Paeds · haematology-oncology-and-transfusion
Thrombocytopenia and immune thrombocytopenia
Also known as Immune thrombocytopenia · ITP · Idiopathic thrombocytopenic purpura · Immune thrombocytopenic purpura · Acute childhood ITP · Chronic ITP · Primary immune thrombocytopenia
Fellowship guide to thrombocytopenia and immune thrombocytopenia in children. Covers the isolated thrombocytopenia with a platelet count under 100 times ten to the nine per litre in an otherwise well child, the typical preschool presentation with bruising and petechiae one to four weeks after a viral illness, and the ASH 2019 framework that recommends observation over treatment for the child with no bleeding or mild skin-only bleeding regardless of the platelet count, with first-line IVIG at 0.8 to 1 g per kg as a single dose, a short course of corticosteroids, or anti-D at 50 to 75 micrograms per kg when treatment is needed, second-line thrombopoietin-receptor agonists such as eltrombopag for the chronic phase beyond twelve months, and the now-rare splenectomy.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A healthy two-year-old who was entirely well last week is brought in covered in bruises and pinhead red spots, and the blood test shows a platelet count that has fallen to single digits. The child looks well, is running around the waiting room, and the rest of the blood count is normal. This is the classic face of childhood immune thrombocytopenia, and it is one of the commonest and most reassuring diagnoses in paediatric haematology once it is properly understood. Immune thrombocytopenia, always abbreviated ITP, is an autoimmune disorder in which the body makes antibodies against its own platelets, the antibody-coated platelets are destroyed in the spleen, and the platelet count falls until the bruising and the petechiae appear. [3][4]
The name itself carries a lesson, because it has changed. The condition was once called idiopathic thrombocytopenic purpura, meaning a purpura of unknown cause, and the fellow may still see the abbreviation ITP used that way in older texts. The international working group of Rodeghiero and colleagues, reporting in Blood in 2009, retired the word idiopathic and replaced it with immune, because the autoimmune mechanism is now firmly established, and the word purpura was dropped, because many children bruise without true purpura. The abbreviation ITP survives, but it now means immune thrombocytopenia. [4]
Three ideas make this topic central to the paediatric exam, and each is a deliberate decision rather than a reflex. The first is that the diagnosis is clinical and is made by exclusion, because ITP is defined as an isolated thrombocytopenia with a platelet count under 100 times ten to the nine per litre in a child who is otherwise well, with no other cytopenias and no blasts on the blood film. The second is the ASH 2019 observation-first principle, which holds that a child with no bleeding or only mild skin bleeding is managed by observation regardless of how low the count has fallen. The third is the distinction between the phases of the disease, because the child whose thrombocytopenia persists beyond twelve months moves into chronic ITP and into a different set of second-line drugs. [1][4]
Classification
ITP is sorted in two ways, by how long it has lasted and by whether a cause has been found, and each sort changes the management. The classification by time is the one the fellow must know cold, because it is the Rodeghiero standard of 2009 and it is the framework the ASH 2019 guideline is built on. The disease runs in three phases, and the cut-offs are three months and twelve months. Newly diagnosed ITP runs from the day of diagnosis to three months, persistent ITP runs from three months to twelve months, and chronic ITP is defined by persistence beyond twelve months. The reason the fellow learns these cut-offs is that the prognosis and the treatment change at each boundary, because most children remit within the first three months, the persistent phase is a watching brief, and the chronic phase is where the second-line drugs enter. [4]

The classification by cause separates primary from secondary ITP, and the distinction matters because a secondary ITP is managed by treating the underlying disorder alongside the platelet count. Primary ITP, which is the standalone form that this topic addresses, is the diagnosis when no underlying cause is found, and it accounts for the great majority of childhood cases. Secondary ITP is the diagnosis when the thrombocytopenia is driven by another condition, and the common ones in childhood are the connective tissue diseases such as systemic lupus erythematosus, the immunodeficiencies such as common variable immunodeficiency, the chronic infections such as HIV and hepatitis C, and the lymphoproliferative and immune-dysregulation syndromes such as Evans syndrome, in which ITP and autoimmune haemolytic anaemia run together. [2][3]
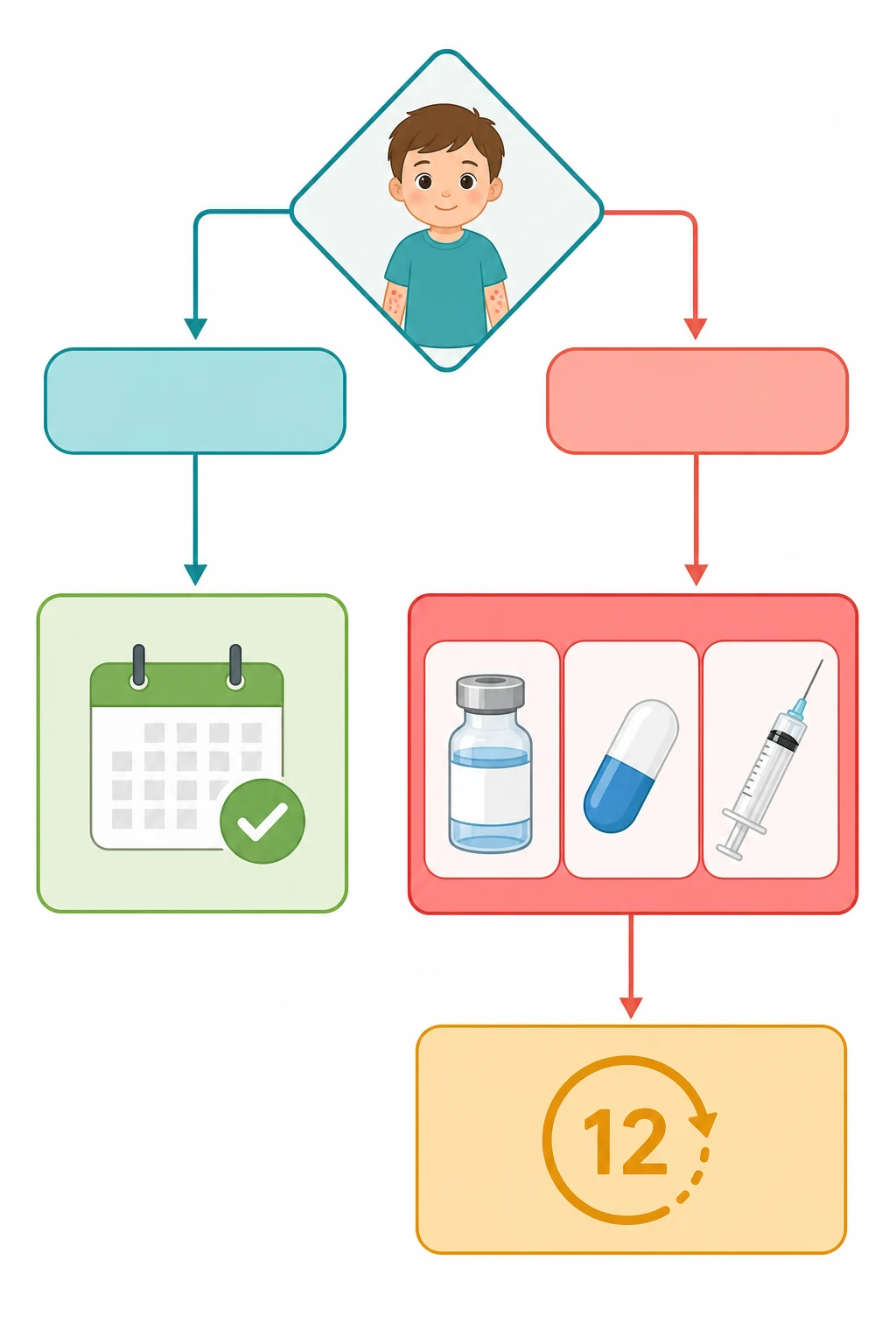
A third classification, by bleeding severity rather than by the disease itself, drives the day-to-day decision, and it is the one the ASH 2019 guideline enforces. Bleeding is graded as none, mild, moderate, or severe, and the grade is what decides whether the child is observed or treated. The child with no bleeding or with mild skin-only bleeding, meaning bruising and petechiae but no mucosal wet bleeding, is observed regardless of the count. The child with moderate or severe bleeding, meaning mucosal bleeding such as epistaxis or menorrhagia, wet purpura, or any life-threatening bleed, is treated. This grading is the reason the platelet count alone never drives the decision, and it is the single point examiners test most often. [1]
Epidemiology & Risk Factors
ITP is the commonest cause of isolated thrombocytopenia in an otherwise well child, and the fellow will see it many times in a career. The incidence sits at roughly 1.9 to 6.4 children per 100,000 per year across the high-income registries, with the variation reflecting ascertainment and the threshold for testing. The peak age is the preschool years, between two and five years, and there is a slight male predominance in the youngest children that equalises and then reverses toward a female predominance in adolescence, because the adolescent form overlaps with the adult female-skewed autoimmune pattern. [3][5]
The single most consistent feature of the history is the preceding viral illness, and the fellow asks for it directly. Roughly half to two-thirds of children have had a viral infection in the one to four weeks before the bruising appeared, and the classic triggers are the common exanthems, varicella, measles, rubella, and mumps, the herpesviruses Epstein-Barr, cytomegalovirus, and human herpesvirus 6, and the respiratory and gastrointestinal viruses. A few drugs, vaccines, and the parvovirus B19 and hepatitis C infections are also recognised triggers. The mechanism is a break in immune tolerance, in which the antibody response raised against the virus cross-reacts with the platelet surface glycoproteins. [3]
The natural history is the reason the observation strategy exists, and it is built on the Intercontinental Cooperative Immune Thrombocytopenia Study Group, or ICIS, registries, which together enrolled many thousands of children. The prospective registry of 2540 children reported by Kühne and colleagues in 2003, and the twelve-month follow-up registry I reported by Imbach and colleagues in 2006, established that the great majority of children remit spontaneously. About 70 to 80 percent remit within six months, and the proportion that reaches the chronic phase beyond twelve months is about 20 to 25 percent. The intracranial haemorrhage, the feared complication, is rare, with an incidence estimated below one percent, and it is the central reason the fellow must balance observation against the rare catastrophic bleed. [5][6]
Pathophysiology
The simplest way to picture ITP is that the body mistakes its own platelets for a virus and clears them, and this picture is correct as far as it goes, but it is incomplete, and the incompleteness is what the fellowship-level answer must address. The autoantibodies, which are mostly of the immunoglobulin G class, are directed against the major platelet surface glycoproteins, glycoprotein IIb/IIIa and glycoprotein Ib/IX, the very complexes that drive platelet aggregation and adhesion. Once the antibodies coat the platelet, the antibody-coated platelet is recognised by the Fc receptors on the splenic macrophages, and it is engulfed and destroyed. The spleen is therefore both the site of antibody production and the site of platelet clearance, which is why the spleen sits at the centre of the disease and at the centre of the second-line surgery. [3]
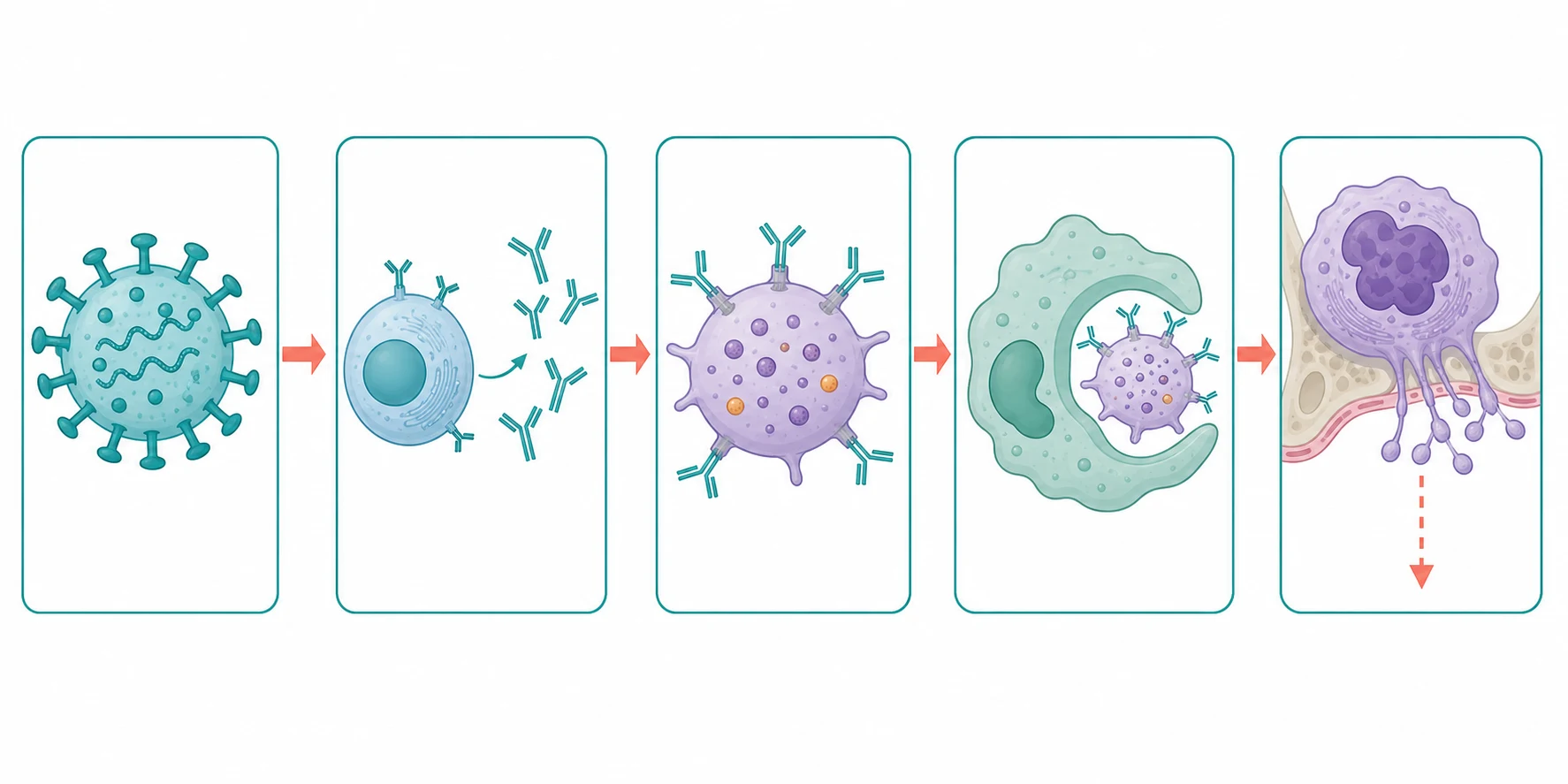
The half of the story that the older texts missed, and that the fellow must now include, is the impaired production. For decades ITP was taught as a pure destruction disease, in which the platelets were lost faster than the marrow could make them, and the megakaryocyte was assumed to be working flat out. The measurement of platelet production, by reticulated platelet counts and by megakaryocyte imaging, showed that this assumption was wrong. The autoantibodies and the cytotoxic T-cells also injure the megakaryocytes, so the platelet production is impaired as well as the destruction increased, and the low count is the sum of both. This is the reason the thrombopoietin-receptor agonists, which stimulate the megakaryocyte, work so well, and it is the reason they are a logical second-line therapy rather than a desperate measure. [3][11]
The break in immune tolerance that sets the whole process in motion is linked to the preceding viral illness, and it has both a humoral and a cellular arm. The antibody response is driven by autoreactive B-cells, and the clearance is driven by the splenic macrophages, but cytotoxic T-cells also directly kill the platelets and the megakaryocytes, which explains why the B-cell-depleting drug rituximab works in only some patients. The autoantibody targets, glycoprotein IIb/IIIa and glycoprotein Ib/IX, are the same targets that the drug-induced antibodies hit, which is why ITP shares features with the drug-induced thrombocytopenias. The fellow who holds the dual mechanism, the destruction and the impaired production, and who can name the antibody targets, has the pathophysiology the examiner wants. [3]
Clinical Presentation
The child with newly diagnosed ITP looks well, and the contrast between the alarming blood test and the well-looking child is the single most reassuring feature of the presentation. The parents bring the child in because they have noticed, over hours to a few days, a sudden crop of bruises and pinhead red spots, the petechiae, across the skin, and they are frightened because there has been no major injury. There may be bleeding from the gums, a nosebleed, or, in the menstrual girl, a heavy period. The history is that of an entirely well child one to four weeks after a viral illness, and there is no fever, no bone pain, no weight loss, and no pallor at the time of presentation. [5]
The examination confirms the well child with the isolated skin and mucosal bleeding. The skin shows the bruising and the petechiae, and the mucosae may show the wet purpura, which is the blood blisters on the buccal mucosa and the palate that signal a higher risk of major bleeding. The lymph nodes are not enlarged, the liver and the spleen are not palpable, and the rest of the examination is normal. The presence of hepatosplenomegaly or significant lymphadenopathy is a red flag that excludes ITP and points to acute leukaemia or another infiltrative disorder, and the fellow who finds an enlarged spleen in a supposed ITP stops and reconsiders the diagnosis. [5]
The well preschool child with isolated bruising, petechiae, and a platelet count under 100 times ten to the nine per litre is the universal face of ITP, and it is recognised the same way in Australia, Aotearoa New Zealand, the United Kingdom, the United States, and Canada. The threshold for testing, the access to haematology referral, and the availability of IVIG and the thrombopoietin-receptor agonists vary by health system, but the ASH 2019 and the international consensus framework are followed across the English-speaking fellowship regions. The fellow should know the local pathway for urgent haematology review and the local supply of anti-D, which has been withdrawn in several countries. [1][2]
The bleeding severity, not the platelet count, is what the fellow grades, because it is the grade that drives the decision. Mild bleeding is the bruising and the petechiae confined to the skin, and it is by far the commonest presentation. Moderate bleeding adds the clinically significant mucosal bleeding, such as a nosebleed that is hard to stop or a heavy period. Severe bleeding is the life-threatening bleed, and the feared one is the intracranial haemorrhage, which presents with a severe headache, vomiting, altered consciousness, or a focal deficit. The severe bleed is rare, but it is the reason the fellow teaches the family the safety-net, and it is the one presentation that demands urgent combined treatment. [1]
The chronic ITP, the phase beyond twelve months, presents differently in tempo but not in kind. The child has lived with the diagnosis for a year, the bleeding has been recurrent but rarely severe, and the burden has shifted from the acute event to the chronic impact on school, sport, and quality of life. The adolescent girl who has reached the chronic phase carries the added burden of the heavy menstrual bleeding, and the fellow addresses contraception, the menstrual suppression, and the iron deficiency that the chronic blood loss brings. The chronic child is the one for whom the second-line drugs and the multidisciplinary support are built. [11][12]
Differential Diagnosis
The diagnosis of ITP is a diagnosis of exclusion, and the differential is the set of disorders that also cause a low platelet count, each of which the fellow must be able to separate from ITP at the bedside and on the blood film. The first and the most important is acute leukaemia, because the child with leukaemia may also present with bruising and petechiae, and the cost of missing it is catastrophic. The separation rests on the blood count and the film: ITP is an isolated thrombocytopenia with a normal haemoglobin, a normal white cell count, and no blasts, while leukaemia shows other cytopenias and blast cells on the film, and it is often accompanied by the fever, the bone pain, and the organomegaly that ITP lacks. [3][5]
Immune thrombocytopenia
isolated thrombocytopenia, well child
- Isolated platelet count under 100 times ten to the nine per litre
- Normal haemoglobin and white cell count, no blasts
- Large platelets on the film, no schistocytes
- No hepatosplenomegaly, no lymphadenopathy, often post-viral
Acute leukaemia
marrow infiltration
- Thrombocytopenia with anaemia and neutropenia or blasts
- Blast cells on the blood film
- Fever, bone pain, hepatosplenomegaly, lymphadenopathy
- Bone marrow aspirate confirms the diagnosis
Aplastic anaemia
marrow failure
- Pancytopenia affecting all three lineages
- Hypocellular bone marrow on the aspirate
- Insidious onset, no organomegaly
- Congenital and acquired forms
Haemolytic uraemic syndrome
microangiopathic
- Thrombocytopenia with fragmented schistocytes on the film
- Acute kidney injury and hypertension
- Preceding diarrhoeal illness, often Shiga-toxin
- Microangiopathic haemolytic anaemia
The inherited thrombocytopenias are the second group the fellow must hold, because they are rarer but they are missed, and the miss carries consequences. Wiskott-Aldrich syndrome is the classic example, an X-linked disorder of small platelets, eczema, and immunodeficiency, and the small mean platelet volume on the full blood count is the clue that separates it from ITP, because the ITP platelets are large. Congenital amegakaryocytic thrombocytopenia presents in the neonate with an isolated severe thrombocytopenia and a near-absent megakaryocyte lineage, and the type 2B von Willebrand disease causes a thrombocytopenia with a family history of bleeding and a gain-of-function von Willebrand factor. The fellow checks the mean platelet volume, the family history, and the onset, because the inherited disorders do not remit and they are not treated as ITP. [3]
The secondary immune thrombocytopenias are the third group, and they are sought when the history or the examination raises the suspicion. Systemic lupus erythematosus presents with the thrombocytopenia alongside the other features of the connective tissue disease, and the adolescent girl with ITP is screened for the antinuclear antibody if there is any clinical pointer. Evans syndrome is the combination of ITP and autoimmune haemolytic anaemia, and it is identified by a positive direct antiglobulin test and a falling haemoglobin with a high reticulocyte count. HIV and hepatitis C are tested when the risk factors are present, and the common variable immunodeficiency is sought when the immunoglobulin levels are low. The neonatal thrombocytopenia is a separate entity, because the neonatal alloimmune thrombocytopenia and the neonatal autoimmune thrombocytopenia from a mother with ITP have their own mechanisms and management. [2][3]
Clinical & Bedside Assessment
The assessment of the child with suspected ITP has two goals, to grade the bleeding and to exclude the alternative diagnoses, and the fellow does both at the same encounter. The history establishes the tempo, the preceding viral illness, and the absence of the red flags. The bleeding is graded by asking about the sites and the severity: the bruising and the petechiae are mild, the nosebleeds and the heavy periods are moderate, and the severe headache or the altered consciousness is severe. The family is asked about the family history of bleeding, because a positive family history points away from ITP and toward an inherited disorder, and about the drug history, because several drugs cause thrombocytopenia. [5]
The examination is a focused search for the features that confirm the well child and for the features that exclude ITP. The skin and the mucosae are inspected for the bruising, the petechiae, and the wet purpura. The lymph nodes are palpated for the adenopathy that points to leukaemia or a lymphoproliferative disorder. The abdomen is palpated for the liver and the spleen, because the hepatosplenomegaly excludes ITP and points to infiltration or a storage disorder. The developmental and the general examination confirms the otherwise well child, because the child with the eczema and the small platelets of Wiskott-Aldrich syndrome, or the dysmorphic child, is not the straightforward ITP. [5]
The structured assessment of the child with suspected ITP
Grade the bleeding as none, mild skin-only, moderate mucosal, or severe life-threatening
Ask about the preceding viral illness, the family bleeding history, and the drug history
Inspect the skin and the mucosae for bruising, petechiae, and wet purpura
Palpate the lymph nodes and the abdomen to exclude hepatosplenomegaly and adenopathy
Confirm the isolated thrombocytopenia on the full blood count and the normal film
Decide observation or treatment by the bleeding grade, not by the platelet count
The safety-netting advice is a clinical act as important as any investigation, because the child managed by observation is sent home, and the family must know when to return. The family is taught that the bruising and the petechiae are expected, that the child should avoid the contact sports and the medications that interfere with platelets such as the non-steroidal anti-inflammatories, and that any severe headache, persistent bleeding, or altered consciousness is an emergency that demands immediate assessment. The fellow gives the advice in writing, because the family that has the written plan is the family that acts on it. The safety-net is the price of the observation strategy, and it is the reason the strategy is safe. [1]
Investigations
The investigation of the typical ITP is deliberately minimal, and the fellow who over-investigates the straightforward case has misunderstood the diagnosis. The full blood count is the essential test, and it shows the isolated thrombocytopenia with a normal haemoglobin, a normal white cell count, and a normal differential. The blood film is the essential partner, and it shows the large platelets, the absence of blast cells, the absence of schistocytes, and the absence of the other abnormalities that point to an alternative diagnosis. The combination of the isolated thrombocytopenia and the reassuring film, in the well child with the typical history, is the diagnosis, and no further testing is needed for the primary case. [1][3]
The bone marrow aspirate is the test that the fellow is most often asked about, and the ASH 2019 guideline is explicit on when it is and is not indicated. The guideline recommends against a routine bone marrow aspirate or biopsy in the child with the typical presentation, because the typical ITP is a clinical diagnosis and the marrow is normal. The marrow is indicated when the presentation is atypical, meaning when there are other cytopenias, when the film shows blasts or other abnormalities, when the examination shows organomegaly, when the thrombocytopenia is refractory, or when the disease follows an atypical course. The fellow who orders a marrow for every ITP over-investigates, and the fellow who never orders one misses the leukaemia that masqueraded as ITP. [1]
[1]The additional tests are selective and driven by the history and the examination. The direct antiglobulin test is done when anti-D is being considered, because anti-D is given only to the Rh-D positive child with a negative direct antiglobulin test and an intact spleen. The blood group and the Rh type are checked for the same reason. The HIV and the hepatitis C serology are done when the risk factors are present, and the immunoglobulin levels are checked when a common variable immunodeficiency is suspected. The antinuclear antibody and the lupus screen are done for the adolescent girl with any clinical pointer to a connective tissue disease. None of these is routine for the straightforward preschool presentation. [2]
The measurement of the platelet-associated antibodies and the thrombopoietin level are not useful in routine practice, because the antibody assays are neither sensitive nor specific, and they do not change the management. The bleeding time and the platelet function assays are also not part of the routine workup. The fellow who holds the minimal investigation of the typical case, the count and the film, and the selective addition of the secondary-cause testing, has the investigation strategy the guideline endorses. [1][2]
Management — Resuscitation
ITP is rarely a resuscitation, but it carries one true emergency, and the fellow must meet it with the same discipline as any resuscitation. The emergency is the life-threatening bleed, and the feared example is the intracranial haemorrhage. A child with ITP who develops a sudden severe headache, vomiting, altered consciousness, or a focal neurological deficit has an intracranial haemorrhage until proven otherwise, and the response is the immediate combined administration of IVIG and high-dose corticosteroids together with a platelet transfusion, alongside the urgent imaging and the neurosurgical and critical-care referral. The aim is to raise the platelet count as fast as possible, because the IVIG works within twenty-four to forty-eight hours and the platelet transfusion, though the transfused platelets are also destroyed, provides a temporary bridge. [1][3]
The same combined approach is used for any other life-threatening bleed, such as a severe gastrointestinal bleed or a massive epistaxis that does not stop. The combination of IVIG and corticosteroids is the most rapid way to suppress the antibody-mediated destruction, the platelet transfusion is the temporary bridge, and the child is managed in the high-dependency or the intensive-care setting. The decision to give all three together, rather than to wait for the response to one, is the decision that the severe bleed demands, because the cost of waiting is the catastrophic progression of the bleed. [1]
[1]The child with the moderate bleed, such as the uncontrolled epistaxis or the heavy menorrhagia, is treated but not resuscitated, and the treatment is the first-line therapy described below. The distinction between the resuscitation of the severe bleed and the treatment of the moderate bleed is the distinction between the combined emergency therapy and the single first-line agent, and it rests on the clinical judgement of the bleeding severity. The fellow who treats the moderate bleed with the same urgency as the severe bleed, and who observes the mild bleed, has the calibration the guideline asks for. [1]
Management — Definitive & Stepwise
The definitive management of ITP is the decision between observation and treatment, and it is the decision that the ASH 2019 guideline has reshaped. The guideline recommends, as a strong recommendation based on moderate-quality evidence, that the child with newly diagnosed ITP and no bleeding or only mild skin bleeding is managed by observation, regardless of the platelet count. This is the single most important and most testable point in the topic, because it reverses the older reflex of treating every child with a count under a certain threshold. The rationale is that the bleeding risk in the child with no or mild bleeding is low, that the treatments carry their own harms, and that the natural history is toward remission in the great majority. [1]
The observation is active, not passive, and it carries the safety-net described above. The family is taught the danger signs, the child is reviewed in the clinic within a week to ten days, and the count is monitored until it rises. The child avoids the contact sports and the platelet-interfering medications, and the family is given the written emergency plan. The observation continues as long as the bleeding remains mild, and it shifts to treatment if the bleeding worsens. The platelet count is monitored, but it is not the trigger for treatment, because the trigger is the bleeding. [1]
[1]The first-line treatment, when the bleeding warrants it, is a choice among three agents, and the ASH 2019 guideline sets out the doses and the selection logic. The first agent is IVIG, given as a single dose of 0.8 to 1 g per kg, and it is chosen when a rapid rise in the platelet count is wanted, because it works within twenty-four to forty-eight hours. The second agent is a short course of corticosteroids, and the usual regimen is oral prednisolone or prednisone at a dose of 1 to 2 mg per kg per day for a short course, with the dose tapered and stopped, because the long-course steroids carry the growth, the mood, and the bone risks that the short course avoids. The third agent is anti-D, given as a single dose of 50 to 75 micrograms per kg, and it is an option only for the Rh-D positive child with a negative direct antiglobulin test and an intact spleen. [1][8]
IVIG for childhood ITP
Dose
0.8 to 1 g per kg as a single intravenous dose, with a second dose the next day if the response is inadequate
Corticosteroids for childhood ITP
Dose
Oral prednisolone 1 to 2 mg per kg per day, or 4 mg per kg per day for four days, as a short course that is then tapered and stopped
Anti-D for childhood ITP
Dose
50 to 75 micrograms per kg as a single intravenous dose over three to five minutes, for the Rh-D positive child with a negative direct antiglobulin test and an intact spleen
The selection among the three first-line agents is driven by the clinical context, and the ASH 2019 guideline offers the logic. IVIG is suggested over corticosteroids when a rapid rise in the platelet count is wanted, such as in the child with the moderate or severe bleed. Corticosteroids are suggested over IVIG when the cost and the intravenous access favour the oral route, such as in the child with the moderate bleed who can swallow the tablets. Anti-D is an option for the eligible child, and it works almost as quickly as IVIG, but the haemolysis risk and the withdrawal of anti-D in several countries have reduced its place in practice. The fellow names the agent, the dose, and the rationale, and the fellow checks the Rh type and the direct antiglobulin test before the anti-D. [1][8]
The second-line therapy is reserved for the persistent and the chronic ITP, the phases beyond three to twelve months, and it is where the thrombopoietin-receptor agonists, the rituximab, and the now-rare splenectomy live. The thrombopoietin-receptor agonists are the major advance of the last decade, and they work by stimulating the megakaryocyte to produce more platelets, which addresses the impaired-production half of the mechanism. Eltrombopag, an oral thrombopoietin-receptor agonist, was evaluated in the PETIT trial reported by Bussel and colleagues in 2015, a randomised placebo-controlled study in children with persistent and chronic ITP, and it showed a durable platelet response in a substantial proportion. The eltrombopag dose in the child aged six years and over is 25 to 75 mg once daily, and in the child under six years it is 12.5 to 50 mg once daily, titrated to the response. Romiplostim, the subcutaneous thrombopoietin-receptor agonist, is the alternative. [10][11]
Eltrombopag for chronic childhood ITP
Dose
25 to 75 mg orally once daily for the child aged six years and over, and 12.5 to 50 mg once daily for the child aged one to five years, titrated to maintain the platelet count over 50 times ten to the nine per litre
Rituximab, the anti-CD20 monoclonal antibody that depletes the B-cells, is the second second-line option, and it works in a proportion of the chronic cases, though the response is often transient and the relapse is common. Splenectomy, once the second-line treatment of choice, is now rare, because the thrombopoietin-receptor agonists and the rituximab have largely replaced it, and because the splenectomy carries the lifelong risk of the overwhelming post-splenectomy infection. When a splenectomy is still considered, it is deferred until the chronic phase, often well beyond twelve months, and it is preceded by the pneumococcal, the meningococcal, and the Haemophilus influenzae type b vaccination and the lifelong penicillin prophylaxis. The fellow who holds the second-line ladder, the thrombopoietin-receptor agonist first, the rituximab, and the rare splenectomy last, has the modern strategy. [11][12]
Specific Subtypes & Scenarios
The chronic ITP, the phase beyond twelve months, is its own clinical entity, and the fellow manages it as a chronic disease rather than as an acute event. The child has lived with the thrombocytopenia for a year or more, the bleeding has been recurrent but rarely severe, and the burden has shifted to the quality of life, the school and the sport, and the anxiety of the family. The management is built around the thrombopoietin-receptor agonist, which is given as a long-term maintenance therapy to keep the platelet count above the bleeding threshold, and around the regular review of the bleeding, the blood count, and the side effects. The child is supported by the multidisciplinary team, the haematology nurse, the psychologist, and the school liaison, because the chronic disease is as much a psychological as a haematological burden. [11]
The splenectomy, now rare, is the scenario the fellow is most often asked to appraise. The historical place of the splenectomy was as the second-line treatment of choice, because the spleen is the site of both the antibody production and the platelet clearance, and the removal of the spleen cured a proportion of the chronic cases. The modern place of the splenectomy is as a last resort, because the thrombopoietin-receptor agonists have taken over the second-line space, and because the splenectomy carries the lifelong risk of the overwhelming post-splenectomy infection. When a splenectomy is considered, the child is vaccinated against the pneumococcus, the meningococcus, and Haemophilus influenzae type b at least two weeks before the surgery, the lifelong penicillin prophylaxis is begun, and the family is taught the fever-as-an-emergency rule. [12]
The first two years of a child diagnosed with ITP
The secondary ITP, the thrombocytopenia driven by an underlying disorder, is managed by treating the underlying disorder alongside the platelet count. The child with the systemic lupus erythematosus and the ITP is managed jointly with the rheumatology team, and the immunosuppression that treats the lupus also treats the thrombocytopenia. The child with the common variable immunodeficiency and the ITP receives the immunoglobulin replacement that addresses the immunodeficiency, and the thrombocytopenia is monitored. The child with the Evans syndrome, the combination of ITP and autoimmune haemolytic anaemia, is often more refractory, and the second-line therapy is begun earlier. The fellow who recognises the secondary ITP and who involves the relevant specialty early, avoids the error of treating the count in isolation. [2][3]
The neonatal and the adolescent ITP differ from the typical preschool presentation, and the fellow holds the differences. The neonatal thrombocytopenia is a separate entity, because the neonatal alloimmune thrombocytopenia, in which the mother makes antibodies against a paternally-inherited platelet antigen the fetus carries, and the neonatal autoimmune thrombocytopenia, in which the mother has ITP and the antibody crosses the placenta, have their own mechanisms and their own management. The adolescent ITP carries the higher chronicity, the heavy menstrual bleeding, and the lifestyle and the reproductive implications, and the fellow addresses the contraception, the menstrual suppression, and the iron deficiency, alongside the haematological management. [3]
Complications & Pitfalls
The first pitfall is treating by the platelet count rather than by the bleeding severity, and it is the error that the ASH 2019 guideline is built to correct. The fellow who treats every child with a count under a certain threshold, because the count looks frightening, exposes the child to the harms of the treatment without the benefit, because the child with no or mild bleeding is not at risk of a major bleed. The count is monitored, but the decision is driven by the bleeding, and the fellow who holds this distinction has the central principle of the modern management. [1]
The second pitfall is missing the acute leukaemia on the blood film, and it is the error with the highest cost. The child with the leukaemia may present with the bruising and the petechiae, and if the film is not examined, or if the other cytopenias are overlooked, the diagnosis is missed, and the delay in the leukaemia treatment is the catastrophic consequence. The fellow examines the film personally, checks the other cell lines, and orders the bone marrow aspirate whenever the presentation is atypical. The corticosteroids given for a presumed ITP can transiently mask the leukaemia, which is why the marrow is done before the steroids in the atypical case. [5]
[1]The third pitfall is the haemolysis of the anti-D, and it is the reason anti-D has been withdrawn or restricted in several countries. The anti-D works by coating the Rh-D positive red cells, which are then cleared in the spleen alongside the antibody-coated platelets, and the platelet count rises because the competing red-cell clearance spares the platelets. The cost is the clinically significant intravascular haemolysis, which has caused severe anaemia and acute kidney injury, and the fellow who gives the anti-D checks the haemoglobin and the urine afterwards, and is prepared to transfuse. The direct antiglobulin test, the Rh type, and the spleen check are the gatekeepers that prevent the wrong child from receiving the anti-D. [8]
The fourth pitfall is the chronic-disease burden, the psychological impact of the years of the thrombocytopenia, the restrictions on the sport and the activity, the anxiety of the family, and the school absences. The child with the chronic ITP is supported by the multidisciplinary team, the psychologist, the school liaison, and the peer support, because the chronic disease is a whole-child burden, and the fellow who treats only the count misses the child. The quality-of-life measures, and the transition planning for the adolescent, are part of the management, and they are the reason the chronic ITP is managed in the specialist centre. [11][12]
Prognosis & Disposition
The outlook for the child with ITP is overwhelmingly favourable, and this is the central fact the fellow carries into the counselling. The great majority, about 70 to 80 percent, remit within six months, and the proportion that reaches the chronic phase beyond twelve months is about 20 to 25 percent. The intracranial haemorrhage is rare, with an incidence estimated below one percent, and the mortality, in the modern era, is very low. The family is counselled on the benign natural history, on the observation strategy, and on the safety-net, because the reassurance is as important as the treatment. [5][6]
The factors that predict the development of the chronic ITP are the older age, the adolescent onset, the gradual rather than the sudden onset, the female sex in adolescence, and the absence of the preceding viral illness. The preschool child with the sudden post-viral presentation is the child most likely to remit, and the adolescent girl with the gradual onset is the child most likely to persist. The fellow who recognises the chronic-risk profile counsels the family early, sets up the multidisciplinary support, and begins the second-line planning. [6][7]
The disposition of the child is shared between the primary care team and the haematology service, and the fellow coordinates the two. The child with the newly diagnosed ITP and the mild bleeding is managed in the outpatient setting, with the haematology review within a week to ten days and the primary care team holding the safety-net. The child with the moderate or the severe bleed is managed in the hospital, with the first-line therapy and the inpatient observation. The child with the chronic ITP is followed in the specialist haematology clinic, with the multidisciplinary support and the second-line therapy. The discharge from the clinic is appropriate when the count has normalised and the bleeding has resolved, and the family is given the written advice and the open access for the future. [1]
Special Populations
The neonate with the thrombocytopenia is a separate population, and the fellow holds the two mechanisms. The neonatal alloimmune thrombocytopenia occurs when the mother, who lacks a platelet-specific antigen that the fetus has inherited from the father, makes antibodies against the fetal platelet antigen, and the antibody crosses the placenta and destroys the fetal platelets. The neonatal autoimmune thrombocytopenia occurs when the mother has ITP, and her anti-platelet antibody crosses the placenta. The two are distinguished by the maternal platelet count and the antibody specificity, and the management differs, because the alloimmune thrombocytopenia may need the matched-antigen-negative platelet transfusion, while the autoimmune thrombocytopenia is managed by the IVIG if the bleeding warrants it. [3]
The adolescent with the ITP is the second special population, and the fellow addresses the higher chronicity, the heavy menstrual bleeding, and the lifestyle and the reproductive implications. The adolescent is more likely than the preschool child to develop the chronic ITP, and the second-line therapy is begun earlier if the bleeding is troublesome. The heavy menstrual bleeding is managed by the menstrual suppression, the norethisterone or the levonorgestrel intrauterine system, and the iron replacement, because the chronic blood loss causes the iron deficiency. The contraception and the reproductive counselling are addressed, because the adolescent girl with the ITP who becomes pregnant carries the added risks, and the thrombopoietin-receptor agonists and the rituximab are reviewed for the reproductive safety. [11]
ITP
The child with the inherited thrombocytopenia, such as the Wiskott-Aldrich syndrome or the congenital amegakaryocytic thrombocytopenia, is the third special population, and the fellow distinguishes these from the ITP because they do not remit and they are not treated as ITP. The small platelets of the Wiskott-Aldrich syndrome, the eczema, and the immunodeficiency are the pointers, and the mean platelet volume is the simple test that separates the small-platelet inherited disorders from the large-platelet ITP. The child with the secondary ITP, driven by the systemic lupus erythematosus or the common variable immunodeficiency, is the fourth population, and the management is the joint management of the underlying disorder and the platelet count. [3]
The child in the rural or the remote setting, far from the specialist centre, is managed by the local team in the partnership with the haematology service through the telehealth and the shared protocols. The local team holds the safety-net, the written emergency plan, and the first-line therapy, and it ships the child to the centre for the second-line and the multidisciplinary care. The fellow who works in the rural setting values the telehealth link, because the child who is connected to the centre through it does as well as the child who lives next door to it. [1]
Evidence, Guidelines & Regional Differences
The evidence base for the management of childhood ITP rests on the ICIS registries, the treatment trials, and the ASH 2019 guideline, and the fellow who holds these three has the framework. The ICIS prospective registry of 2540 children reported by Kühne in 2003, and the twelve-month follow-up registry I reported by Imbach in 2006, established the natural history, the remission rate, and the proportion that reaches the chronic phase. The Blanchette trial of 1994, a randomised comparison of IVIG, anti-D, and oral prednisone in the childhood acute ITP, established the first-line agents and their relative speed, and it remains the classic first-line trial. The PETIT trial of eltrombopag reported by Bussel in 2015 established the thrombopoietin-receptor agonist in the chronic paediatric ITP. [5][6][8][10]
ASH 2019 guideline - Neunert 2019
Key finding
The American Society of Hematology 2019 guideline recommended, as a strong recommendation, observation over treatment for the child with newly diagnosed ITP and no bleeding or only mild skin bleeding, regardless of the platelet count, and it set out the first-line therapy doses of IVIG 0.8 to 1 g per kg, short-course corticosteroids, and anti-D 50 to 75 micrograms per kg.
PETIT trial - eltrombopag, Bussel 2015
Key finding
The randomised placebo-controlled PETIT trial showed that eltrombopag produced a durable platelet response in a substantial proportion of children with persistent and chronic ITP, at a dose of 25 to 75 mg once daily in the six-and-over and 12.5 to 50 mg in the under-six.
The guidelines that translate this evidence into practice are the ASH 2019 guideline of Neunert and colleagues, and the updated international consensus report of Provan and colleagues, both published in Blood Advances in 2019, and together they define the modern management. The ASH guideline is the most testable, because it sets the observation-first recommendation and the first-line doses, and it is the source the fellow quotes in the exam. The international consensus report adds the secondary-cause workup, the bleeding-score assessment, and the chronic-disease management, and it complements the ASH framework. The local guidelines, the British Committee for Standards in Haematology, the Canadian, and the Australian and New Zealand pathways, follow the same principles with the regional variations in the drug availability. [1][2]
The chief controversies are the place of the anti-D, the duration of the thrombopoietin-receptor agonist therapy, and the rare splenectomy. The anti-D has been withdrawn or restricted in several countries because of the clinically significant haemolysis, and the fellow checks the local availability before the anti-D is considered. The duration of the thrombopoietin-receptor agonist therapy is open, because the long-term safety is still being followed, and some children are treated for years while others are tapered. The splenectomy is now rare, because the thrombopoietin-receptor agonists have taken over, but it remains an option for the refractory case after the vaccination and the penicillin prophylaxis. The fellow follows the local guideline and explains the choice to the family. [11][12]
Exam Pearls
The single most testable fact is the definition, and the fellow learns it word for word. Immune thrombocytopenia is an isolated thrombocytopenia, a platelet count under 100 times ten to the nine per litre, in an otherwise well child, with a normal blood film showing large platelets and no blasts, and no hepatosplenomegaly. The fellow who can state the count threshold, the isolated nature, the well child, and the normal film, has the diagnosis. The term immune has replaced idiopathic, and the term purpura has been dropped, per the Rodeghiero standard of 2009. [4]
The ASH 2019 observation recommendation is the second most testable fact, and it is asked as a principle. The child with newly diagnosed ITP and no bleeding or only mild skin bleeding is managed by observation, regardless of the platelet count, and the decision to treat is driven by the bleeding severity. The fellow who treats every low count, or who cannot state the observation principle, loses the marks. The rationale is the low bleeding risk in the mild case, the treatment harms, and the favourable natural history. [1]
[4] [6]The first-line doses round out the high-yield set. IVIG is 0.8 to 1 g per kg as a single dose, the corticosteroids are a short course of oral prednisolone at 1 to 2 mg per kg per day, and the anti-D is 50 to 75 micrograms per kg for the Rh-D positive child with a negative direct antiglobulin test and an intact spleen. The bone marrow aspirate is not routine for the typical presentation. The thrombopoietin-receptor agonist eltrombopag is 25 to 75 mg once daily for the six-and-over and 12.5 to 50 mg for the under-six. The fellow who holds these four sets of numbers, the definition, the observation principle, the first-line doses, and the phase cut-offs, carries the whole topic. [1][10]
References
- [1]Neunert C, Terrell DR, Arnold DM American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv, 2019.PMID 31794604
- [2]Provan D, Arnold DM, Bussel JB Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv, 2019.PMID 31770441
- [3]Cooper N, Ghanima W Immune Thrombocytopenia. N Engl J Med, 2019.PMID 31483965
- [4]Rodeghiero F, Stasi R, Gernsheimer T Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood, 2009.PMID 19005182
- [5]Kühne T, Buchanan GR, Zimmerman S A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr, 2003.PMID 14615730
- [6]Imbach P, Kühne T, Müller D Childhood ITP: 12 months follow-up data from the prospective registry I of the Intercontinental Childhood ITP Study Group (ICIS). Pediatr Blood Cancer, 2006.PMID 16086422
- [7]Kühne T, Berchtold W, Michaels LA Newly diagnosed immune thrombocytopenia in children and adults: a comparative prospective observational registry of the Intercontinental Cooperative Immune Thrombocytopenia Study Group. Haematologica, 2011.PMID 21880634
- [8]Blanchette V, Imbach P, Andrew M Randomised trial of intravenous immunoglobulin G, intravenous anti-D, and oral prednisone in childhood acute immune thrombocytopenic purpura. Lancet, 1994.PMID 7915773
- [9]Mithoowani S, Arnold DM First-Line Therapy for Immune Thrombocytopenia. Hamostaseologie, 2019.PMID 31170773
- [10]Bussel JB, de Miguel PG, Despotovic JM Eltrombopag for the treatment of children with persistent and chronic immune thrombocytopenia (PETIT): a randomised, multicentre, placebo-controlled study. Lancet Haematol, 2015.PMID 26688484
- [11]Neunert CE, Grace RF Thrombopoietin-receptor agonists in children with immune thrombocytopenia. Lancet, 2015.PMID 26231462
- [12]Grace RF, Despotovic JM, Bennett CM Physician decision making in selection of second-line treatments in immune thrombocytopenia in children. Am J Hematol, 2018.PMID 29659042