index, and stage the puberty for the delayed growth of anterior pituitary iron loading
Examine the conjunctivae for pallor, the abdomen for liver and spleen size, and the heart for a gallop or murmur
Review the pre-transfusion haemoglobin, the ferritin, and the most recent cardiac T2 star and liver iron results
Confirm the chelation adherence and the adverse-effect monitoring, and book the annual cardiac magnetic resonance and the endocrine review
The teaching of the family to recognise the danger signs is a clinical act as important as any examination finding. The parents are taught to bring the child in at once for fever, because the splenectomised child is at risk of overwhelming sepsis, and to watch for increasing pallor, breathlessness or abdominal swelling that may signal a haemolytic crisis or a hypersplenic sequestration. The family on deferiprone is taught that any fever is an emergency until the neutrophil count is checked, because agranulocytosis can be fatal within hours. Adherence to chelation is the single behaviour most tied to survival, so the fellow asks about it, troubleshoots it, and frames it as the heart of home care. [11]
Investigations
The diagnosis of a thalassaemia syndrome is a laboratory diagnosis, made by separating the haemoglobins and by counting the genes. The first test in the symptomatic child is the full blood count and film, which shows the severe microcytic, hypochromic anaemia, a high red cell count, target cells and basophilic stippling, and a reticulocyte count that is inappropriately low for the degree of haemolysis because of the ineffective erythropoiesis. The picture points to a thalassaemia, but it does not settle the genotype, which is the work of the haemoglobin separation. [12]
The haemoglobin is separated by one of three methods, high-performance liquid chromatography, haemoglobin electrophoresis or capillary electrophoresis, and each identifies the relative proportions of haemoglobin A, haemoglobin F and haemoglobin A2. In beta-thalassaemia major, before any transfusion, the haemoglobin F is markedly raised, often to 70 percent or more, the haemoglobin A is absent in the beta-zero form or reduced in the beta-plus form, and the haemoglobin A2 is raised. A transfused child carries the donor haemoglobin A, so the diagnosis may require waiting for the transfused cells to clear or testing the family, which is why the electrophoresis is most informative when it is done before the first transfusion. [12]
The approach to identifying carriers and affected fetuses varies by region, but the principle is the same everywhere: find the couples at risk before the affected child is born. Australia, Aotearoa New Zealand, the United Kingdom, the United States and Canada offer carrier screening by blood count and electrophoresis, often at the pre-pregnancy or antenatal visit, and they offer prenatal diagnosis by chorionic villus sampling or amniocentesis to the couple at one in four risk. In high-prevalence regions of South-East Asia, universal screening targets the alpha-thalassaemia deletions that cause Hb Bart hydrops fetalis. The fellow should know the local screening pathway and the local prenatal diagnostic test. [12]
The monitoring of the transfused child is the investigation that runs for life, and it tracks the iron, the organs and the growth. The ferritin is measured at every visit as a simple marker of iron load, though it is an acute-phase reactant and an imperfect guide to tissue iron. The cardiac T2 star magnetic resonance imaging measures the myocardial iron directly, with a value of over 20 milliseconds normal and a value under 10 milliseconds marking severe loading and a high risk of heart failure. The liver iron concentration, measured by magnetic resonance imaging, tracks the hepatic load, and the annual endocrine review tracks the growth hormone axis, the gonadal axis and the glucose tolerance that iron slowly erodes. [8][11]
Management — Resuscitation
Thalassaemia major is a chronic disease, but it carries a small set of acute, time-critical dangers that the fellow must meet as emergencies. The first is the presentation in acute heart failure from iron cardiomyopathy, which is the chief cause of death in the disease and which may be the event that first brings a poorly chelated adolescent to hospital. The response is the standard treatment of acute heart failure with diuretics, afterload reduction and inotropic support, together with intensive chelation, often with intravenous deferoxamine combined with oral deferiprone, because deferiprone is the chelator with the strongest evidence for removing cardiac iron. [7][8]
The second danger is agranulocytosis from deferiprone, which is the chelation toxicity most likely to kill quickly. The drug is given at 75 mg per kg per day in three divided doses, and it carries a small but real risk of a profound fall in the neutrophil count to under 0.5 times ten to the nine per litre, which leaves the child defenceless against gram-negative and gram-positive sepsis. The absolute neutrophil count is checked weekly for as long as the drug is taken, and any fever on deferiprone is treated as a medical emergency: the drug is stopped at once, a blood culture is taken, and broad-spectrum parenteral antibiotics that cover the gram-negatives are given without waiting for the count. [11]
[7] [11]The third danger is overwhelming sepsis in the child who has had a splenectomy, which is offered for hypersplenism that drives the transfusion need. The splenectomised child is functionally or anatomically asplenic, and so is at risk of overwhelming encapsulated sepsis in the same way as the child with sickle cell disease. The response is a prompt parenteral broad-spectrum antibiotic, such as ceftriaxone, after a blood culture, for any fever above 38.5 degrees Celsius. Splenectomy is delayed until after five to six years of age wherever possible, because the risk of post-splenectomy sepsis is highest in the youngest child, and the child is fully immunised against the pneumococcus, the meningococcus and Haemophilus influenzae type b before the spleen is removed. [6]
Management — Definitive & Stepwise
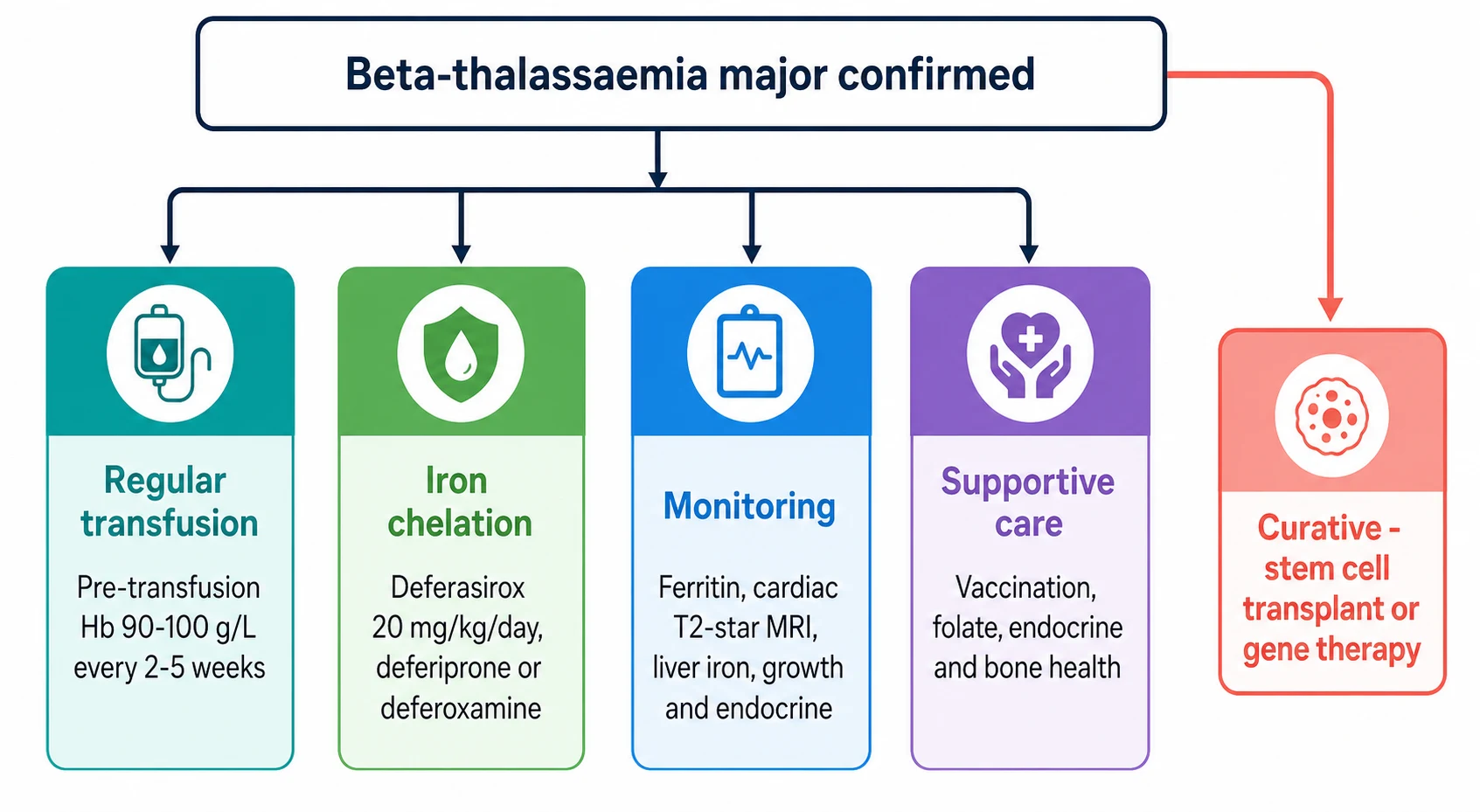
The definitive management of beta-thalassaemia major rests on three pillars that begin in infancy and run for life. The first pillar is the regular transfusion programme, the intervention that transforms the child from the wasted, bony, failing infant into a child who grows and goes to school. The aim is to keep the pre-transfusion haemoglobin above 90 to 100 g per litre, because this level suppresses the ineffective erythropoiesis, halts the marrow expansion and the skeletal change, and lets the child grow normally. The transfusion is given every two to five weeks, using leucodepleted, extended-phenotype-matched red cells, at a volume of 10 to 20 mL per kg over two to four hours. [6]
Regular transfusion programme for beta-thalassaemia major
Dose
Leucodepleted, extended-phenotype-matched packed red cells every two to five weeks to keep the pre-transfusion haemoglobin at 90 to 100 g per litre, at a volume of 10 to 20 mL per kg over two to four hours, aiming for a post-transfusion haemoglobin around 140 to 150 g per litre
The second pillar is iron chelation, the price the child pays for the transfusions that keep them alive, and the intervention that more than any other decides whether the child reaches adulthood. Chelation is started once the ferritin exceeds 1000 micrograms per litre or after about 10 to 20 transfusions, usually around two to three years of age. The oral chelator deferasirox transformed chelation by allowing a once-daily tablet in place of the nightly subcutaneous infusion, and the phase 3 trial of Cappellini and colleagues, published in 2006, established it as equivalent to deferoxamine at removing liver iron in transfused thalassaemia. The starting dose is 20 mg per kg per day orally once daily, titrated to the iron load, with renal and hepatic function monitored. [4][5]
Deferasirox for iron chelation in thalassaemia
Dose
Starting 20 mg per kg per day orally once daily, titrated up to 30 to 40 mg per kg per day to the iron load, for transfusional iron overload once the ferritin exceeds 1000 micrograms per litre
The older chelators retain a place, and the fellow must know them because the cardiac iron and the intolerances often demand their use. Deferoxamine is given by subcutaneous infusion over 8 to 12 hours, five to seven nights a week, at 20 to 40 mg per kg per day, and it is the chelator with the longest safety record but the heaviest burden of adherence. Deferiprone is given at 75 mg per kg per day in three divided doses, and it is the chelator with the strongest evidence for removing cardiac iron, which makes it the drug of choice for the child with a low cardiac T2 star. The chief toxicity of deferiprone is agranulocytosis, which demands a weekly absolute neutrophil count, and arthropathy, while the chief toxicity of deferoxamine is ocular and auditory toxicity and local skin reactions. [11][7]
[6] [4]The third pillar is the surveillance of iron and its target organs, because the iron kills silently and the only way to keep it safe is to measure it. The cardiac T2 star magnetic resonance imaging is done annually from around the age of five, because the myocardium loads more slowly than the liver and the cardiac failure is the event to prevent. A T2 star of over 20 milliseconds is normal, a value between 10 and 20 milliseconds indicates mild to moderate loading, and a value under 10 milliseconds marks severe loading and a high short-term risk of heart failure. The liver iron and the ferritin are tracked at each visit, the endocrine axes are reviewed annually, and the bone density is monitored because osteoporosis is a common and disabling late effect. [8][11]
The curative options are offered alongside the lifelong programme, and they have transformed the outlook for the child with a suitable donor or access to a gene therapy programme. Allogeneic haematopoietic stem cell transplant from a matched sibling donor is the longest-established cure, with survival over 90 percent in the well-chelated, young child, and it remains the standard of cure where a matched sibling is available. Betibeglogene autotemcel gene therapy adds a functional beta-globin gene to the child's own stem cells, removing the need for a donor, and the long-term data of Kwiatkowski and colleagues show durable transfusion independence. The pioneer trial of Thompson and colleagues, published in 2018, established the proof of concept that an ex vivo gene addition could free a transfusion-dependent child from transfusion. [1][3]
A disease-modifying drug that is not curative has added a fourth option for the transfusion-dependent adult and adolescent. Luspatercept is a recombinant fusion protein that promotes late-stage red cell maturation, and in the BELIEVE trial, reported in its final form by Cappellini and colleagues in 2025, it reduced the transfusion burden and the iron load in transfusion-dependent beta-thalassaemia. It is given by subcutaneous injection every three weeks, it does not replace transfusion or chelation, and it offers a meaningful reduction in transfusion demand for the child or young adult in whom curative therapy is not available or not chosen. [2]
Luspatercept for transfusion-dependent beta-thalassaemia
Dose
1.0 to 1.25 mg per kg by subcutaneous injection every three weeks, titrated to the haemoglobin and the transfusion response, for transfusion-dependent beta-thalassaemia
Specific Subtypes & Scenarios
Beta-thalassaemia intermedia is the form that sits between the trait and the major, and the child who has it presents later and runs a milder course. The haemoglobin typically sits between 70 and 100 g per litre without regular transfusion, the spleen is enlarged, and the child manages for years on folate and occasional transfusion for illness or pregnancy. The complications are different from those of the major, because the ineffective erythropoiesis is left unchecked: extramedullary haematopoiesis produces masses that can compress the spine, leg ulcers appear from chronic anaemia, the iron loads from increased gut absorption even without transfusion, and the high platelet and red cell turnover raises the risk of thrombosis and pulmonary hypertension. The decision to start a regular transfusion is made when the complications or the growth demand it. [12]
Haemoglobin H disease is the moderate alpha-thalassaemia of three-gene deletion, and the fellow meets it in the clinic and in the exam. The child has a chronic haemolytic anaemia with a haemoglobin between 70 and 100 g per litre, intermittent jaundice and splenomegaly, and the blood film shows the characteristic target cells and the supravital-stain inclusions of haemoglobin H, the beta-chain tetramers that precipitate in the red cell. The anaemia worsens with infection, with oxidant drugs such as sulphonamides, and in pregnancy, and the child occasionally needs a transfusion for an acute fall. Splenectomy is sometimes offered for the severe hypersplenic form, and the parents are counselled to avoid the oxidants that precipitate a haemolytic crisis. [9][10]
The first decade of a child with beta-thalassaemia major
Hb Bart hydrops fetalis is the lethal end of the alpha-thalassaemia ladder, and it is the form that the obstetric and the neonatal teams meet together. The fetus that inherits four alpha-gene deletions cannot make any functional haemoglobin, because the alpha chain is shared by both fetal and adult haemoglobin, so the gamma chains join in fours to form Hb Bart, a haemoglobin that binds oxygen so tightly it releases none to the tissues. The fetus becomes hydropic, with massive oedema and a huge placenta, and is stillborn or dies in the neonatal period unless intrauterine transfusions are begun. Survivors are transfusion-dependent for life and need chelation, and the prevention rests on carrier screening of couples from high-prevalence regions and prenatal diagnosis of the at-risk pregnancy. [9]
Complications & Pitfalls
The first pitfall is the failure to chelate, or the failure to chelate adequately, and it is the pitfall that leads directly to the cardiac death that still claims the lives of young adults with thalassaemia. The deferoxamine infusion is burdensome, the oral chelators have toxicities, and the iron accumulates silently for years before the heart fails, so the adolescent who stops the chelation in the years of rebellion presents a few years later in acute heart failure. The fellow who tracks the ferritin and the cardiac T2 star, who asks about adherence at every visit, and who switches chelator when the cardiac iron rises, is the fellow who prevents this death. [7][8]
[7] [8]The second pitfall is the agranulocytosis of deferiprone, which is the chelation toxicity most likely to be missed until it is too late. The drug is given to the child with the worst cardiac iron, precisely because it removes it best, and the weekly absolute neutrophil count is the monitoring that stands between the child and a fatal neutropenic sepsis. The fellow who omits the weekly count, or who fails to stop the drug at the first fever, runs the child into a gram-negative bacteraemia. The message is that deferiprone is a powerful drug that demands a powerful discipline of monitoring, and any fever on it is agranulocytosis until the count proves otherwise. [11]
The third pitfall is endocrine failure, which is the slow injury that iron does to the glands that govern growth, puberty and glucose. The anterior pituitary loads early, so the child is short and the puberty is late, the gonadal axis fails so the adolescent is infertile without hormone replacement, the pancreas loads so the glucose tolerance falls and the young adult develops diabetes, and the parathyroid and the thyroid may fail in turn. The annual endocrine review, with the growth charts, the pubertal staging, the oral glucose tolerance test and the hormone panels, is the surveillance that catches these injuries early and replaces the hormones before the damage is complete. [11][12]
The fourth pitfall is alloimmunisation and transfusion-transmitted infection, which are the risks inseparable from a life of transfusion. The child forms antibodies against the donor red cell antigens, which makes future cross-matching harder and can cause haemolytic transfusion reactions, so the child is phenotyped for the major antigens before the first transfusion and is matched for them wherever possible. The blood must be screened for the hepatitis viruses and the human immunodeficiency virus, and in regions of variable blood safety the transfusion itself is a risk that the fellow weighs, which is why the sourcing and the testing of the blood are part of the care as much as the dose. [6]
Prognosis & Disposition
The modern outlook for a child with beta-thalassaemia major, born into a health system with safe blood and chelation, is one of survival into adulthood, and this is the central fact the fellow carries into every counselling. The cohort of Borgna-Pignatti and colleagues showed that survival improved steadily across the decades of deferoxamine and transfusion, and the analysis of Modell and colleagues linked that improving survival directly to the cardiac T2 star magnetic resonance imaging that detected the myocardial iron before it killed. The death that was once common in the second decade, from iron cardiomyopathy, is now preventable in the child who stays on the programme. [6][8]
The disposition of the child is shared between the primary care team and the thalassaemia centre, and the fellow coordinates the two. The primary care team runs the routine immunisations, the growth and the acute presentations, while the thalassaemia centre runs the transfusion, the chelation, the iron surveillance, the endocrine review and the curative decision. The child is followed in the centre from diagnosis, the transfusion and the chelation are titrated at each visit, and the curative options of transplant and gene therapy are discussed as the child grows and as the programmes become available. [12]
The transition to adult care is the junction that decides whether the young adult stays alive, because the years around transfer are the years the chelation is most often dropped. The fellow plans the crossing over years, hands over a young adult who knows the diagnosis and carries the chelation, and connects the young person to an adult thalassaemia service rather than to an emergency department. The aim is a young person who reaches adult care connected to a service, who keeps the chelation going through the transition, and who carries the curative decision forward into the adult years. [12]
Special Populations
The migrant, the refugee and the family from a high-prevalence region carry the double burden of the gene and the access, and the fellow who works in a mixed population plans for both. The child who arrives from a country without screening may present late, with the bony changes of untreated disease or with the cardiac failure of unchelated iron, so the fellow has a low threshold to test the haemoglobins of any child from a Mediterranean, Middle Eastern, South Asian or South-East Asian family with an unexplained microcytic anaemia. The family with limited language or limited trust needs the transfusion and the chelation organised around them, with interpreter support and a clear written plan, because the adherence that decides survival is the behaviour most easily lost to a barrier of access. [12]
The couple at risk of Hb Bart hydrops fetalis is the special population the fellow meets in the antenatal clinic, and the counselling is the intervention that prevents the lethal outcome. Two carriers of the cis alpha-thalassaemia deletion, common in South-East Asia, have a one in four chance of a fetus with four-gene deletion in each pregnancy, and the prenatal diagnosis by chorionic villus sampling or amniocentesis is offered to identify the affected fetus. Where intrauterine transfusion is available and chosen, a surviving infant is transfusion-dependent for life, and the counselling lays out the choices honestly, from termination to intrauterine transfusion to expectant management. [9]
GLOBIN
The child in a rural or remote setting, far from a thalassaemia centre, is managed by the local team in partnership with the centre through shared protocols and telehealth. The local team holds the transfusion schedule, the chelation adherence and the emergency plan for fever and for heart failure, and it ships the child to the centre for the annual cardiac magnetic resonance and the endocrine review. The fellow who works in such a setting values the local network and the shared blood supply, because the child who is connected to the centre through it does as well as the child who lives next door to it. [6]
Evidence, Guidelines & Regional Differences
The evidence base for thalassaemia major is built on the chelation and transfusion trials that transformed the disease from a fatal anaemia of childhood into a chronic disease of adulthood. The phase 3 deferasirox trial of Cappellini and colleagues, published in 2006, established the oral chelator as equivalent to deferoxamine, and freed a generation of children from the nightly subcutaneous infusion. The cardiac survival analysis of Modell and colleagues linked the improving survival to the T2 star magnetic resonance imaging, and the cardiac morbidity study of Borgna-Pignatti and colleagues set the benchmark for the chelation that prevents the heart failure. [4][8][7]
Deferasirox phase 3 - Cappellini 2006
Key finding
Once-daily oral deferasirox at 20 or 30 mg per kg per day reduced liver iron concentration to a similar degree as deferoxamine in transfused beta-thalassaemia patients over one year.
BELIEVE - luspatercept, Cappellini 2025
Key finding
Subcutaneous luspatercept every three weeks produced a reduction in transfusion burden, with about a fifth to a third of transfusion-dependent beta-thalassaemia patients achieving at least a 33 percent reduction over 48 weeks.
Betibeglogene autotemcel gene therapy - Kwiatkowski 2026
Key finding
Ex vivo addition of a functional beta-globin gene to the patient's own stem cells produced durable transfusion independence in the majority of transfusion-dependent beta-thalassaemia patients, with a stable haemoglobin above 90 g per litre off transfusion.
The guidelines that translate this evidence into practice differ by region, and the fellow names the one being followed. The Thalassaemia International Federation guideline is the most widely used comprehensive standard, and it sets the transfusion target, the chelation thresholds and the surveillance schedule. The 2026 primer of Piel and colleagues synthesises the current evidence across the syndromes. The British Society for Haematology sets the practice in the United Kingdom, the local newborn and carrier screening programmes set the diagnosis, and the regional access to safe blood and chelation sets the ceiling of what is achievable. [12]
The chief controversy is the timing and the choice of the curative pathway. Allogeneic transplant from a matched sibling donor remains the standard of cure where a donor is available, and it carries the lowest long-term risk, but most children lack a matched sibling. Gene therapy removes the donor barrier and the graft-versus-host risk, but it carries the cost and the conditioning-related infertility, and the longest-term data are still accruing. Luspatercept sits alongside both as a burden-reducing drug rather than a cure. The fellow follows the local programme, weighs the donor against the gene therapy, and explains the choice to the family as the child grows. [1][3][2]
Exam Pearls
The single most testable fact is the mechanism, so learn it word for word. Beta-thalassaemia arises from reduced or absent beta-globin synthesis, the unpaired alpha chains precipitate in the red cell precursors, and the cells die in the marrow in a process called ineffective erythropoiesis, which is the dominant cause of the anaemia. The fellow who can state this sequence exactly, and who can explain that the high fetal haemoglobin is the compensating feature and the reason the newborn is well, has the molecular heart of the disease. [12]
The transfusion and the chelation numbers are the second most testable set, and they are asked as a pair. The pre-transfusion haemoglobin target is 90 to 100 g per litre, given every two to five weeks, and chelation starts once the ferritin exceeds 1000 micrograms per litre. Deferasirox starts at 20 mg per kg per day orally once daily, deferiprone at 75 mg per kg per day in three divided doses, and deferoxamine at 20 to 40 mg per kg per day by subcutaneous infusion. The fellow who holds these numbers carries the management of the disease. [4][6]
[7] [8]The alpha-thalassaemia ladder is the third high-yield set, and it turns on the number of alpha genes deleted. One deletion is the silent carrier, two is the trait, three is haemoglobin H disease with its moderate haemolysis, and four is Hb Bart hydrops fetalis, lethal without intrauterine transfusion. The fellow who can recite the ladder, and who can name Hb Bart and Hb H as the abnormal tetramers of gamma and beta chains, has the alpha side of the topic. The curative options of transplant and betibeglogene gene therapy, and the BELIEVE luspatercept trial, round out the high-yield set. [9][1]
References
- [1]Thompson AA, Walters MC, Kwiatkowski J Gene therapy in patients with transfusion-dependent beta-thalassemia. N Engl J Med, 2018.PMID 29669226
- [2]Cappellini MD, Viprakasit V, Georgiev P Long-term efficacy and safety of luspatercept for the treatment of anaemia in patients with transfusion-dependent beta-thalassaemia (BELIEVE): final results from a phase 3 randomised trial. Lancet Haematol, 2025.PMID 39947215
- [3]Kwiatkowski JL, Thompson AA, Schneiderman J Long-term efficacy and safety results of betibeglogene autotemcel gene therapy for transfusion-dependent beta-thalassemia. Blood, 2026.PMID 41525466
- [4]Cappellini MD, Cohen A, Piga A A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood, 2006.PMID 16352812
- [5]Piga A, Galanello R, Forni GL Randomized phase II trial of deferasirox (Exjade, ICL670), a once-daily, orally-administered iron chelator, in comparison to deferoxamine in thalassemia patients with transfusional iron overload. Haematologica, 2006.PMID 16818273
- [6]Borgna-Pignatti C, Rugolotto S, De Stefano P Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica, 2004.PMID 15477202
- [7]Borgna-Pignatti C, Cappellini MD, De Stefano P Cardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassemia major. Blood, 2006.PMID 16373663
- [8]Modell B, Khan M, Darlison M Improved survival of thalassaemia major in the UK and relation to T2* cardiovascular magnetic resonance. J Cardiovasc Magn Reson, 2008.PMID 18817553
- [9]Vichinsky EP Clinical manifestations of α-thalassemia. Cold Spring Harb Perspect Med, 2013.PMID 23543077
- [10]Lal A, Viprakasit V, Vichinsky E Disease burden, management strategies, and unmet needs in alpha-thalassemia due to hemoglobin H disease. Am J Hematol, 2024.PMID 39037279
- [11]Hoffbrand AV, Taher A, Cappellini MD How I treat transfusional iron overload. Blood, 2012.PMID 22919029
- [12]Piel FB, de Montalembert M, Das R Thalassaemia. Nat Rev Dis Primers, 2026.PMID 42426018