Paeds · haematology-oncology-and-transfusion
Wilms tumour and renal malignancies
Also known as Nephroblastoma · Childhood kidney tumour · Clear cell sarcoma of kidney · Malignant rhabdoid tumour of the kidney · Congenital mesoblastic nephroma · Paediatric renal cell carcinoma
Fellowship guide to Wilms tumour and the paediatric renal malignancies. Covers nephroblastoma as the commonest renal malignancy of childhood arising from the nephrogenic blastema with the WT1 and 11p15 genetic hits and the syndromic predisposition of WAGR, Beckwith-Wiedemann and Denys-Drash, the peak age of three to four years and the classic presentation of a painless abdominal mass with haematuria and hypertension, the clear cell sarcoma and the malignant rhabdoid tumour and the congenital mesoblastic nephroma of the infant, the first-line ultrasound with Doppler of the renal vein and the inferior vena cava, the computed tomography or magnetic resonance imaging and the chest imaging for staging, the Children's Oncology Group do-not-biopsy strategy of the upfront radical nephrectomy against the SIOP strategy of preoperative chemotherapy, the risk-adapted vincristine, dactinomycin and doxorubicin chemotherapy and the selective radiotherapy, and the late effects of cardiotoxicity and renal irradiation.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
Overview & Definition
A mother finds a lump in her three-year-old's belly at bath time, the child is otherwise well, and the question at the bedside is whether this is an enlarged spleen, a distended bladder, or a kidney tumour. Wilms tumour, the nephroblastoma, is the commonest renal malignancy of childhood, and the single thing that separates the child who is cured from the child who is harmed is how fast the clinician recognises the painless abdominal mass, reaches the ultrasound, and refers to the specialist centre without biopsying the mass on the way. The tumour is an embryonal neoplasm that arises from the persistent nephrogenic blastema, the immature kidney-forming tissue that should have disappeared before birth, and it grows silently inside the kidney until a parent or a doctor feels it. [1]
The paediatric renal malignancies are a family rather than a single disease, and the histology and the age change everything. Wilms tumour dominates, peaking at three to four years of age and accounting for the great majority of the childhood renal tumours, while the clear cell sarcoma of the kidney, the malignant rhabdoid tumour and the congenital mesoblastic nephroma occupy the corners and carry the different prognoses. The renal cell carcinoma of the adolescent sits at the other end of the age range. Holding these tumours together, with the Wilms at the centre, is what allows the fellow to answer the questions the boards ask about the renal mass in a child. [1][3]
The first task at the bedside is not to name the tumour but to judge whether the child is in danger from a tumour rupture, a hypertension crisis, or a tumour thrombus in the inferior vena cava. The second task is to image the kidney and the cava, and the third is to build the multidisciplinary plan that runs paediatric oncology, paediatric surgery, radiation oncology, nephrology and cardiology together. The reason the topic sits at the heart of the fellowship examination is that the candidate who can read the pattern, reach the scan, and navigate the surgery-first pathway while protecting the future kidney and heart is demonstrating exactly the reasoning the boards reward. [4]
Classification
The most useful way to classify a paediatric renal mass at the bedside is by the age of the child, because the age predicts the histology, the behaviour and the prognosis. Wilms tumour is the school-age and the preschool child's tumour, peaking at three to four years, and it is the commonest of the group. The congenital mesoblastic nephroma is the tumour of the newborn and the young infant, and it is the one renal tumour that is largely benign and cured by the resection. The malignant rhabdoid tumour and the clear cell sarcoma of the kidney are the aggressive tumours of the young child that demand the bone and the brain imaging and the intensive therapy. The renal cell carcinoma is the tumour of the adolescent, biologically closer to the adult disease. [1][3]

A parallel classification runs through the histology and the behaviour of the Wilms tumour itself, and it sorts the tumours into the favourable and the unfavourable in a way that changes the intensity of the therapy. The favourable histology is the classic triphasic pattern of the blastemal, the stromal and the epithelial components, and it carries the excellent prognosis. The anaplastic histology is the unfavourable pattern, defined by the enlarged, hyperchromatic and multipolar nuclei, and it is divided into the focal and the diffuse anaplasia, with the diffuse anaplasia carrying the worst prognosis of the Wilms family. The molecular layer, with the loss of the WT1, the 11p15 imprinting and the TP53 mutation of the anaplasia, now sits on top of the histology and refines the risk. [2][8]
The staging classification is the one that drives the treatment, and it runs from the stage one to the stage five. The Children's Oncology Group stages the tumour after the upfront nephrectomy, with the stage one confined to the kidney and completely resected, the stage two beyond the kidney but completely resected, the stage three with the residual abdominal disease or the lymph node involvement, the stage four with the haematogenous metastases, and the stage five the bilateral disease. The SIOP stages the tumour after the preoperative chemotherapy, and it pairs the local stage with the histological response to drive the postoperative therapy. [4][5]
Favourable-histology Wilms
classic triphasic
- Blastemal, stromal and epithelial components
- The commonest pattern, around ninety percent of cases
- Cured in around ninety percent with the modern therapy
- Treated with the vincristine and the dactinomycin, the doxorubicin for the higher stage
Anaplastic Wilms
unfavourable
- Enlarged multipolar hyperchromatic nuclei
- Focal versus the diffuse anaplasia
- Diffuse anaplasia carries the worst prognosis of the Wilms family
- Treated with the more intensive regimen including the doxorubicin and the radiotherapy
Clear cell sarcoma
bone and brain spread
- The aggressive tumour of the young child
- Metastasises to the bone and the brain, unlike the Wilms
- Needs the bone scan and the brain imaging
- Treated with the doxorubicin-based intensive regimen
Malignant rhabdoid tumour
infant, central nervous system
- The tumour of the infant and the very young child
- Associated with the SMARCB1 INI1 loss and the central nervous system tumours
- Carries the worst prognosis of the renal malignancies
- Treated with the intensive multimodal therapy
Epidemiology & Risk Factors
The epidemiology of the Wilms tumour is the story of a common and curable childhood cancer. Wilms tumour is the commonest renal malignancy of childhood and one of the commonest solid tumours of childhood, with an annual incidence of around eight to ten per million children under fifteen, and a lifetime risk of roughly one in ten thousand. The peak age is three to four years, with the great majority diagnosed between one and five years of age, and there is a slight female predominance and a higher incidence in children of African ancestry and a lower incidence in those of East Asian ancestry. Around five to ten percent of the children present with the bilateral disease, which reshapes the entire surgical strategy around the kidney preservation. [1]
The risk factors cluster around the inherited overgrowth and the WT1 syndromes, and the fellow who knows them can both anticipate the tumour and counsel the family. The Beckwith-Wiedemann syndrome and the isolated hemihypertrophy carry the 11p15 imprinting abnormality and the macrosomia, the macroglossia and the abdominal wall defect, and they raise the Wilms risk enough to warrant the surveillance ultrasound. The WAGR syndrome, the deletion of the 11p13 that carries the WT1 gene, combines the Wilms, the aniridia, the genitourinary anomalies and the intellectual disability. The Denys-Drash syndrome pairs the WT1 mutation with the mesangial sclerosis and the pseudohermaphroditism. These syndromes are sought in every child with a Wilms tumour because they change the counselling and the surveillance of the family. [2][6]
The molecular epidemiology is the modern layer that explains the biology of the tumour, and it is built around the WT1 gene and the 11p15 locus. The WT1 gene on the 11p13 is the classic Wilms tumour suppressor, lost in the WAGR and the Denys-Drash syndromes and in a fraction of the sporadic tumours. The 11p15 locus is the Beckwith-Wiedemann locus, and its loss of the imprinting drives the overgrowth and the Wilms predisposition. The WTX gene, the AMER1, on the X chromosome is mutated in a further fraction. The genetic landscape is now mapped in detail, and it links the developmental biology of the kidney to the clinical behaviour of the tumour. [2][8]
The environmental and the parental risk factors are weak and inconsistent, and the fellow should not overstate them. The bulk of the Wilms tumours arise from the developmental error of the persistent nephrogenic rest rather than from an identifiable exposure, and the strong risk factors are the inherited syndromes. The one modifiable element is the surveillance of the syndromic child, with the serial abdominal ultrasound that catches the tumour early. The lesson for the exam is that the inherited syndromes are the risk factors worth naming, and the environmental causes are the ones worth questioning. [6]
Pathophysiology
The pathophysiology of the Wilms tumour is best understood as the failed completion of the kidney development, with the immature tissue that should have matured instead persisting and acquiring the genetic hits that turn it malignant. The kidney forms from the metanephric blastema, and the immature cells normally differentiate into the nephrons and disappear before birth. In the child who develops a Wilms tumour, a cluster of these cells persists after birth as a nephrogenic rest, and a second genetic hit, the loss of the WT1 or the 11p15 or the WTX, releases the rest from its growth control and drives it into the tumour. [2][3]
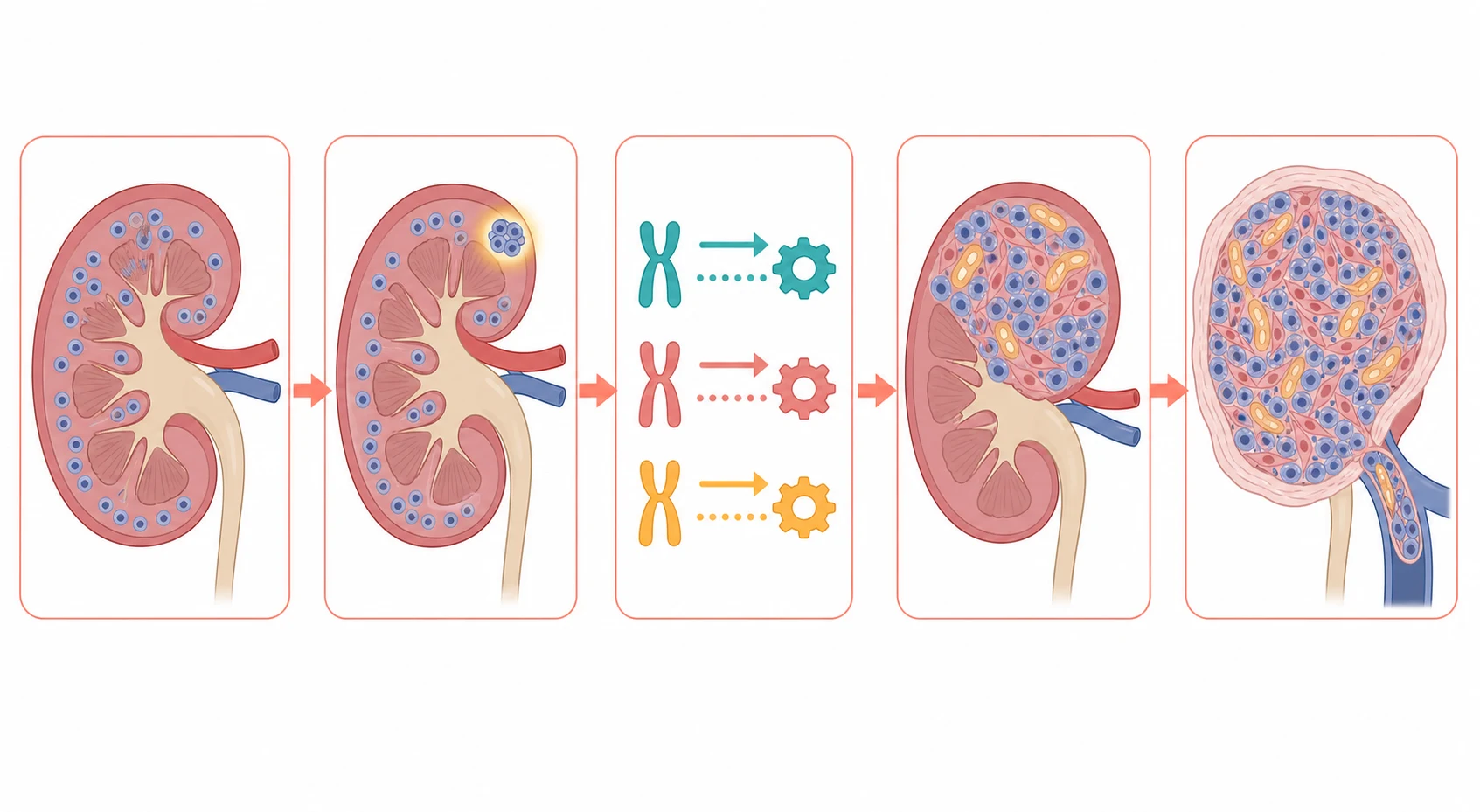
A second mechanism explains why the Wilms tumour looks the way it does under the microscope. The tumour retains the capacity to attempt the differentiation, so that it grows as the classic triphasic mixture of the small blue blastemal cells, the spindle stromal cells and the epithelial tubules and glomeruloid structures. This triphasic pattern is the hallmark of the favourable histology, and it reflects the embryonal origin of the tumour. The anaplasia, by contrast, is the marker of the genomic instability and the resistance to the chemotherapy, and it carries the worse prognosis because the anaplastic cells fail to die under the treatment. [8]
The clinical consequences of the tumour flow from its anatomy within the kidney. The growing mass distends the renal capsule and presents as the painless mass, and it invades the collecting system to produce the haematuria. The tumour compresses or the ischaemic kidney releases the renin, and the renin-angiotensin cascade produces the hypertension that is found in around a fifth of the children. The tumour extends into the renal vein and then into the inferior vena cava and even the right atrium in a minority, and this intravascular extension is the pathophysiology that changes the surgical plan and that demands the Doppler ultrasound before the operating room. [1][9]
The pathophysiology of the late effects is the cost of the therapy that cures the child, and the fellow must hold it alongside the cure. The doxorubicin produces the dose-dependent cardiotoxicity, the congestive heart failure that can declare decades later, and it is the reason the cardiac surveillance is built into the survivorship. The abdominal radiotherapy injures the remaining kidney and the musculoskeletal and the reproductive tissues, and it is the reason the radiotherapy is reserved for the higher stage. Understanding these mechanisms is what allows the survivorship plan to anticipate and to mitigate the harm rather than to react after it appears. [4]
Clinical Presentation
The child with a Wilms tumour presents in one of several ways, and the recognition of the pattern at the bedside is the skill that decides the outcome. The dominant presentation is the painless abdominal mass, found by the parent at the bath time or the dressing, or by the doctor at a routine examination, and it is typically smooth, firm and confined to one flank without crossing the midline. The mass may grow rapidly over weeks, and the child is often otherwise well, which is the feature that can lull the clinician into the reassurance. The haematuria and the hypertension are the associated features that point to the kidney. [1]
The history holds the diagnosis more often than the examination, and the tempo and the associated features are the discriminators. The parent who describes a lump that appeared over a few weeks, with the occasional blood in the urine or the complaint of the abdominal discomfort, is describing the Wilms tumour until the imaging settles it. The hypertension is often asymptomatic and discovered at the examination, but it can declare as the headache or the irritability. The fever and the abdominal pain can accompany a tumour that has undergone the internal haemorrhage or the necrosis. [4]
The atypical presentations are the ones that catch the unwary clinician, and the fellow must hold them. The acute abdominal pain and the abdominal distension can be the presentation of the tumour rupture, which spills the tumour into the peritoneal cavity and upstages the disease, and it is the emergency that demands the urgent imaging and the oncology referral. The varicocele in the boy, particularly the right-sided varicocele, can be the presentation of the cava obstruction by the tumour thrombus. The syndromic child, the Beckwith-Wiedemann or the hemihypertrophy, can have the tumour found on the surveillance ultrasound before it is ever felt. [9]
The other paediatric renal tumours present at their own ages and with their own patterns, and the fellow must separate them. The congenital mesoblastic nephroma presents in the newborn and the young infant as a large, firm abdominal mass, and it is the largely benign tumour cured by the resection. The clear cell sarcoma and the malignant rhabdoid tumour present in the young child with the abdominal mass and the metastases to the bone and the brain, which is why they demand the bone scan and the brain imaging. The renal cell carcinoma presents in the adolescent with the haematuria and the flank pain, biologically closer to the adult disease. [3]
Differential Diagnosis
The differential of the child with the abdominal mass is built around the site and the character of the mass, and the imaging is the test that resolves it. The Wilms tumour is a flank mass that arises from the kidney, and it is distinguished from the neuroblastoma, the adrenal mass that typically crosses the midline and pushes the kidney down. The hydronephrosis and the multicystic dysplastic kidney present as the cystic flank masses, and the polycystic kidney disease as the bilateral enlarged kidneys. The splenomegaly and the hepatomegaly are the upper abdominal masses that the careful examination separates from the renal mass. [1]
The benign mimics that must be excluded, because treating them as a tumour is as harmful as missing a tumour, are the renal cyst, the mesoblastic nephroma of the infant, the multilocular cystic nephroma and the angiomyolipoma of the tuberous sclerosis. The imaging, with the ultrasound and the computed tomography or the magnetic resonance imaging, separates the solid from the cystic lesion and the fat-containing angiomyolipoma from the Wilms. The multilocular cystic nephroma is the cystic lesion of the young child that is largely benign but that overlaps with the cystic Wilms, and it is managed with the surgery. [3]
The chief diagnostic pitfalls for the fellow are the cases in which the diagnosis is delayed or the management is derailed because the pattern is misread. The Wilms mass that is labelled an enlarged liver or an enlarged spleen, and whose flank origin is missed, is the classic delayed diagnosis, and the lesson is to image any unexplained abdominal mass in a child. The hydronephrosis that is labelled a tumour, and whose cystic character is not recognised, is the second. The Wilms that is biopsied before the nephrectomy, and that is upstaged by the spill, is the third, and it is the one the examination tests most heavily. [4]
The separation of the Wilms tumour from the neuroblastoma is the single most examined differential, because the two tumours share the age and the abdominal presentation. The neuroblastoma is the adrenal mass that crosses the midline, that calcifies, that elevates the urinary catecholamines, and that pushes the kidney downward rather than arising from it. The Wilms tumour is the renal mass that stays on one side, that is smooth and solid, and that displaces rather than crosses. The imaging, with the ultrasound and the cross-sectional scan, and the urinary catecholamines, resolve the question in almost every case. [1]
Clinical & Bedside Assessment
The bedside assessment of the child with the suspected renal mass is a search for the mass itself, the signs of the complications, and the stigmata of the syndromes. The assessment begins with the airway, the breathing and the circulation, and the blood pressure, because the hypertension is common and the tumour rupture is the emergency. Once the child is stable, the focused history turns to the onset and the growth of the mass, the haematuria, the abdominal pain, the family history of the cancer, and the birth history of the macrosomia or the hemihypertrophy. [1]
The examination is systematic and takes only a few minutes, but each finding carries weight. The mass is examined for the size, the consistency, the mobility and the crossing of the midline, because the Wilms tumour is the smooth, firm, mobile flank mass that does not cross the midline, while the neuroblastoma is the hard, fixed mass that does. The blood pressure is measured and plotted, because the hypertension is found in around a fifth and it requires the control before the surgery. The urinalysis is checked for the haematuria, and the skin is examined for the stigmata of the Beckwith-Wiedemann and the hemihypertrophy. [4]
The focused examination of the child with the suspected renal mass
Measure and plot the blood pressure, because the hypertension is found in around a fifth of the children with a renal tumour
Examine the mass for the size, the consistency, the mobility and the crossing of the midline, distinguishing the smooth flank Wilms from the hard crossing neuroblastoma
Check the urinalysis for the haematuria, the common associated feature of the renal tumour
Inspect the skin and the growth for the stigmata of the Beckwith-Wiedemann, the hemihypertrophy and the aniridia of the WAGR
Examine for the varicocele in the boy, the sign of the inferior vena cava obstruction by the tumour thrombus
Assess the child for the signs of the tumour rupture, the abdominal distension, the peritonism and the instability
The severity of the presentation and the danger of the complications are judged at the bedside, and they decide the speed of the scan and the start of the blood pressure control. The child with the abdominal pain, the distension and the peritonism has the tumour rupture until the imaging proves otherwise, and the child is moved to the urgent scan and the oncology referral. The child with the right-sided varicocele or the cava obstruction signs has the intravascular tumour thrombus, and the Doppler of the cava is performed without delay. The child with the severe hypertension has the blood pressure controlled before the surgery, with the careful titration of the antihypertensive. [9]
The stigmata of the inherited syndromes are sought in every child with a Wilms tumour, because they change the counselling and the surveillance. The aniridia, the absence of the iris, points to the WAGR syndrome and the 11p13 deletion. The macrosomia, the macroglossia, the ear creases and the abdominal wall defect point to the Beckwith-Wiedemann syndrome and the 11p15 locus. The asymmetrical growth, the hemihypertrophy, points to the overgrowth syndrome and the Wilms predisposition. The genital ambiguity points to the Denys-Drash syndrome and the WT1 mutation. These findings are the bridge to the genetic counselling and the surveillance of the family. [6]
Investigations
The investigation of a suspected renal mass moves in two steps, the imaging that confirms and localises the tumour, and the surgical and the histological studies that name it. The first and the decisive test is the abdominal ultrasound with the Doppler of the renal vein and the inferior vena cava, because it confirms the renal origin, it shows the solid character of the mass, and it defines the tumour thrombus that changes the surgical approach. The ultrasound is the test that every child with an abdominal mass receives first, and it is the one that decides the next step. [1]
The cross-sectional imaging refines the picture and stages the tumour. The computed tomography or the magnetic resonance imaging of the abdomen defines the size, the local extension, the relationship to the renal vessels and the contralateral kidney, and the presence of the intravascular thrombus. The chest computed tomography is performed in every child, because the lung is the commonest site of the haematogenous metastasis and the presence of the pulmonary disease moves the tumour to the stage four. The bone scan and the brain magnetic resonance imaging are added for the clear cell sarcoma and the malignant rhabdoid tumour, because both metastasise to the bone and the brain. [4][9]
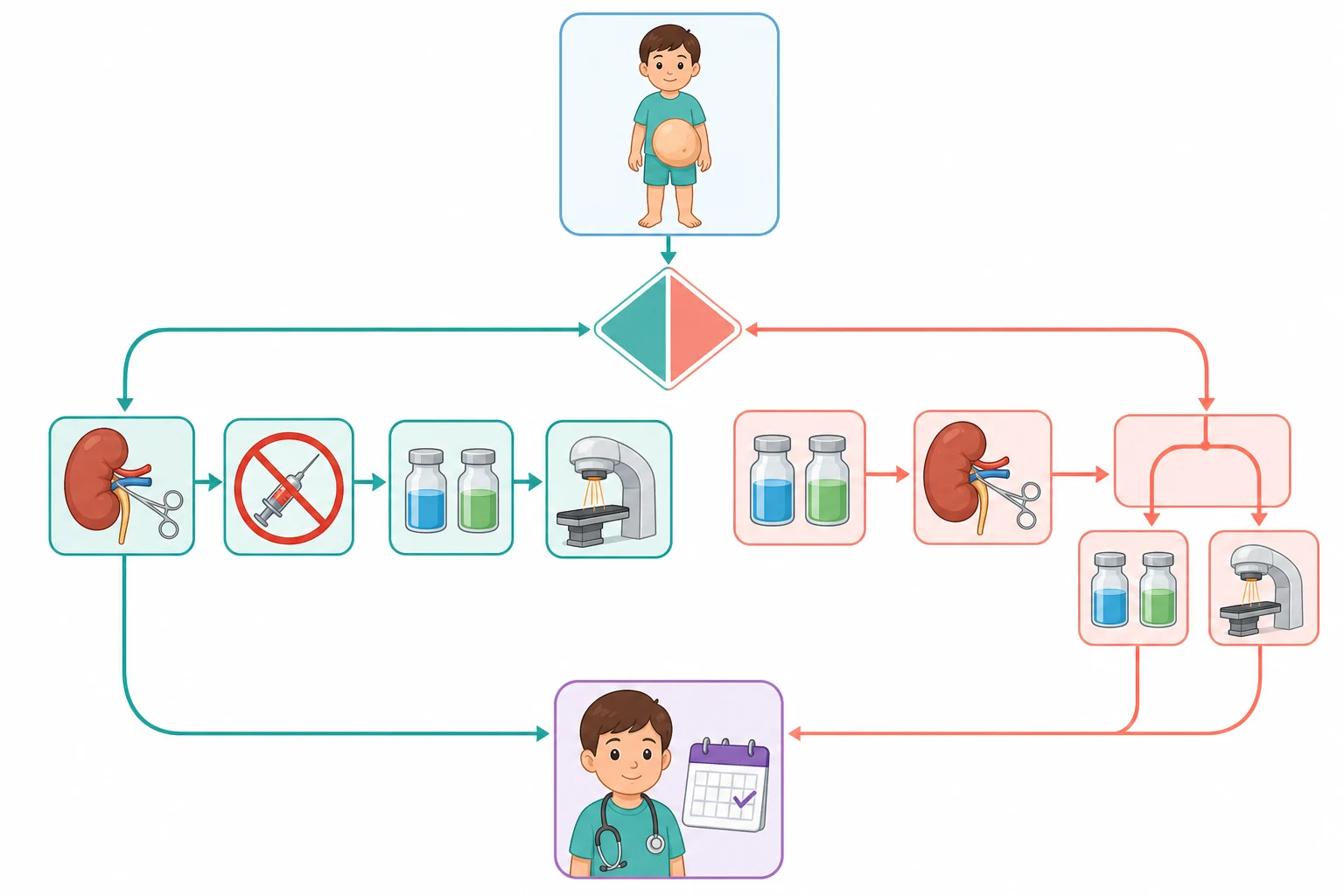
Across Australia, Aotearoa New Zealand, the United Kingdom, Europe, the United States and Canada, the ultrasound with the Doppler and the cross-sectional imaging of the abdomen and the chest are the standard diagnostic tests for a suspected paediatric renal tumour. The policy on the biopsy divides the two great traditions, the Children's Oncology Group that avoids the biopsy of the typical tumour and proceeds to the upfront nephrectomy, and the SIOP that gives the preoperative chemotherapy to the typical tumour and resects it afterwards. The fellow should know the local protocol, because it changes the sequence of the care.
[4][5]The histological and the molecular diagnosis is made on the tumour tissue, obtained at the nephrectomy in the Children's Oncology Group approach or after the preoperative chemotherapy in the SIOP approach. The histology grades the tumour as the favourable or the anaplastic, with the focal and the diffuse anaplasia distinguished, and the molecular studies add the WT1, the 11p15 and the TP53 alterations that refine the risk. The loss of the INI1, the SMARCB1, defines the malignant rhabdoid tumour and separates it from the Wilms. The histology and the molecular layer together drive the risk-adapted therapy. [2][8]
The preoperative blood tests are requested once the tumour is confirmed, and they anticipate the surgical and the medical complications. The full blood count, the coagulation, the electrolytes and the renal function are sent before the surgery, because the renal function and the coagulation guide the operative and the anaesthetic plan. The urinalysis confirms the haematuria. The blood pressure is monitored and controlled, and the renin and the aldosterone are occasionally sent for the hypertension workup. The tumour markers are not useful for the Wilms tumour itself, but the urinary catecholamines are sent when the neuroblastoma is in the differential. [1]
Management — Resuscitation
The child with the renal tumour is rarely the dramatic resuscitation, but the complications demand the immediate response. The tumour rupture, with the abdominal pain, the distension and the peritonism, is the emergency that spills the tumour into the peritoneal cavity and upstages the disease, and the child is kept at rest, the imaging is performed urgently, and the oncology team is alerted before any further handling. The severe hypertension is controlled with the careful titration of the antihypertensive, and the child is kept calm and pain-free to avoid the hypertensive surges. [9]
The intravascular tumour thrombus is the complication that changes the surgical plan, and it is defined before the operating room. The thrombus that extends into the renal vein alone is managed at the nephrectomy with the ligation of the vein. The thrombus that extends into the inferior vena cava, above the hepatic veins, or into the right atrium demands the more complex surgery, sometimes the cardiopulmonary bypass, and occasionally the preoperative chemotherapy to shrink the thrombus first. The Doppler ultrasound and the cross-sectional imaging define the cephalad extent of the thrombus, and the plan is built around it. [9]
The hypertension is managed with the antihypertensive that is titrated to the response, and the choice is built around the calcium channel blocker or the beta-blocker, with the angiotensin-converting enzyme inhibitor avoided in the single kidney. The tumour itself is the source of the renin, and the definitive surgery or the chemotherapy resolves the hypertension in most. The blood pressure is monitored through the surgery, because the anaesthetic and the surgical manipulation can provoke the surges, and the hypotension after the tumour removal is anticipated as the renin source is removed. [1]
The disposition of the child is to the specialist paediatric oncology centre, and the regional or the rural hospital is responsible for the recognition, the initial imaging and the safe transfer. The child with the stable tumour is transferred for the definitive imaging and the surgery, and the child with the rupture or the cava thrombus is stabilised and transferred with the oncology and the surgical teams informed. The family is counselled on the plan, and the child is prepared in an age-appropriate way for the hospital stay and the surgery. [1]
Management — Definitive & Stepwise
The definitive management of the Wilms tumour is built around the three modalities of the surgery, the chemotherapy and the radiotherapy, and the combination is tailored to the stage, the histology and the molecular subtype. The overarching principle is that the complete surgical resection is the foundation of the cure, that the chemotherapy is added for the higher stage, and that the radiotherapy is reserved for the residual abdominal disease and the metastases. The two great traditions, the Children's Oncology Group and the SIOP, differ on the sequence but share the goal. [4]
[4] [5]The Children's Oncology Group strategy is the upfront radical nephrectomy for the typical resectable tumour, followed by the stage and the histology-based chemotherapy. The surgery removes the kidney and the tumour en bloc through the transverse abdominal incision, with the sampling of the lymph nodes and the inspection of the contralateral kidney and the peritoneum. The stage is then assigned from the surgical findings and the postoperative scan, and the chemotherapy and the radiotherapy are built around it. The avoidance of the biopsy is the cornerstone, because the intact capsule and the clean resection are what keep the tumour at the lower stage. [4]
The SIOP strategy is the preoperative chemotherapy for the radiologically typical tumour, followed by the nephrectomy and the histology-based therapy. The child receives the four weeks of the vincristine and the dactinomycin before the surgery, which shrinks the tumour and reduces the rupture risk, and the postoperative histological response then drives the risk stratification into the low, the intermediate and the high risk. The two strategies achieve the similar survival, and the choice between them is the regional protocol rather than the individual decision. The fellow must know both and defend each. [5][7]
Risk-adapted chemotherapy for the favourable-histology Wilms tumour
Dose
Stage one and two favourable histology receive the vincristine and the dactinomycin, the two-drug regimen, with no radiotherapy. Stage three favourable histology receives the vincristine, the dactinomycin and the doxorubicin, the three-drug regimen, with the abdominal radiotherapy. Stage four favourable histology receives the three-drug regimen with the radiotherapy to the metastatic sites, typically the lungs
The anaplastic Wilms tumour and the high-risk renal tumours receive the more intensive therapy, because they are the ones that fail the standard regimen. The diffuse anaplasia is treated with the regimen that adds the cyclophosphamide, the etoposide and the carboplatin to the vincristine, the dactinomycin and the doxorubicin, along with the radiotherapy. The clear cell sarcoma of the kidney is treated with the doxorubicin-based intensive regimen and the radiotherapy, and it requires the bone and the brain imaging at the diagnosis and the follow-up. The malignant rhabdoid tumour is treated with the intensive multimodal therapy and carries the worst prognosis of the renal malignancies. [3][8]
The bilateral Wilms tumour, the stage five disease, is the scenario that reshapes the surgery around the kidney preservation, because the bilateral radical nephrectomy would commit the child to the dialysis. The contemporary strategy is the preoperative chemotherapy to shrink both tumours, followed by the bilateral nephron-sparing surgery that removes the tumours while preserving the renal parenchyma. The goal is the cure with the least renal loss, and the renal function is monitored for the life, because the chronic kidney disease is the long-term risk. The fellow who can hold the cure and the kidney preservation together demonstrates the reasoning the boards reward. [10]
Specific Subtypes & Scenarios
The favourable-histology Wilms tumour
The favourable-histology Wilms tumour is the paradigm of the curable paediatric cancer, and it is the tumour the fellow must know in detail. It presents with the classic triphasic histology of the blastemal, the stromal and the epithelial components, it grows within the kidney, and it is treated with the surgery and the stage-based chemotherapy. The stage one and two disease receive the two-drug regimen of the vincristine and the dactinomycin with no radiotherapy, and the survival exceeds ninety percent. The stage three disease receives the three-drug regimen with the radiotherapy, and the survival sits around eighty to ninety percent. The stage four disease, with the lung metastases, receives the three-drug regimen and the lung radiotherapy, and the survival is around seventy to eighty percent. [4][9]
The anaplastic Wilms tumour
The anaplastic Wilms tumour is the unfavourable pattern, and it is the one that fails the standard therapy. The anaplasia is defined by the enlarged, the hyperchromatic and the multipolar nuclei, and it is divided into the focal anaplasia, confined to the discrete focus within the tumour, and the diffuse anaplasia, which is the widespread change that carries the worst prognosis. The diffuse anaplasia is treated with the intensive regimen that adds the cyclophosphamide, the etoposide and the carboplatin, along with the radiotherapy. The TP53 mutation underlies much of the anaplasia, and it is the molecular marker of the chemotherapy resistance. The fellow must distinguish the focal from the diffuse anaplasia, because it changes the intensity and the prognosis. [8]
The clear cell sarcoma and the malignant rhabdoid tumour
The clear cell sarcoma of the kidney and the malignant rhabdoid tumour are the aggressive renal tumours of the young child, and they are distinguished from the Wilms by the histology, the behaviour and the metastatic pattern. The clear cell sarcoma, named for its clear cells, is the tumour that metastasises to the bone and the brain, which is why it demands the bone scan and the brain imaging that the Wilms does not. The malignant rhabdoid tumour, defined by the loss of the INI1 or the SMARCB1, is the tumour of the infant that is associated with the synchronous central nervous system tumours and that carries the worst prognosis of the renal malignancies. Both are treated with the intensive multimodal therapy. [3][8]
RENAL
The bilateral and the syndromic Wilms tumour
The bilateral Wilms tumour, the stage five disease, is managed with the kidney preservation as the central goal, because the bilateral radical nephrectomy would commit the child to the dialysis. The contemporary strategy is the preoperative chemotherapy to shrink both tumours, followed by the bilateral nephron-sparing surgery. The syndromic child, with the Beckwith-Wiedemann or the WAGR, may present with the bilateral disease and requires the long-term renal surveillance. The fellow who balances the cure against the kidney preservation demonstrates the reasoning that the long case and the viva reward. [10]
Complications & Pitfalls
The complications of the Wilms tumour divide into the disease-related and the treatment-related, and the fellow must hold both because the iatrogenic harm can rival the disease. The disease-related complications are the tumour rupture, the intravascular thrombus, the hypertension and the metastases, and they are the complications that drive the resuscitation and that are anticipated from the moment the tumour is seen on the scan. The tumour rupture, with the spill of the tumour into the peritoneum, is the one that upstages the disease and that is prevented by the gentle handling and the preoperative chemotherapy in the SIOP approach. [9]
The surgical complications begin at the nephrectomy, and the most consequential is the tumour spill and the upstaging. The spill, whether from the rupture before the surgery or from the breach at the operation, converts a lower stage into a stage three and commits the child to the more intensive chemotherapy and the abdominal radiotherapy. The other surgical complications are the injury to the renal vessels, the injury to the contralateral kidney, the small-bowel obstruction and the wound infection. The thorough inspection of the contralateral kidney at the surgery is the safeguard against the missed bilateral disease. [4]
The treatment-related complications are the costs of the cure, and they shape the survivorship. The doxorubicin produces the dose-dependent cardiotoxicity, the congestive heart failure that can declare decades later, and it is the reason the cardiac surveillance with the echocardiography is built into the survivorship. The abdominal radiotherapy injures the remaining kidney, the musculoskeletal growth and the reproductive tissues, and the radiation to the lung field carries the pulmonary fibrosis. The secondary malignancy follows the radiation and the chemotherapy years later, and the renal failure follows the nephrectomy in the bilateral disease. [4]
Prognosis & Disposition
The prognosis of a child with a Wilms tumour is one of the great successes of the paediatric oncology, and it is determined by the stage, the histology, the molecular profile and the completeness of the resection. The favourable-histology Wilms tumour has the survival of around ninety percent overall, with the stage one disease exceeding ninety five percent and the stage four disease sitting around seventy to eighty percent. The anaplastic Wilms, the clear cell sarcoma and the malignant rhabdoid tumour carry the worse prognosis, with the diffuse anaplasia and the rhabdoid tumour the worst of the group. The molecular profile refines the risk further. [4][9]
The disposition of the child is to the specialist paediatric oncology centre, and the management is delivered in the tertiary centre with the paediatric surgery, the radiation oncology and the paediatric intensive care. The regional or the rural hospital is responsible for the recognition, the initial imaging and the safe transfer, and the long distances and the retrieval times are the reason the early recognition is so heavily weighted in the exam. The child with the palliative disease, the relapsed or the refractory tumour, is managed with the salvage therapy and the palliative care, with the local services supported by the specialist centre. [1]
In Australia and Aotearoa New Zealand, the child with a newly diagnosed Wilms tumour is managed in a tertiary paediatric oncology centre, with the paediatric retrieval services transferring the child from the regional or the rural hospital. The surgery and the radiotherapy are delivered in the specialised centres, and the family is supported by the social work and the educational liaison. The long distances are the reason the early recognition and the safe transfer from the referring hospital are so heavily weighted in the exam.
[1]The long-term surveillance of the survivor is the reward and the burden of the cure, because the late effects of the therapy are common and they accumulate with the time. The survivor has the annual imaging for the recurrence in the early years, the annual renal function and the blood pressure for the chronic kidney disease, the echocardiography for the doxorubicin cardiotoxicity, and the attention to the growth and the fertility. The transition to the adult late-effects service is prepared in the adolescence, with the reproductive and the genetic counselling, the secondary malignancy surveillance and the pregnancy risk assessment. The fellow who builds the survivorship plan demonstrates the care that extends beyond the cure. [4]
Special Populations
The infant and the very young child hold a special position in this topic, because the differential shifts towards the congenital mesoblastic nephroma and the malignant rhabdoid tumour, and the imaging is less reliable. The infant under six months with the renal mass is the one child in whom the biopsy is more often considered, because the mesoblastic nephroma and the rhabdoid tumour are more common and the preoperative imaging cannot reliably distinguish them. The mesoblastic nephroma of the newborn is the largely benign tumour cured by the resection, and it is the reason the infant renal mass is approached with the different strategy. [3]
The syndromic child, with the Beckwith-Wiedemann, the WAGR or the hemihypertrophy, is the special population that changes the surveillance and the counselling. The Beckwith-Wiedemann syndrome and the isolated hemihypertrophy warrant the serial abdominal ultrasound, every three months through the period of the highest risk, to catch the Wilms tumour early. The WAGR and the Denys-Drash syndromes carry the WT1 mutation, and the family is counselled on the inheritance and the surveillance. These syndromes are sought in every child with a Wilms tumour because they change the plan for the child and the siblings. [6]
Socioeconomic disadvantage, the remoteness and the migrant or the refugee status shape the access to the diagnosis and the treatment, and they are the reason the early recognition in the primary care and the regional hospital is so heavily emphasised. A child far from the specialist centre may first present to the clinician who sees few such cases, and the abdominal mass that flags the tumour is the bridge to the retrieval and the specialist care. The language and the cultural barriers are addressed with the interpreter and the cultural support, and the family is supported through the long and the unfamiliar treatment. [1]
The adolescent and the young adult with the renal tumour, often the renal cell carcinoma rather than the Wilms, is prepared for the transition to the adult service with the counselling and the documentation that make it safe. The reproductive and the genetic counselling, the fertility preservation before the gonadotoxic therapy, and the late-effects surveillance are addressed before the handover. The young person leaves the paediatric service with the survivorship plan and the named adult provider, and the transition is a clinical act as important as the diagnosis. [3]
Evidence, Guidelines & Regional Differences
The landmark evidence that underpins the modern treatment of the Wilms tumour is the product of the successive National Wilms Tumor Study and the Children's Oncology Group trials in North America and the SIOP trials in Europe, and it is the reason the survival has risen from the low rate of the pre-chemotherapy era to the around ninety percent of today. The trials refined the chemotherapy from the vincristine alone to the vincristine and the dactinomycin and then the doxorubicin for the higher stage, and they defined the role of the radiotherapy for the stage three and the metastatic disease. The avoidance of the radiotherapy for the stage one and two disease was the milestone that reduced the late effects without sacrificing the survival. [4][5]
The contemporary trials test the further de-escalation of the therapy for the low-risk disease and the intensification for the high-risk disease, balanced against the late effects. The trials of the reduced vincristine and the dactinomycin for the very low-risk stage one disease, and the molecular risk stratification that separates the low-risk from the high-risk within the favourable histology, are the contemporary research. The doxorubicin cardiotoxicity and the secondary malignancy are the late effects that drive the de-escalation, and the renal preservation is the goal that drives the nephron-sparing surgery in the bilateral and the syndromic disease. [4][8]
The Children's Oncology Group and the SIOP strategies divide the global practice, with the Children's Oncology Group prevailing in North America and the SIOP across Europe, and both achieve the similar survival. The Children's Oncology Group proceeds to the upfront radical nephrectomy for the typical tumour and stages it after, while the SIOP gives the four weeks of the preoperative vincristine and the dactinomycin and stages the histological response after. Australia and Aotearoa New Zealand follow the Children's Oncology Group strategy. The fellow should know the local protocol and defend the choice.
[4][5]The controversies and the open questions are the live ones. The optimal management of the bilateral and the syndromic disease, balanced between the cure and the kidney preservation, is one. The role of the molecular risk stratification in the de-escalation of the therapy for the low-risk favourable-histology disease is another. The place of the nephron-sparing surgery in the unilateral disease with the favourable biology is being explored. The fellow holds these as the open questions and cites the trials and the guidelines rather than the dogma, and the honest acknowledgement of the uncertainty is the mark of the mature candidate. [7][10]
Exam Pearls
The high-yield facts for the exam are the ones that change a decision at the bedside, and they are worth carrying as the sharp statements. Wilms tumour is the commonest renal malignancy of childhood, peaking at three to four years, and it presents with the painless abdominal mass that does not cross the midline. The triad of the abdominal mass, the haematuria and the hypertension points to the renal tumour, and the first test is the ultrasound with the Doppler of the cava. The biopsy of the typical tumour is avoided in the Children's Oncology Group approach because the spill upstages it to the stage three. [1][4]
The favourable-histology Wilms is cured in around ninety percent with the surgery and the vincristine and the dactinomycin, with the doxorubicin and the radiotherapy added for the stage three and the stage four. The anaplastic Wilms, the clear cell sarcoma and the malignant rhabdoid tumour carry the worse prognosis and the more intensive therapy. The clear cell sarcoma metastasises to the bone and the brain, and the malignant rhabdoid tumour is associated with the central nervous system tumours and the loss of the INI1. The congenital mesoblastic nephroma is the largely benign tumour of the infant. [3][8]
The final pearls are the ones that catch the candidate who has learned the headline and forgotten the corner. The neuroblastoma crosses the midline and the Wilms does not, and the urinary catecholamines separate them. The tumour thrombus into the cava is defined before the surgery, and it changes the operative plan. The doxorubicin cardiotoxicity declares decades later, and the echocardiography is the lifelong surveillance. The congenital mesoblastic nephroma is the tumour of the newborn and the largely benign one. The message for the exam is that the corners are where the marks are won, and the reasoning that holds the histology and the staging at the centre is the one the boards reward. [9]
References
- [1]Spreafico F, Fernandez CV, Brok J Wilms tumour Nat Rev Dis Primers, 2021.PMID 34650095
- [2]Treger TD, Chowdhury T, Pritchard-Jones K The genetic changes of Wilms tumour Nat Rev Nephrol, 2019.PMID 30705419
- [3]Perotti D, Williams RD, Wegert J Hallmark discoveries in the biology of Wilms tumour Nat Rev Urol, 2024.PMID 37848532
- [4]Dome JS, Mullen EA, Dix DB Impact of the First Generation of Children's Oncology Group Clinical Trials on Clinical Practice for Wilms Tumor J Natl Compr Canc Netw, 2021.PMID 34416705
- [5]Graf N, Tournade MF, de Kraker J The role of preoperative chemotherapy in the management of Wilms' tumor. The SIOP studies Urol Clin North Am, 2000.PMID 10985144
- [6]Kalish JM, Becktell KD, Bougeard G Update on Surveillance for Wilms Tumor and Hepatoblastoma in Beckwith-Wiedemann Syndrome and Other Predisposition Syndromes Clin Cancer Res, 2024.PMID 39320341
- [7]Vujanic GM, D'Hooghe E, Graf N Prognostic significance of histopathological response to preoperative chemotherapy in unilateral Wilms' tumor: an analysis of 899 patients treated on the SIOP WT 2001 protocol Int J Cancer, 2021.PMID 34109628
- [8]Gadd S, Huff V, Walz AL A Children's Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor Nat Genet, 2017.PMID 28825729
- [9]Benedetti DJ, Varela CR, Renfro LA Treatment of children with favorable histology Wilms tumor with extrapulmonary metastases: a report from the COG studies AREN0533 and AREN03B2 and NWTSG study NWTS-5 Cancer, 2024.PMID 37933882
- [10]Ehrlich P, Chi YY, Chintagumpala MM Results of the First Prospective Multi-institutional Treatment Study in Children With Bilateral Wilms Tumor (AREN0534): a report from the Children's Oncology Group Ann Surg, 2017.PMID 28795993