Paeds · infectious-diseases
Prolonged, recurrent and periodic fever
Also known as Periodic fever in children · Recurrent fever · Fever of unknown origin in children · PFAPA syndrome · Hereditary periodic fever syndromes · Autoinflammatory recurrent fever · Cyclic fever
Fellowship guide to the child with prolonged, recurrent or periodic fever: separate the three patterns, build the differential for each, run a staged fever-of-unknown-origin workup, recognise PFAPA from the Marshall and Thomas clinical criteria with normal between-episode health, distinguish PFAPA from the hereditary periodic fever syndromes (FMF, HIDS/MVK, TRAPS, CAPS) and cyclic neutropenia by attack duration and signature features, and deliver evidence-based care (corticosteroid to abort PFAPA attacks, cimetidine prophylaxis, tonsillectomy supported by three RCTs, colchicine for FMF, IL-1 blockade for CAPS/TRAPS/HIDS) while never missing malignancy, endocarditis, Kawasaki disease, HLH and immunocompromised-host fever.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
P.R.P. — pattern the fever first
Overview & Definition
A child with fever that "will not go away" or "keeps coming back" is one of the most common reasons a general paediatrician is asked for an opinion. The trap is to lump these children together and start investigating blindly. The discipline Samuel Long set out was to let the fever pattern do the triage before ordering a single test, because the pattern — not the height of the fever — points to the diagnosis. [1]
Three patterns matter. Prolonged fever is a single continuous febrile illness that has outlasted the expected course of a self-limited infection; in practical terms, fever for more than 7 to 14 days (and classically beyond two weeks) without a diagnosis after an appropriate initial workup is termed fever of unknown origin. Recurrent fever is two or more discrete febrile episodes separated by fever-free intervals, with each episode resolving spontaneously and the child well between attacks. Periodic fever is the special subset of recurrent fever in which attacks recur at predictable, stereotyped (clockwork) intervals. [1] [13]
Your job at first contact is therefore narrow and decisive: confirm the pattern with a careful history and diary, screen for the dangerous mimics, and only then decide how deeply to investigate. Most recurrent fever in young children is repeated common infection — a pattern, not a disease — and the art is recognising the minority who have something serious, inherited or autoinflammatory. [1] [10]
Classification
The causes fall out of the pattern. Prolonged fever is sorted into occult infection, inflammatory disease, malignancy and a miscellaneous group. Recurrent fever adds repeated common infection, immunodeficiency and the periodic fever syndromes. Periodic fever is itself a short list dominated by PFAPA, with the hereditary periodic fever syndromes and cyclic neutropenia behind it. [1] [5]
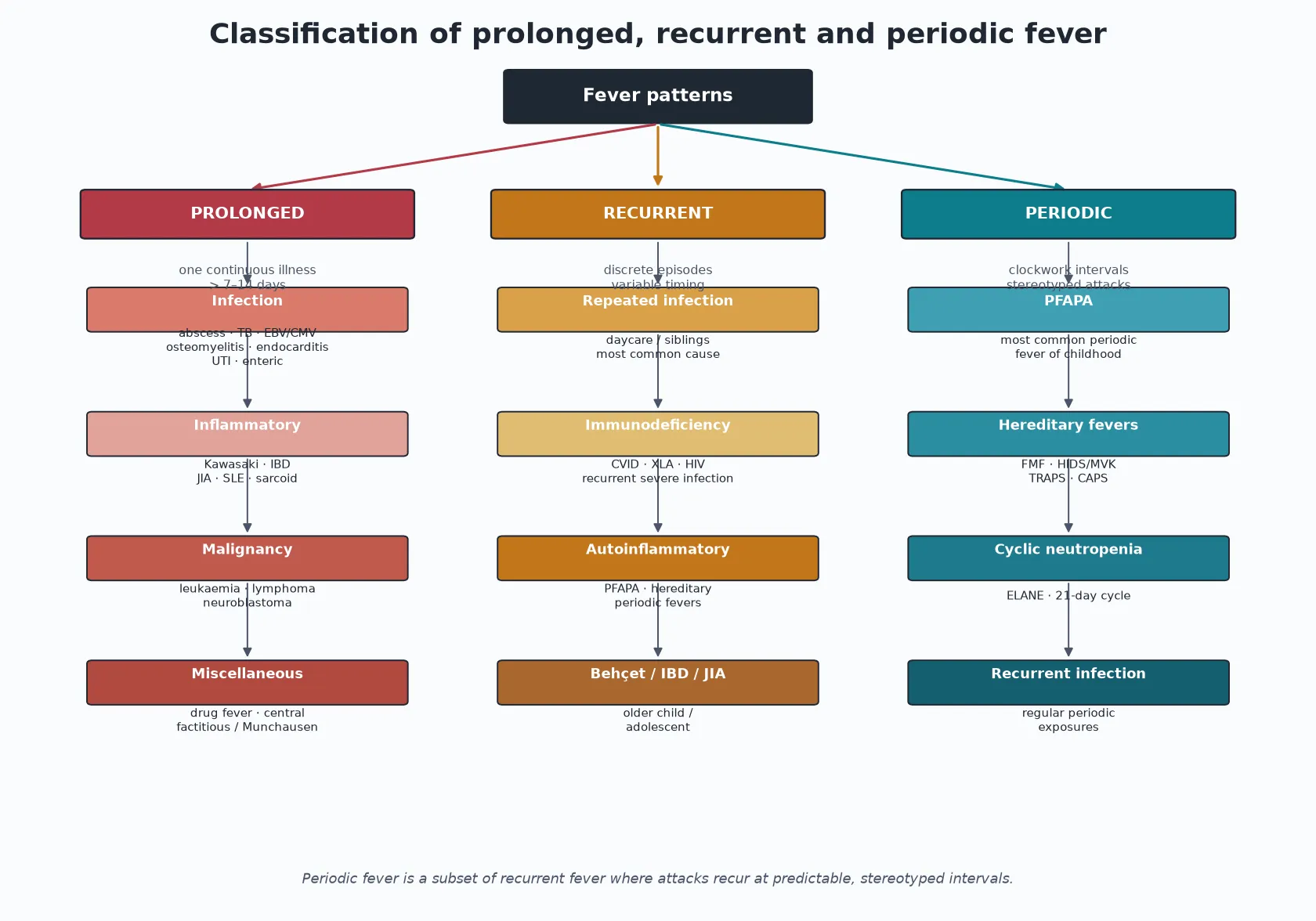
The three patterns, with the question each one asks
Prolonged (FUO)
- One continuous illness beyond 7 to 14 days
- Ask: what smouldering infection, inflammation or malignancy have I missed?
- Infection, inflammatory, malignancy, miscellaneous
- Triage the dangerous mimics first
Recurrent
- Two or more discrete episodes, well between
- Ask: is this just repeated common infection, or something inherited?
- Repeated infection, immunodeficiency, periodic syndromes
- The commonest cause overall
Periodic
- Clockwork, stereotyped intervals
- Ask: which periodic fever syndrome is this?
- PFAPA is by far the commonest
- FMF, HIDS, TRAPS, CAPS, cyclic neutropenia
Epidemiology & Risk Factors
Repeated viral and bacterial infection is the commonest cause of recurrent fever in childhood, amplified by daycare attendance, siblings, household crowding and exposure in the winter respiratory season. It is a normal developmental phenomenon: a young child in care can have eight to ten febrile illnesses a year, each clinically distinct and each resolving on its own. Recognising this normal pattern prevents a great deal of unnecessary investigation. [1] [13]
PFAPA at a glance
The hereditary periodic fevers are rarer but carry a higher stakes. Familial Mediterranean fever (FMF) is the most prevalent and tracks with Mediterranean ancestry — Armenian, Turkish, Sephardi-Jewish and Arab populations — reflecting its autosomal recessive MEFV inheritance. Hyperimmunoglobulin D syndrome (HIDS, mevalonate kinase deficiency) is also autosomal recessive and is concentrated in northern European (especially Dutch) families. TRAPS (TNF receptor-associated periodic syndrome) and CAPS (cryopyrin-associated periodic syndrome) are autosomal dominant. These ethnic and family clues are cheap, powerful and easy to forget in clinic. [5] [11] [12]
The dangerous causes of prolonged fever — malignancy (leukaemia, lymphoma, neuroblastoma), infective endocarditis, tuberculosis and haemophagocytic lymphohistiocytosis — are individually uncommon but collectively the reason this topic exists. Fever in the immunocompromised or neutropenic host is a separate emergency and is not classified as FUO: it gets prompt empiric broad-spectrum antibiotics first, questions later. [13] [14]
Pathophysiology
The periodic fever syndromes are autoinflammatory, a word worth unpacking because it is the key to the whole family. In an autoimmune disease the adaptive immune system attacks the self through autoantibodies or autoreactive T cells. In an autoinflammatory disease the problem sits one layer down: the innate immune system is switched on constitutively, producing recurrent, self-limited inflammatory attacks with no infection and no autoantibodies. [5] [10]
The hereditary periodic fevers are monogenic. A gain-of-function mutation in MEFV, MVK, TNFRSF1A or NLRP3 drives over-activation of the inflammasome, with surges of interleukin-1-beta, interleukin-18 and interleukin-6. Each attack is the clinical face of that cytokine surge — fever, serositis, rash, lymphadenopathy — and each attack resolves as the surge subsides, leaving the child entirely well and the inflammatory markers back to normal. PFAPA is polygenic rather than monogenic, but it shares the same innate dysregulation centred on the tonsil, with elevated interleukin-1-beta and interleukin-18 during attacks. [2] [4] [5]
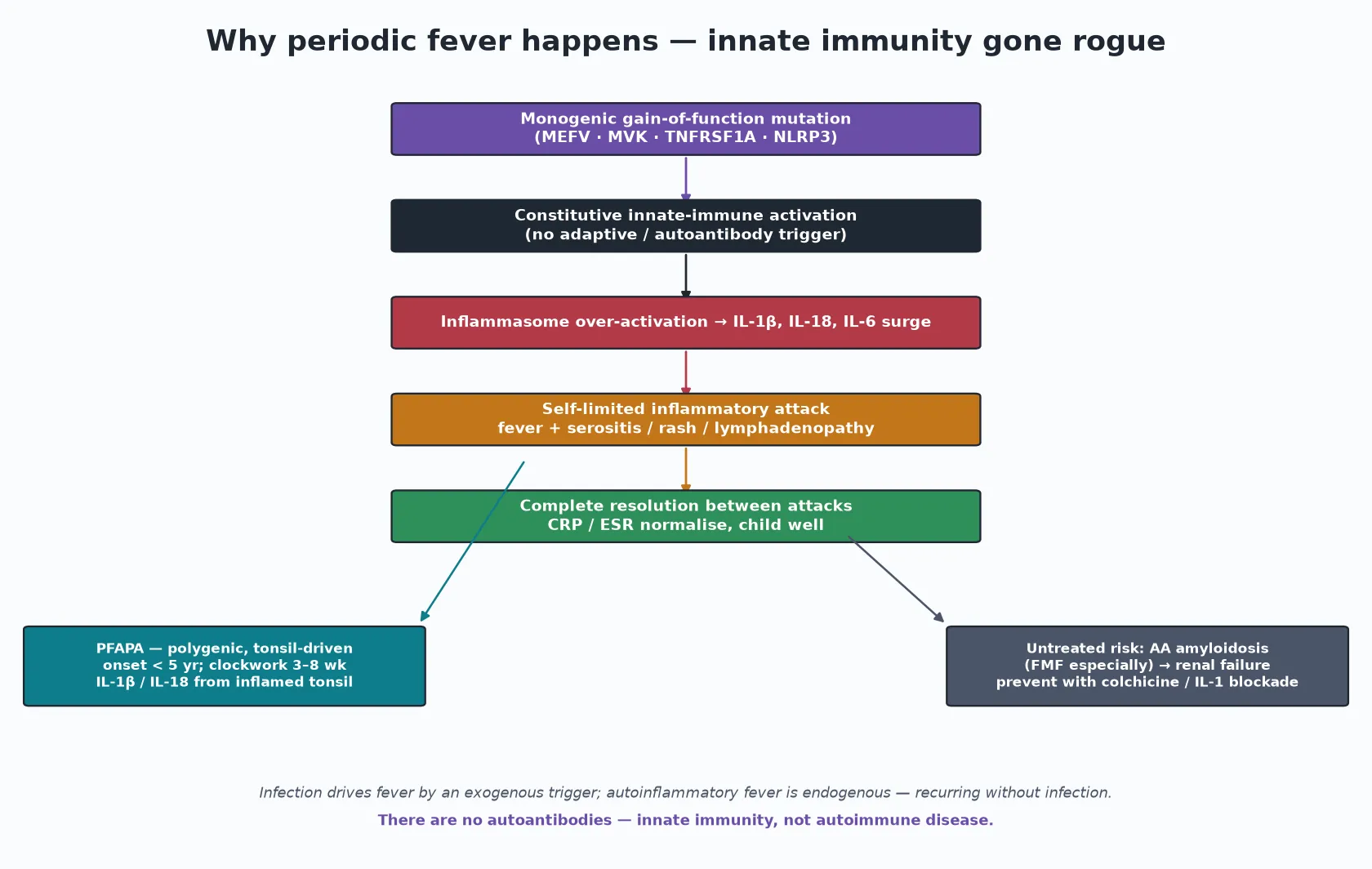
The mechanism explains both the treatment and the long-term risk. Blocking interleukin-1 with canakinumab switches off the attack in CAPS, TRAPS and HIDS, which is the most direct proof that the cytokine is the engine. In FMF the recurrent serositis damages serum amyloid A handling, so untreated FMF seeds AA amyloidosis in the kidney and causes renal failure — the single most important reason not to "watch and wait" when FMF is on the table. Colchicine prevents both the attacks and the amyloidosis. PFAPA, in contrast, never causes amyloidosis. [5] [11]
Clinical Presentation
The presentation is the pattern, so the history is the examination. Ask when each fever started and stopped, how high it ran, what time of day it peaked (an evening rise hints at TB or malignancy), how the child looked between episodes, and whether anyone else at home was unwell. Document a fever diary across at least two suspected attacks: the interval, the duration, and the features that accompany each one. A stereotyped, clockwork diary is the single most powerful clue to PFAPA. [1] [3]
PFAPA announces itself with the tetrad Marshall described in 1987: periodic fever plus pharyngitis plus aphthous stomatitis plus cervical adenitis. The fever is high — 39 to 40 degrees Celsius — and abrupt, lasting 3 to 6 days and recurring every 3 to 8 weeks. The throat and mouth findings appear with the fever, the cervical nodes are tender and enlarged, and — critically — between attacks the child is completely well and growing normally. Onset is before 5 years in the great majority. [2] [3] [4]
The hereditary periodic fevers each have a signature that breaks the PFAPA mould. FMF produces short, one- to three-day attacks of fever with serositis — peritonitis that can mimic an acute abdomen, pleuritis, and a monoarthritis — together with an erysipelas-like rash, usually in a child of Mediterranean ancestry. HIDS attacks last 3 to 7 days, are often triggered by vaccination, and bring a maculopapular rash with generalised lymphadenopathy, hepatosplenomegaly, abdominal pain and aphthae. TRAPS breaks the pattern in a different way: the attacks are long, lasting one to three weeks, with striking periorbital oedema and a migratory rash that travels with underlying myalgia. [5] [11] [12]
Two presentations are dangerous rather than curious. A child with prolonged fever plus weight loss, night sweats, pallor, bruising or hepatosplenomegaly is not a recurrent-fever puzzle — that is malignancy, endocarditis or HLH until proven otherwise. And fever for more than 5 days with conjunctivitis, a polymorphous rash, mucous-membrane change and extremity change is Kawasaki disease, where the clock is ticking toward day 10 for IVIG to protect the coronary arteries. [13] [14]
Differential Diagnosis
For prolonged fever, work through four buckets in order. Occult infection heads the list: a hidden abscess (peritonsillar, intra-abdominal, retroperitoneal or psoas), osteomyelitis, infective endocarditis, tuberculosis, Epstein-Barr or cytomegalovirus, urinary tract infection, and enteric fever in the returned traveller. Inflammatory disease follows — Kawasaki disease, inflammatory bowel disease, juvenile idiopathic arthritis, systemic lupus erythematosus. Malignancy is the bucket you cannot miss: leukaemia, lymphoma and neuroblastoma. The miscellaneous bucket holds drug fever, central fever, and fabricated or induced illness (Munchausen-by-proxy), which is easy to overlook. [1] [13] [14]
For recurrent fever, the first question is whether the episodes are clinically distinct (pointing to repeated infection) or stereotyped (pointing to a periodic syndrome). Repeated common infection is the default and needs no workup beyond a careful history. Reach for immunodeficiency — common variable immunodeficiency, X-linked agammaglobulinaemia, complement deficiency or HIV — when infections are severe, persistent, caused by unusual organisms, or accompanied by failure to thrive. Behcet disease, inflammatory bowel disease and juvenile idiopathic arthritis enter the list in older children and adolescents. [1] [10]
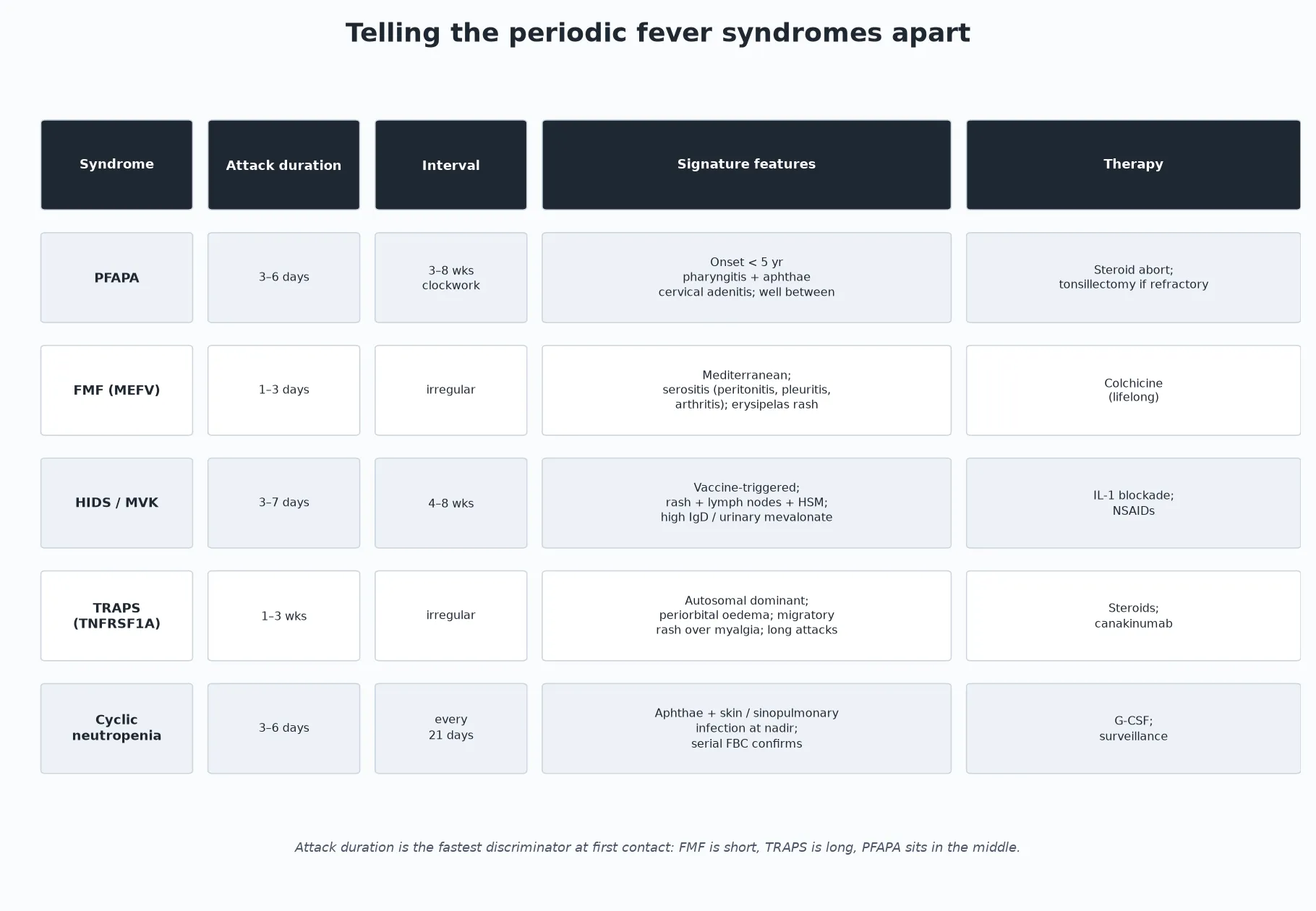
The periodic fever syndromes are best separated by two numbers and one feature. The attack duration sorts them fast: FMF is short at 1 to 3 days, PFAPA sits in the middle at 3 to 6 days, HIDS runs 3 to 7 days, and TRAPS is the outlier at 1 to 3 weeks. The interval regularity sorts them again: PFAPA is clockwork, cyclic neutropenia is a 21-day metronome, while FMF and HIDS are irregular. Then the signature feature confirms: periorbital oedema with migratory myalgia is TRAPS; serositis with an erysipelas rash in a Mediterranean child is FMF; aphthae and infection at a countable nadir is cyclic neutropenia. [4] [5] [12]
Clinical & Bedside Assessment
Begin with the fever diary and a full developmental and growth history. Plot the growth, because normal growth between attacks is a core PFAPA criterion and faltering growth redirects you toward malignancy, chronic infection, immunodeficiency or inflammatory bowel disease. Ask specifically about travel (enteric fever, malaria, brucellosis, Q fever, TB), animal, food and water exposures, TB contacts, sick household contacts, immunisation status and trigger patterns such as vaccination in HIDS. [1] [12] [13]
A targeted family history is disproportionately useful in the periodic fevers. Ask about Mediterranean, Armenian, Turkish, Sephardi-Jewish or Arab heritage for FMF; recurrent abdominal pain or "appendicitis" attacks in relatives (FMF serositis); recurrent unexplained fevers in parents or siblings; and consanguinity. A family history of recurrent aphthous ulcers, uveitis or renal failure (amyloidosis) can change the whole diagnostic direction. [5] [11]
The examination is systems-based and deliberately looks for the dangerous mimics. Check the skin for rash, erysipelas, erythema nodosum, splinter haemorrhages and petechiae; the oropharynx for aphthae, dental sepsis and tonsillar disease; lymph nodes for generalised enlargement; the abdomen for hepatosplenomegaly and masses; the heart for a new murmur; and the joints for arthritis. Between-episode wellness with entirely normal examination and growth is itself a diagnostic finding for PFAPA. [1] [14]
A useful bedside manoeuvre for PFAPA is to measure inflammatory markers during a documented attack and again when the child is well. A CRP and ESR that rise sharply with the fever and normalise completely between attacks is a PFAPA signature; persistently elevated markers between episodes point instead toward chronic infection, inflammatory disease or malignancy and argue against uncomplicated PFAPA. [3] [4]
Investigations
Stage the workup to the pattern, and do not over-investigate classic PFAPA. For prolonged fever, begin with tier-one bloods: a full blood count with differential and film, CRP and ESR, blood cultures taken before any antibiotics, urinalysis and urine culture, electrolytes, liver function tests and albumin, and a lactate dehydrogenase as a malignancy screen. Add EBV and CMV serology, an interferon-gamma release assay for tuberculosis, an HIV test, an antistreptolysin-O titre and a chest X-ray as the history directs. [13] [14]
Move to targeted imaging and microbiology only as the clinical lead dictates. Ultrasound, CT or MRI hunts for an occult abscess (especially head and neck, intra-abdominal, retroperitoneal and psoas) and for osteomyelitis. Echocardiography is indicated when endocarditis is possible and is also the key investigation for Kawasaki disease. Bone marrow examination is reserved for suspected haematological malignancy — typically when there are unexplained cytopenias or blasts on the blood film — not as a routine FUO test. [13] [14]
For suspected periodic fever syndromes, send an autoinflammatory gene panel covering MEFV, MVK, TNFRSF1A and NLRP3, together with serum IgD and urinary mevalonic acid when HIDS is plausible. Serial full blood counts over several weeks confirm cyclic neutropenia by showing the characteristic 21-day oscillation in neutrophil count. Apply the Livneh clinical criteria for FMF and use the Steichen clinical criterion to exclude HIDS when a child has long, vaccination-triggered attacks with rash and lymphadenopathy. [5] [11] [12]
PFAPA has no confirmatory laboratory test. The diagnosis is clinical, built on the Marshall and Thomas criteria and a documented diary, and made only after infection and the hereditary fevers have been excluded. The supporting evidence is the rise-and-fall of CRP during attacks and complete wellness between them. The commonest error is the opposite — running serial batteries on a textbook PFAPA child in place of recognising the pattern and aborting the attacks. [2] [3] [10]
Management — Resuscitation
Stabilise the acutely unwell child first. A child who is toxic, shocked, in heart failure or carrying the stigmata of malignancy or HLH needs resuscitation and urgent targeted investigation, not a diary. The prolonged-fever and periodic-fever pathways are for the child who is systemically well between episodes — if they are not well between episodes, rethink the diagnosis. [1] [13]
Two thresholds are time-critical and easy to miss. Kawasaki disease is treated with intravenous immunoglobulin and aspirin ideally before day 10 of fever to prevent coronary artery aneurysms, so a child with five days of fever and the Kawasaki features needs an urgent echo and paediatric review, not a recurrent-fever plan. And any fever in a neutropenic oncology or immunocompromised child is treated as presumed bacteraemia within the hour. [13] [14]
Management — Definitive & Stepwise

For prolonged fever, run the staged workup described above and treat the cause once it is found. Modern practice adds fluorodeoxyglucose PET-CT for genuinely refractory FUO after targeted testing is negative, where it can localise occult malignancy, vasculitis or hidden infection. The goal is a diagnosis, not a label; a child discharged as "FUO — resolve" should still have a clear safety-net and a review date. [13] [14]
For PFAPA, the management ladder is reassuring and evidence-based. The first move is to abort each attack with a single dose of oral corticosteroid, which ends most attacks within hours, although it can shorten the interval to the next episode. For the child whose attacks are frequent, disruptive or refractory, cimetidine prophylaxis benefits a subset. Tonsillectomy is supported by three randomised trials and is reserved for severe, refractory disease after multidisciplinary discussion. PFAPA resolves spontaneously by adolescence, so the aim is symptom control and sparing avoidable investigation, not cure. [6] [7] [8] [10]
Prednisolone — to abort a PFAPA attack
Dose
1 to 2 mg per kg as a single oral dose at attack onset
The hereditary periodic fevers need disease-specific therapy, which is why distinguishing them from PFAPA matters so much. FMF is treated with lifelong colchicine, which prevents both the attacks and the development of AA amyloidosis — this is one of the few treatments in paediatrics that changes long-term survival. CAPS, TRAPS and HIDS respond to interleukin-1 blockade with canakinumab, the most direct confirmation that the cytokine drives the disease. Cyclic neutropenia is managed with G-CSF and infection surveillance. [5] [11]
The PFAPA management ladder
Step 1 — Confirm the pattern: a documented diary across at least two attacks, well between, CRP rising then falling
Step 2 — Apply the Marshall and Thomas clinical criteria and exclude the hereditary periodic fevers with history and targeted genetic testing
Step 3 — Abort each attack with a single oral dose of prednisolone 1 to 2 mg per kg
Step 4 — For frequent or disruptive attacks, trial cimetidine prophylaxis
Step 5 — For severe refractory disease, refer to ENT for tonsillectomy after multidisciplinary discussion (three RCTs support effectiveness)
Specific Subtypes & Scenarios
PFAPA is managed by the general paediatrician with a shared diary and a clear plan to abort attacks. Reassure the family that the syndrome resolves spontaneously — the median duration from onset to resolution is about 6 to 8 years — and that it does not cause amyloidosis or long-term harm. Education of parents and teachers to recognise attacks and treat early is the cornerstone of outpatient care. [3] [4]
Familial Mediterranean fever is the prototype hereditary periodic fever and the one with the highest stakes. Apply the Livneh clinical criteria, screen the family, and start lifelong colchicine once the diagnosis is secure. The goal is not just fewer attacks but prevention of AA amyloidosis and renal failure, so adherence matters as much as diagnosis. Warn about the acute abdominal attacks that can mimic a surgical abdomen and may have been labelled "appendicitis" in relatives. [5] [11]
HIDS (mevalonate kinase deficiency) presents with vaccination-triggered attacks, a maculopapular rash, generalised lymphadenopathy and hepatosplenomegaly. Use the Steichen clinical criterion — immunisation-triggered attacks that are long and accompanied by rash and lymph nodes — to decide whether to pursue the diagnosis, and treat with interleukin-1 blockade when attacks are severe. TRAPS is the great mimic: its long attacks, periorbital oedema and migratory rash overlying myalgia break the PFAPA pattern, and it responds to steroids and canakinumab. Cyclic neutropenia runs a 21-day metronome, with aphthae and sinopulmonary infection at the countable nadir, and is confirmed on serial full blood counts; it is managed with G-CSF and infection surveillance. [5] [12]
Complications & Pitfalls
The gravest pitfall is labelling a serious illness as "just recurrent fever" and missing the dangerous mimics — malignancy, endocarditis, tuberculosis, Kawasaki disease and HLH. A close second is the mirror image: over-investigating textbook PFAPA with repeated blood panels, imaging and antibiotic courses, medicalising a benign syndrome and traumatising the family. Both errors flow from the same source — failing to pattern the fever first. [1] [10]
Two medication pitfalls deserve emphasis. Withholding colchicine from a child with FMF allows preventable attacks and the development of AA amyloidosis with its risk of renal failure — a treatable disease left untreated. And assuming every periodic fever is PFAPA can delay the diagnosis of TRAPS, HIDS or cyclic neutropenia, each of which has a different and effective therapy. The hereditary fevers are uncommon, but they are the ones where the right treatment changes the outcome. [5] [11] [12]
Prognosis & Disposition
PFAPA has an excellent prognosis. Attacks resolve spontaneously, typically by adolescence, with a median symptom duration of about 6 to 8 years from onset and no long-term sequelae, no amyloidosis and no effect on growth or development. The family can be reassured with confidence once the pattern is confirmed and the dangerous mimics excluded. [3] [4]
Recurrent fever driven by repeated common infection improves as the child's immunity and age mature, and it is not a disease at all. The disposition is reassurance, a fever diary, and clear safety-netting for the features that would prompt review. FMF prognosis is determined by colchicine adherence and the prevention of AA amyloidosis; untreated FMF carries a real risk of renal failure, which is why the diagnosis, family screening and treatment are not optional. [5] [11]
Most PFAPA is managed by the general paediatrician with a shared diary and an attack-abort plan. Refer to paediatric infectious diseases and/or a rheumatology-autoinflammation service for complex, refractory or gene-positive periodic fever syndromes, and to ENT for tonsillectomy when the attacks are severe and refractory. Always provide a written safety-net covering the red flags that should trigger urgent review. [1] [10]
Special Populations
In children of Mediterranean, Armenian, Turkish, Sephardi-Jewish or Arab heritage, keep FMF high on the list: lower the threshold to test MEFV and apply the Livneh criteria early, because colchicine changes the long-term outcome. In refugee, migrant and returned-traveller children, weigh tuberculosis, enteric fever, brucellosis, Q fever and malaria in prolonged fever, and use interferon-gamma release assays and travel-specific serology rather than a generic panel. [5] [11] [13]
The immunocompromised and oncology or neutropenic host breaks all the rules above: fever is an emergency that gets empiric broad-spectrum antibiotics first and a fever-of-unknown-origin workup only second. In Indigenous Australian, Maori and Pacific children in ANZ, the burden of recurrent skin and respiratory infection, acute rheumatic fever and post-streptococcal glomerulonephritis is higher, and the social determinants — overcrowding, access and remoteness — must be addressed alongside the medical plan. [13] [14]
Evidence, Guidelines & Regional Differences
The evidence base for this topic rests on a handful of landmark papers. Marshall described PFAPA in 1987; Thomas characterised the larger American cohort in 1999; and Hofer led the 301-patient international cohort in 2014 that defined the phenotypes and natural history. Gattorno and the Eurofever and PRINTO networks published classification criteria for the autoinflammatory recurrent fevers in 2019, and Livneh set out the clinical diagnostic criteria for FMF in 1997. Steichen provided a practical clinical criterion to exclude HIDS in 2009. [2] [3] [4] [5] [11] [12]
The tonsillectomy evidence is unusually strong for a surgical question in a syndrome that resolves on its own. Three randomised trials — Renko in 2007, Garavello in 2009 and Lantto in 2024 (tonsillotomy) — support effectiveness, and the long-term outcomes were characterised by Licameli in 2012. The controversy is not whether tonsillectomy works but which children benefit and where the threshold lies, since PFAPA eventually resolves regardless. [6] [7] [8] [9]
ANZ. General paediatric practice patterns the fever first, runs a staged FUO workup in the systemically well child, and treats empirically only the time-critical mimics. Give particular weight to migrant and refugee TB and to the higher burden of recurrent infection, rheumatic fever and post-streptococcal glomerulonephritis in Indigenous Australian, Maori and Pacific children, addressing overcrowding and access. Refer gene-positive periodic fever syndromes to paediatric infectious diseases and rheumatology-autoinflammation services. [1] [13]
Exam Pearls
- The first discriminator is the fever pattern: one long illness (prolonged or FUO), several discrete variable episodes (recurrent), or clockwork stereotyped attacks (periodic). [1]
- PFAPA tetrad: periodic fever plus pharyngitis plus aphthous stomatitis plus cervical adenitis, onset before 5 years, every 3 to 8 weeks for 3 to 6 days, completely well between attacks, resolves by adolescence. [2] [3]
- Attack duration sorts the periodic fevers fast: FMF 1 to 3 days (short), PFAPA 3 to 6 days, HIDS 3 to 7 days, TRAPS 1 to 3 weeks (long). [4] [5]
- A single oral dose of prednisolone 1 to 2 mg per kg aborts most PFAPA attacks; three RCTs support tonsillectomy for refractory disease; PFAPA never causes amyloidosis. [6] [10]
- FMF is treated with lifelong colchicine, which prevents both attacks and AA amyloidosis; CAPS, TRAPS and HIDS respond to interleukin-1 blockade (canakinumab). [5] [11]
- Never forget the dangerous mimics in prolonged fever: malignancy, endocarditis, tuberculosis, Kawasaki disease and HLH; neutropenic-host fever is an emergency, not FUO. [13] [14]
References
- [1]Long SS. Distinguishing among prolonged, recurrent, and periodic fever syndromes: approach of a pediatric infectious diseases subspecialist. Pediatr Clin North Am, 2005.PMID 15925664
- [2]Marshall GS, Edwards KM, Butler J, Lawton AR. Syndrome of periodic fever, pharyngitis, and aphthous stomatitis. J Pediatr, 1987.PMID 3794885
- [3]Thomas KT, Feder HM Jr, Lawton AR, Edwards KM. Periodic fever syndrome in children. J Pediatr, 1999.PMID 10393598
- [4]Hofer M, Pillet P, Cochard MM, Berg S, et al. International periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis syndrome cohort: description of distinct phenotypes in 301 patients. Rheumatology (Oxford), 2014.PMID 24505122
- [5]Gattorno M, Hofer M, Federici S, Papadopoulou C, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis, 2019.PMID 31018962
- [6]Renko M, Salo E, Putto-Laurila A, Saxen H, et al. A randomized, controlled trial of tonsillectomy in periodic fever, aphthous stomatitis, pharyngitis, and adenitis syndrome. J Pediatr, 2007.PMID 17719940
- [7]Garavello W, Romagnoli M, Gaini RM. Effectiveness of adenotonsillectomy in PFAPA syndrome: a randomized study. J Pediatr, 2009.PMID 19464029
- [8]Lantto U, Tapiainen T, Pokka T, Koivunen P. Tonsillotomy for Periodic Fever Syndrome: A Randomized and Controlled Trial. Laryngoscope, 2024.PMID 37477273
- [9]Licameli G, Lawton M, Kenna M, Daggis M, et al. Long-term surgical outcomes of adenotonsillectomy for PFAPA syndrome. Arch Otolaryngol Head Neck Surg, 2012.PMID 23069819
- [10]Wang A, Manthiram K, Dedeoglu F. Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) syndrome: A review. World J Otorhinolaryngol Head Neck Surg, 2021.PMID 34430824
- [11]Livneh A, Langevitz P, Zemer D, Zaks N, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum, 1997.PMID 9336425
- [12]Steichen O, van der Hilst J, Simon A, Cuisset L. A clinical criterion to exclude the hyperimmunoglobulin D syndrome (mild mevalonate kinase deficiency) in patients with recurrent fever. J Rheumatol, 2009.PMID 19531764
- [13]Hooft A, Leighton M. Management of prolonged pediatric fever in the emergency department. Pediatr Emerg Med Pract, 2026.PMID 41570317
- [14]Dizi Işık A, Akkoc G, Yilmaz S, et al. Pediatric fever of unknown origin: Diagnostic spectrum revealed by a multidisciplinary retrospective study. Eur J Pediatr, 2025.PMID 41109899