Paeds · nephrology-urology-fluids-and-electrolytes
Acute nephritic syndrome and glomerulonephritis
Also known as Acute glomerulonephritis · Acute nephritic syndrome · Post-streptococcal glomerulonephritis · Nephritic syndrome in children · Rapidly progressive glomerulonephritis · Crescentic glomerulonephritis
Fellowship guide to the acute nephritic syndrome in children: the tetrad of haematuria, proteinuria, oedema and hypertension driven by glomerular inflammation, the serum-complement split that separates post-streptococcal glomerulonephritis from C3 glomerulopathy, lupus and IgA disease, the rapidly progressive crescentic course that demands urgent biopsy and immunosuppression, and supportive care (fluid and salt restriction, loop diuretic, antihypertensive, penicillin eradication) with the red flags that escalate to paediatric intensive care and dialysis.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
A six-year-old boy presents with smoky, cola-coloured urine and puffy eyes. Two weeks earlier he had a sore throat. This is the classic face of acute glomerulonephritis, and it is the pattern every paediatric trainee is expected to reason through at viva. The syndrome itself is simply the visible result of glomerular capillary inflammation: the inflamed filter leaks red cells and protein, holds back salt and water, and, when severe enough, fails. [2]
This page owns the acute nephritic syndrome as a presentation and glomerulonephritis as the underlying inflammation. It treats post-streptococcal GN in enough depth to contrast it with every other nephritic cause, and points to the dedicated post-infectious-glomerulonephritis and IgA-nephropathy-and-IgA-vasculitis-nephritis leaves for their full standalone detail. [1]
Overview & Definition
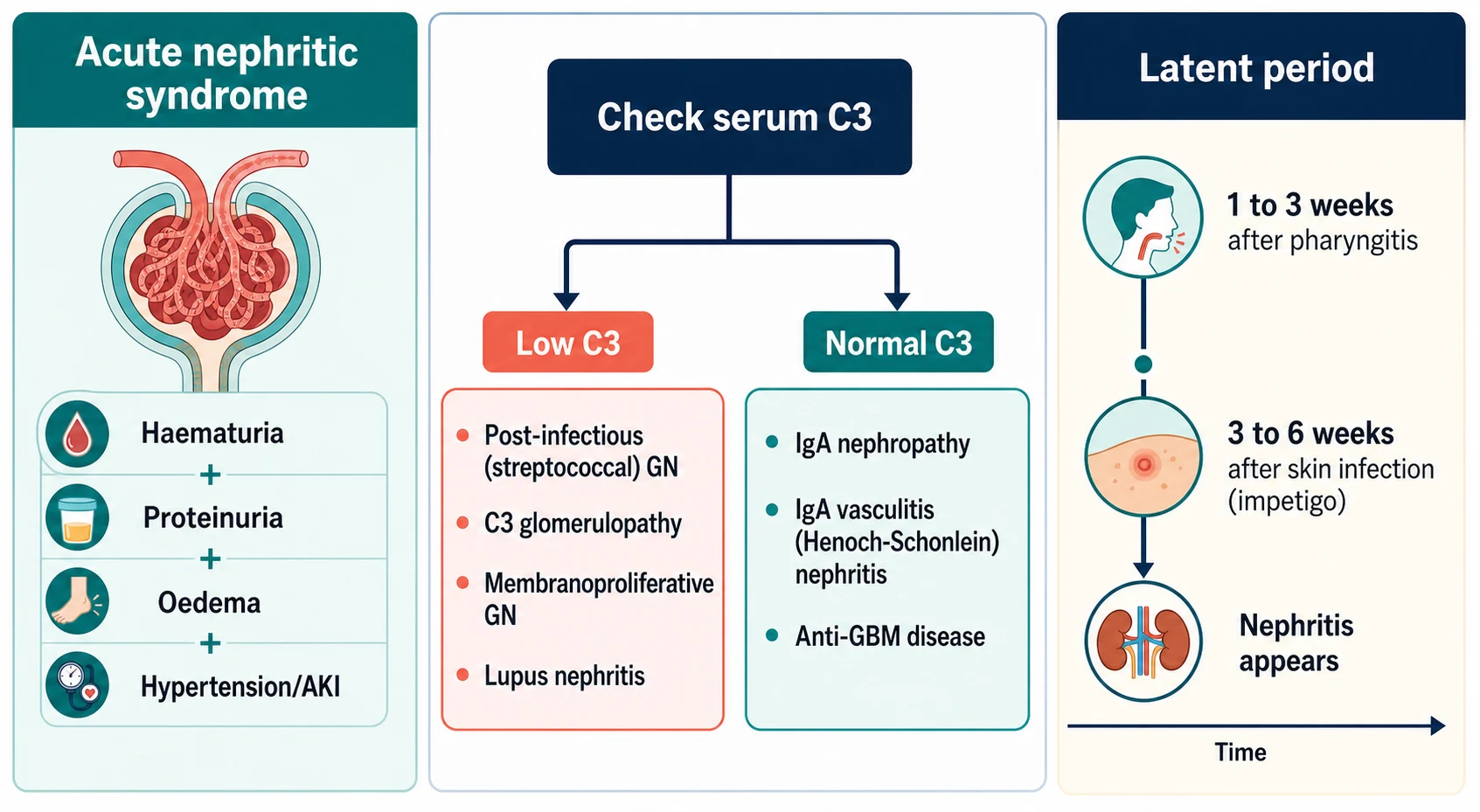
The acute nephritic syndrome is defined by its clinical tetrad: haematuria (gross or microscopic), proteinuria (usually below the nephrotic range), oedema and hypertension, with or without a fall in glomerular filtration rate. It is a syndrome, not a disease — several different glomerular disorders produce it. [1] [2]
Glomerulonephritis (GN) is the histological word behind the syndrome: inflammation of the glomerulus, most often from immune-complex deposition. The KDIGO 2021 Glomerular Diseases guideline frames GN by both its clinical presentation (nephritic, nephrotic, rapidly progressive, or chronic with asymptomatic haematuria) and its immunopathological pattern (immune-complex, pauci-immune, or anti-GBM). The same histology can show up in more than one clinical box, which is why the two frames are not interchangeable. [1]
The key distinction at the bedside is between a self-limiting post-infectious cause (which needs supportive care alone) and a continuing inflammatory disease (which needs biopsy and specific treatment). The serum complement, the tempo of renal failure, and the presence of systemic features are the three levers that move a child from one column to the other. [2] [11]
Classification
Glomerulonephritis is classified along two axes. The first is clinical: nephritic, nephrotic, rapidly progressive, or chronic. The second is immunopathological, based on where immune complexes or antibody settle on the glomerular basement membrane and on the pattern of complement activation. These two views converge at the biopsy. [1]
The complement split is the single most useful bedside classifier, because it sorts the long list of nephritic causes into two manageable groups. [2] [11]
Rapidly progressive glomerulonephritis (RPGN) is its own category, defined as a syndrome rather than a single disease: rapid loss of renal function over days to weeks with crescents (proliferation of parietal epithelial cells and macrophages) occupying Bowman's space on biopsy. KDIGO groups crescentic GN by its immunopathology into anti-GBM disease, immune-complex-mediated GN (which includes post-streptococcal, IgA and lupus), and pauci-immune (ANCA-associated) disease. In children, the immune-complex form is by far the commonest. [1] [2]
Epidemiology & Risk Factors
Post-infectious, especially post-streptococcal, GN is the most common cause of acute glomerulonephritis in children worldwide. It peaks between the ages of two and twelve years, is uncommon under three, and is slightly more frequent in boys. [3] [6]
The trigger matters. Nephritogenic strains of group A streptococcus infect the throat (M types 1 and 12) or the skin (impetigo, M types 49, 55 and 57). The interval between the infection and the nephritis — the latent period — is the fingerprint: one to three weeks after pharyngitis, but three to six weeks after a skin infection. A child with gross haematuria on the same day as a sore throat does not have post-streptococcal GN; that pattern points to IgA nephropathy. [3] [11]
The burden is uneven. Post-streptococcal GN concentrates where overcrowding, poor sanitation and scabies co-infection are common — including remote Aboriginal and Torres Strait Islander communities in northern Australia and tropical low-income regions — and in these settings it is a real contributor to later chronic kidney disease, not a benign one-off. [6] [7]
Several features shift the diagnosis away from classic PSGN before a single blood result is back: an older child or adolescent with a normal complement (IgA nephropathy or IgA vasculitis); an adolescent girl with a low C3 and a low C4 (lupus nephritis); and any child whose C3 fails to recover by eight weeks (C3 glomerulopathy or membranoproliferative GN). [2] [11]
Pathophysiology
Post-streptococcal GN is an immune-complex disease. Nephritogenic streptococcal antigens — nephritis-associated plasmin receptor (NAPlr) and streptococcal pyrogenic exotoxin B (SpeB) — bind circulating antibody, form immune complexes, and deposit in the glomerulus. In-situ formation also contributes. The deposited complexes activate the complement cascade and ignite inflammation. [3] [4]
The cascade then runs in a predictable order. Immune-complex deposition switches on the alternative complement pathway, which is why serum C3 falls while C4 is typically preserved. Activated complement and chemokines draw neutrophils and macrophages into the tuft. The inflammatory cells, together with proliferating endothelial and mesangial cells, swell the capillary lumina (endocapillary proliferation). In the most severe cases the inflammation breaches Bowman's capsule and parietal epithelial cells and macrophages pile up into a crescent. [2] [11]
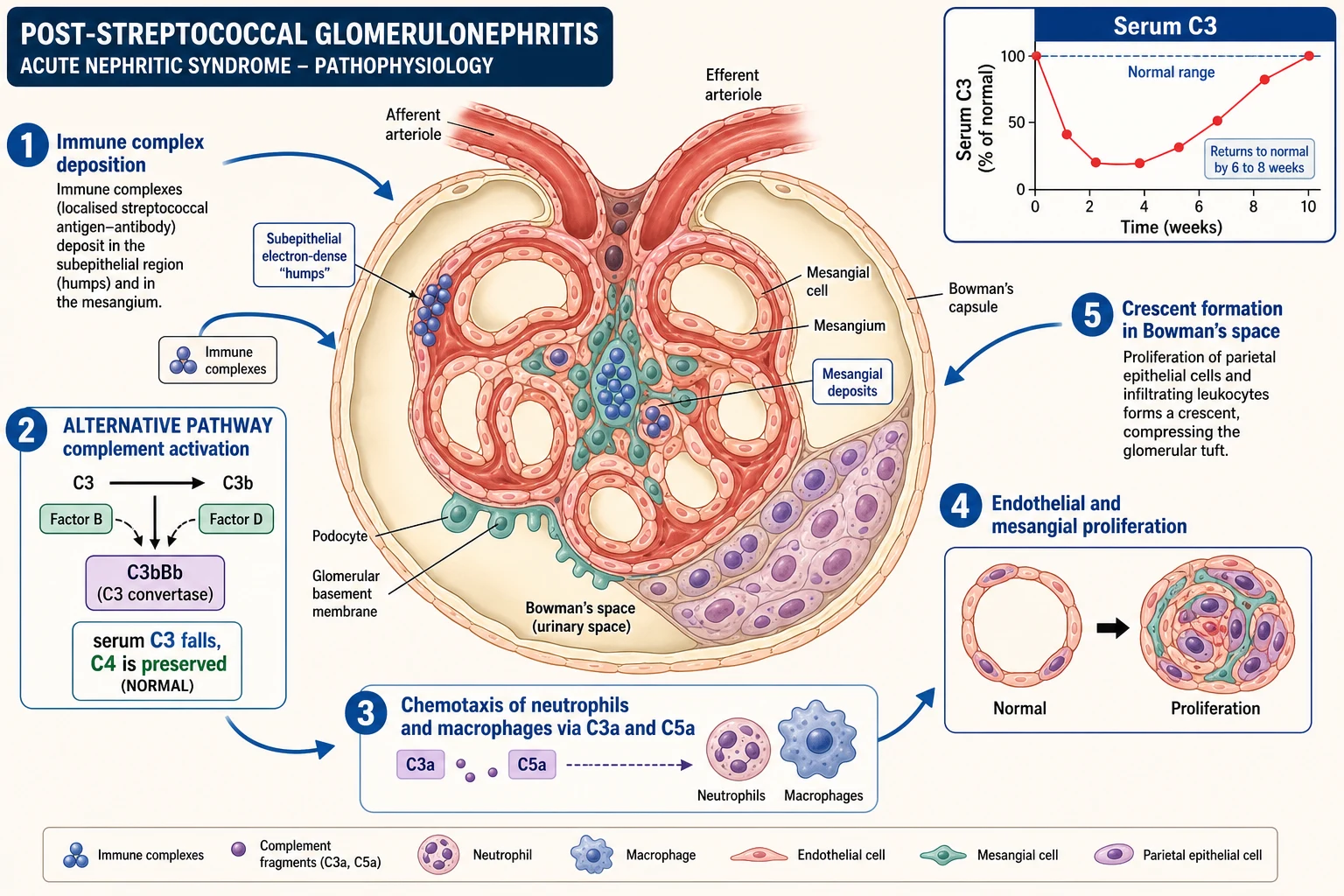
Each clinical feature is the downstream consequence of that inflammation. The damaged filter leaks red cells (haematuria) and protein (proteinuria). Reduced glomerular filtration and secondary tubular salt retention hold onto sodium and water, which produces oedema, oliguria and hypertension. When inflammation is severe enough to form crescents, filtration collapses and the creatinine rises rapidly — the rapidly progressive phenotype. [1] [2]
The time course of the complement is itself diagnostic. Because the immune trigger is transient, the alternative-pathway activation is self-limited: serum C3 falls in the first weeks and normalises within six to eight weeks in classic post-streptococcal GN. Complement that stays low beyond this window is not post-streptococcal — it is ongoing alternative-pathway dysregulation (C3 glomerulopathy) or classical-pathway disease (membranoproliferative GN, lupus), and it demands a biopsy. [3] [11]
Clinical Presentation
The classic presentation is a child between two and twelve years old with cola- or tea-coloured urine, periorbital oedema that is worse in the morning, and oliguria, appearing one to six weeks after a sore throat or a scabbed skin lesion. [6] [3]
Examination reveals facial and dependent oedema and an elevated blood pressure. In the more severe child, salt and water retention becomes clinically apparent as hepatomegaly, a third heart sound or gallop, and pulmonary crackles. The dangerous end of the spectrum is hypertensive encephalopathy — headache, visual disturbance, vomiting, altered consciousness or seizures — driven by the same volume overload that caused the oedema. [2]
Not every child reads the textbook. Microscopic haematuria found on a routine dipstick, an incidental finding of hypertension, or anasarca that mimics nephrotic syndrome can all be the first clue. In older patients, staphylococcal infection of a deep site (endocarditis, osteomyelitis) can produce an IgA-dominant infection-related GN — a reminder that the infecting organism and the host both shape the histology. [5]
Two patterns deserve a specific look. IgA vasculitis (Henoch-Schonlein purpura) nephritis sits alongside a palpable purpura on the lower limbs and buttocks, colicky abdominal pain and arthritis. IgA nephropathy, by contrast, produces synpharyngitic macroscopic haematuria — bleeding during the respiratory infection, not one to six weeks after it — and a normal complement. Holding these temporal patterns side by side is a common viva trap. [9] [10]
Differential Diagnosis
The nephritic differential is long, but it collapses quickly once you know the complement. The first task is always to confirm the bleeding is glomerular. Dysmorphic red cells (especially acanthocytes), red-cell casts, and coexisting proteinuria mark a glomerular source; isomorphic red cells without protein point downstream, to infection, stones, hypercalciuria, trauma or tumour. [1]
A few non-inflammatory causes round out the list. Idiopathic hypercalciuria can present with microscopic haematuria; benign familial haematuria (thin basement membrane nephropathy) gives isolated glomerular haematuria with a family history and normal complement; and Wilms tumour occasionally presents with haematuria. None of these produces the full nephritic tetrad, which is the clue that keeps them separate. [2]
The cardinal rule is to refuse complacency. A rapidly progressive course, nephrotic-range proteinuria, or a normal complement in a presentation that looks classic should never be assumed to be uncomplicated PSGN. Each of these is a biopsy trigger, and treating them as "just post-strep" is the classic error. [1] [11]
Clinical & Bedside Assessment
The focused assessment combines fluid balance, blood pressure and a search for extrarenal clues. Begin with weight, height and body mass index (needed for dosing and for growth trajectory), then move to fluid status: a strict input-output chart, a daily weight taken on the same scales at the same time, and a careful examination for volume overload. [2]
Blood-pressure technique matters, because the diagnosis and the danger both ride on the number. Use a cuff that covers 80 per cent of the upper arm, seat and rest the child for five minutes, measure the right arm first, and check four-limb pressures if coarctation is a possibility (an arm-to-leg gradient would redirect the diagnosis entirely). A single high reading is not enough to call it hypertension, but in a sick, oedematous child it is acted on now, not later. [1]
Examine for the features that change the cause. Look for the healing throat or crusted impetigo of a streptococcal trigger; the palpable lower-limb purpura of IgA vasculitis; the malar rash, oral ulcers, alopecia or arthritis of lupus; and a new murmur suggesting infective endocarditis. Each of these moves the child out of the "uncomplicated PSGN" box. [9] [2]
Identify and act on the bedside signs that escalate care. Respiratory distress, basal crackles and a gallop signal pulmonary oedema. A falling conscious level, papilloedema or focal signs signal hypertensive encephalopathy. Oligoanuria is a marker of severe, possibly crescentic, disease. Any of these warrants high-dependency or intensive care, not the general ward. [2]
Investigations
First-line investigations confirm that the process is glomerular and inflammatory. Send urinalysis (dipstick for blood and protein), urine microscopy for dysmorphic red cells and red-cell casts, serum creatinine and electrolytes, serum albumin, a full blood count, and — the single most informative blood test — serum complement C3 and C4. [1] [2]
Streptococcal serology documents the trigger. The anti-streptolysin O (ASO) titre rises three to five weeks after a throat infection, while anti-DNase B is the more sensitive marker after skin infection and stays positive longer. Either, when positive, supports a recent nephritogenic streptococcal exposure. A throat or skin swab may grow the organism, but treatment of the established GN does not depend on it. [3] [11]
Interpreting the complement is the central investigative skill. A low C3 with a normal or only mildly low C4 favours post-infectious GN or C3 glomerulopathy (alternative pathway). A low C3 with a low C4 favours lupus nephritis, membranoproliferative GN or endocarditis-related GN (classical pathway). A normal complement favours IgA nephropathy or IgA vasculitis nephritis. Reading C3 and C4 together, rather than C3 alone, is what prevents the classic misclassification. [2] [11]
NEPHRITIC
Renal biopsy is not needed for classic post-streptococcal GN, but it is mandatory when the pattern is atypical or severe. The accepted indications are: a rapidly progressive decline in renal function; nephrotic-range proteinuria; persistent hypocomplementaemia beyond eight weeks; a normal complement with no streptococcal evidence; and systemic features suggesting lupus or vasculitis. [1] [11]
When the picture is atypical or rapidly progressive, extend the panel: ANA and anti-dsDNA for lupus; ANCA and anti-GBM antibody for crescentic disease; hepatitis B and C serology and cryoglobulins for membranoproliferative GN; and a renal ultrasound to exclude obstruction or anatomic disease. These are not first-line for every child, but they are the work-up that separates the treatable, continuing diseases from the self-limiting one. [2]
Management — Resuscitation
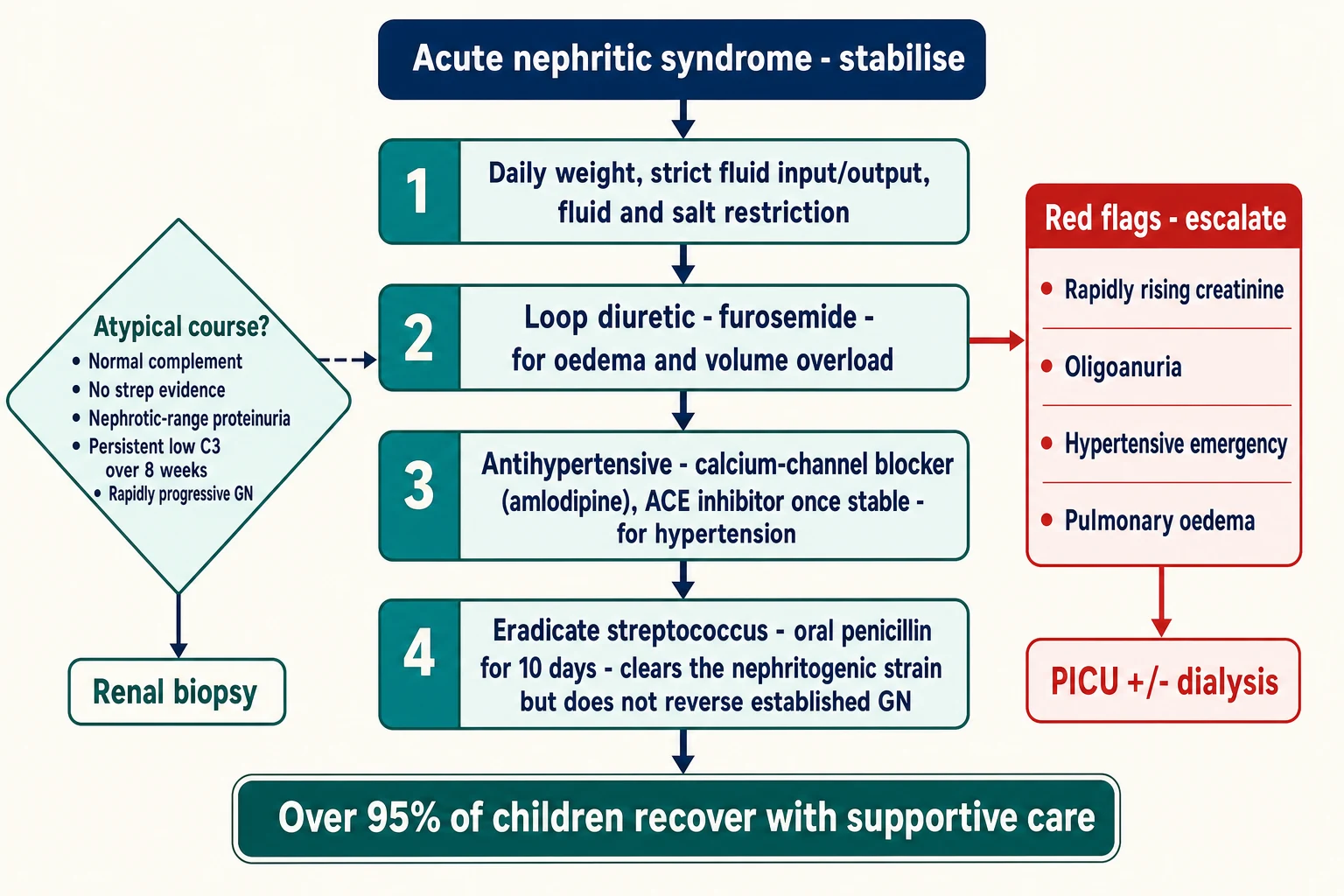
The immediate priorities control the consequences of glomerular inflammation: the salt and water retention, the hypertension, and the falling filtration. Restrict fluid and salt intake, weigh the child daily, and chart fluid balance meticulously. Treat volume overload and oedema with a loop diuretic — furosemide 1 to 2 mg per kilogram orally or intravenously, repeated as needed and guided by the daily weight. [1] [2]
For ordinary hypertension, start an oral agent. A calcium-channel blocker such as amlodipine is a practical first choice; an angiotensin-converting-enzyme inhibitor can be added once renal function and potassium are stable, and is particularly useful when proteinuria is prominent. [1]
Furosemide
Dose
1 to 2 mg/kg/dose
The thresholds for paediatric intensive care and renal replacement therapy are concrete: pulmonary oedema unresponsive to diuretics, hypertensive encephalopathy, severe hyperkalaemia, metabolic acidosis, and oligoanuric renal failure with fluid overload or uraemia. Meeting any one of these moves the child out of the general ward and into intensive care, with dialysis prepared. [1] [2]
Management — Definitive & Stepwise
The definitive bundle is supportive, and its rationale is mechanical. Fluid and salt restriction and loop diuretics unload the retained salt and water that drive oedema and hypertension; antihypertensives protect the brain and the heart; and eradication of the streptococcal strain prevents household spread. None of this reverses the established glomerular inflammation — time does that — but all of it keeps the child safe while the immune process burns out. [1]
Fluid and salt restriction, daily weight, strict input-output charting
Loop diuretic (furosemide 1 to 2 mg/kg) for oedema and volume overload
Antihypertensive: calcium-channel blocker (amlodipine) first; ACE inhibitor once potassium and renal function stable
Streptococcal eradication: penicillin V orally for 10 days, or a single IM dose of benzathine penicillin G
Daily monitoring of blood pressure, weight, urinalysis, creatinine and electrolytes
Plan a 6 to 8 week C3 recheck; biopsy if the course is atypical or rapidly progressive
The eradication regimen clears the nephritogenic strain. A ten-day course of oral phenoxymethylpenicillin (penicillin V), or a single intramuscular dose of benzathine penicillin G — 600,000 units for a child of 27 kg or less, and 1.2 million units for a child over 27 kg — is standard. It is essential to understand that this treats the infection and prevents transmission; it does not shorten the glomerulonephritis, because the immune injury is already established. Penicillin allergy is managed with a cephalosporin or macrolide according to local guidance. [3] [6]
Benzathine penicillin G
Dose
600,000 units if 27 kg or less; 1.2 million units if over 27 kg
Rapidly progressive (crescentic) GN is the exception to supportive care. It demands urgent renal biopsy and prompt immunosuppression, usually high-dose corticosteroids (intravenous methylprednisolone followed by oral prednisolone), with cyclophosphamide, rituximab or plasma exchange added depending on whether the disease is immune-complex-mediated, pauci-immune or anti-GBM. This is paediatric-nephrology territory, started early, because crescents scar quickly. [1] [2]
Monitoring and follow-up close the loop. Track blood pressure, weight, urinalysis, creatinine and C3 until they resolve; recheck C3 at six to eight weeks to confirm recovery. Because a small proportion of children progress to chronic kidney disease over the long term, arrange annual review of blood pressure, urinalysis and renal function even after an apparent full recovery. Advise restriction from contact sport for at least four weeks after the acute illness to protect against a hypertensive spike from fluid shifts. Discharge is appropriate once oedema, hypertension and renal function are improving, with outpatient review at two to four weeks and then every three to six months. [8] [7]
Specific Subtypes & Scenarios
Classic post-streptococcal GN is the benchmark against which every other cause is measured: a low C3 that recovers, a clear latent period, and an excellent prognosis. Staphylococcal infection-related GN is its awkward cousin — seen in older children and adults, often with IgA-dominant deposition, and tied to a deep-seated infection such as endocarditis or osteomyelitis rather than a simple sore throat. Controlling the underlying infection, not blanket immunosuppression, is the priority. [5] [11]
IgA vasculitis (Henoch-Schonlein purpura) nephritis is the most common childhood vasculitis. The nephritis ranges from isolated microscopic haematuria to a full nephritic or nephrotic syndrome, and it is the severity that guides treatment. Renal biopsy and immunosuppression are reserved for nephrotic-range proteinuria, nephritic syndrome with impaired function, or a rapidly progressive course; isolated microscopic haematuria needs only surveillance. Recent systematic reviews support a short course of corticosteroids for significant proteinuria, but not for isolated microscopic haematuria. [9] [10] [12]
C3 glomerulopathy — dense deposit disease and C3 glomerulonephritis — is the key differential when complement will not recover. It presents with a persistently low C3 and a normal C4, is diagnosed on biopsy, and is often progressive. It is the diagnosis to reach for whenever a "post-streptococcal" child still has a low C3 at eight weeks. [11] [2]
Lupus nephritis and membranoproliferative GN complete the persistent-hypocomplementaemia group. The adolescent girl with a low C3 and a low C4, a positive ANA and multisystem features has lupus nephritis until biopsy proves otherwise; the child with a mixed nephritic-nephrotic picture and a persistently low C3 and C4 may have membranoproliferative GN. Both need biopsy and disease-specific immunosuppression, and both are covered in depth in their own leaves. [2] [1]
Complications & Pitfalls
The acute complications are all downstream of salt and water retention and inflammation. Hypertensive encephalopathy and seizures, pulmonary oedema and cardiac failure, acute kidney injury requiring dialysis, and posterior reversible encephalopathy syndrome are the threats that occupy the first days. Each is preventable with disciplined fluid and blood-pressure control. [2]
The classic diagnostic pitfall is assuming that every haematuric child with a recent sore throat has uncomplicated PSGN. A normal complement, a low C4, nephrotic-range proteinuria, a rapidly rising creatinine, or systemic features each point somewhere else — lupus, C3 glomerulopathy, IgA disease, crescentic GN — and each demands investigation rather than reassurance. [11]
The monitoring pitfalls are equally important. Failing to recheck the C3 at six to eight weeks misses persistent hypocomplementaemia. Over-diuresing a volume-overloaded child can tip them into intravascular depletion and worsen the acute kidney injury. Starting an ACE inhibitor in a hyperkalaemic, oliguric child without monitoring the potassium is dangerous. Each of these errors is avoidable with a clear plan and serial review. [1]
The prognostic pitfall cuts the other way. Although more than 95 per cent of children with classic PSGN recover fully, a small but real proportion progress to chronic kidney disease over decades — particularly after epidemic disease. Treating every case as completely benign, and discharging the child from follow-up after the acute illness, loses the few who will need long-term renal protection. [8] [7]
Prognosis & Disposition
More than 95 per cent of children with classic post-streptococcal GN recover. Gross haematuria clears within days to weeks, C3 normalises within six to eight weeks, and proteinuria resolves over months. This favourable natural history is the reason supportive care, rather than immunosuppression, is the standard. [3] [6]
Several markers flag a poorer outcome and should be sought at every review: persistent low complement, nephrotic-range proteinuria, impaired renal function at presentation, crescents on biopsy, and older age or underlying comorbidity. Their presence is the signal to involve a nephrologist early and to plan longer follow-up. [7] [8]
The long-term risk is small but genuine. Twenty-year follow-up of epidemic post-streptococcal GN shows measurable rates of hypertension, proteinuria and reduced filtration — a finding that justifies long-term surveillance of blood pressure and renal function even after an apparent full recovery, and that has particular weight in endemic communities. [8]
Disposition is usually straightforward. Most children are managed on a general paediatric ward with outpatient follow-up. Intensive care or transfer to a renal centre is reserved for hypertensive emergency, pulmonary oedema, dialysis-requiring acute kidney injury, or a rapidly progressive or biopsy-proven atypical course. The decision is made on the clinical trajectory in the first 24 to 48 hours, not on the admission label. [2] [1]
Special Populations
Paediatric dosing is weight-based throughout. Loop diuretics and antihypertensives are calculated per kilogram, and the streptococcal eradication regimen pivots on the 27 kg cut-off for benzathine penicillin. Re-checking every dose against the child's actual weight, not an estimate, is a simple safeguard against error. [3]
In Aboriginal and Torres Strait Islander communities and other high-burden settings, post-streptococcal GN is endemic and tightly linked to scabies co-infection, overcrowding and limited access to primary care. The response is as much public-health as clinical: household screening and treatment of scabies and streptococcal skin sores, single-dose intramuscular benzathine penicillin for cases and contacts, and long-term surveillance because of the documented link to chronic kidney disease. [6] [7]
In the immunocompromised child the differential widens. Opportunistic infection-related GN enters the list, the threshold for biopsy is lower, and nephrotoxic medications (calcineurin inhibitors, antivirals) must be reviewed and held where possible. [11]
Adolescents raise their own issues. Lupus nephritis and pregnancy-related renal disease enter the differential; adherence to long-term immunosuppression is harder to sustain; and any atypical or persistent disease needs a planned transition to adult nephrology. These are the patients most likely to be lost to follow-up, and a deliberate handover protects them. [1]
Evidence, Guidelines & Regional Differences
The KDIGO 2021 Glomerular Diseases guideline is the current international reference. It sets the framework for classifying GN by clinical syndrome and immunopathology, defines the thresholds for investigation and biopsy, and gives the management principles for immune-complex and crescentic disease that this page follows. [1]
Regional practice follows KDIGO with local antibiotic guidance. In ANZ and the United Kingdom, streptococcal eradication follows BNF for Children and the Australian Therapeutic Guidelines; in high-burden regions, eradication is embedded in community secondary-prevention programs that treat whole households rather than individuals. The disease is global, but the operational rules for its prevention are regional. [6]
The main controversy sits in IgA vasculitis nephritis. The role, dose and duration of corticosteroids and other immunosuppression for moderate nephritis is genuinely debated; recent systematic reviews and network meta-analyses support short-course steroids for significant proteinuria but find no benefit for isolated microscopic haematuria. The evidence is firmer for severe, biopsy-proven disease, where steroids and adjunctive agents are standard. [10] [12]
The evidence gap is in prevention and in the harder diseases. There are few large randomised trials in paediatric post-streptococcal GN because supportive care is so effective; the pressing needs are preventing progression in endemic populations and finding effective treatment for C3 glomerulopathy, where current therapy is often inadequate. [7] [11]
Exam Pearls
Nephritic syndrome — high-yield facts
Key finding
The tetrad and the complement rule
The latent-period mnemonic is the most frequently examined temporal pattern: throat one to three weeks, skin three to six weeks, and IgA nephropathy synpharyngitic (concurrent). Getting this backwards in a viva is a common and avoidable error. [3] [9]
Penicillin eradicates the nephritogenic streptococcal strain and prevents household spread, but it does not reverse the established glomerulonephritis — the immune injury is already done. Examiners test this distinction directly. [3]
The poor-prognosis flags are the reverse image of the benign ones: persistent low complement, nephrotic-range proteinuria, impaired function at presentation, and crescents on biopsy. Holding both lists at once is what lets a candidate answer the "is this just post-strep?" question with confidence. [8] [7]
References
- [1]Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int, 2021.PMID 34556256
- [2]Sethi S; De Vriese AS; Fervenza FC Acute glomerulonephritis. Lancet, 2022.PMID 35461559
- [3]Rodriguez-Iturbe B; Musser JM The current state of poststreptococcal glomerulonephritis. J Am Soc Nephrol, 2008.PMID 18667731
- [4]Rodriguez-Iturbe B Autoimmunity in Acute Poststreptococcal GN: A Neglected Aspect of the Disease. J Am Soc Nephrol, 2021.PMID 33531351
- [5]Nasr SH; Fidler ME; Valeri AM; et al Postinfectious glomerulonephritis in the elderly. J Am Soc Nephrol, 2011.PMID 21051737
- [6]Balasubramanian R; Marks SD Post-infectious glomerulonephritis. Paediatr Int Child Health, 2017.PMID 28891413
- [7]Oda T; Yoshizawa N Factors Affecting the Progression of Infection-Related Glomerulonephritis to Chronic Kidney Disease. Int J Mol Sci, 2021.PMID 33477598
- [8]Pinto SW; do Nascimento Lima H; de Abreu TT; et al Twenty-year Follow-up of Patients With Epidemic Glomerulonephritis due to Streptococcus zooepidemicus in Brazil. Kidney Int Rep, 2022.PMID 36090503
- [9]Reamy BV; Servey JT; Williams PM Henoch-Schonlein Purpura (IgA Vasculitis): Rapid Evidence Review. Am Fam Physician, 2020.PMID 32803924
- [10]Mary AL; Clave S; Rousset-Rouviere C; et al Outcome of children with IgA vasculitis with nephritis treated with steroids: a matched controlled study. Pediatr Nephrol, 2023.PMID 37154959
- [11]Iyengar A; Kamath N; Radhakrishnan J; et al Infection-Related Glomerulonephritis in Children and Adults. Semin Nephrol, 2023.PMID 38242806
- [12]Wang Y; He Y; Cheng F; et al Optimal drug treatment for children with IgA vasculitis nephritis: a systematic review and network meta-analysis. Transl Pediatr, 2025.PMID 41502882