Paeds · nephrology-urology-fluids-and-electrolytes
Calcium, magnesium and phosphate disorders
Also known as Hypocalcaemia · Hypercalcaemia · Hypomagnesaemia · Hypermagnesaemia · Hypophosphataemia · Hyperphosphataemia · Mineral and bone disorder · Disorders of divalent cations and phosphate
Fellowship guide to disorders of calcium, magnesium and phosphate in children: corrected calcium and ionised fractions, neonatal early and late hypocalcaemia, hypoparathyroidism and DiGeorge syndrome, vitamin D deficiency rickets, hypercalcaemia with immobilisation and Williams syndrome, hypomagnesaemia as the cause of refractory hypocalcaemia and hypokalaemia, refeeding hypophosphataemia, X-linked hypophosphataemic rickets with excess FGF23, tumour lysis hyperphosphataemia with hypocalcaemia, and the emergency doses of intravenous calcium gluconate and magnesium sulfate.
On this page & tools
Your progress
Saved locally on this device.
Practise this topic
Target exams
Red flags
Life stages
Care settings
Clinical exam formats
Board mappings
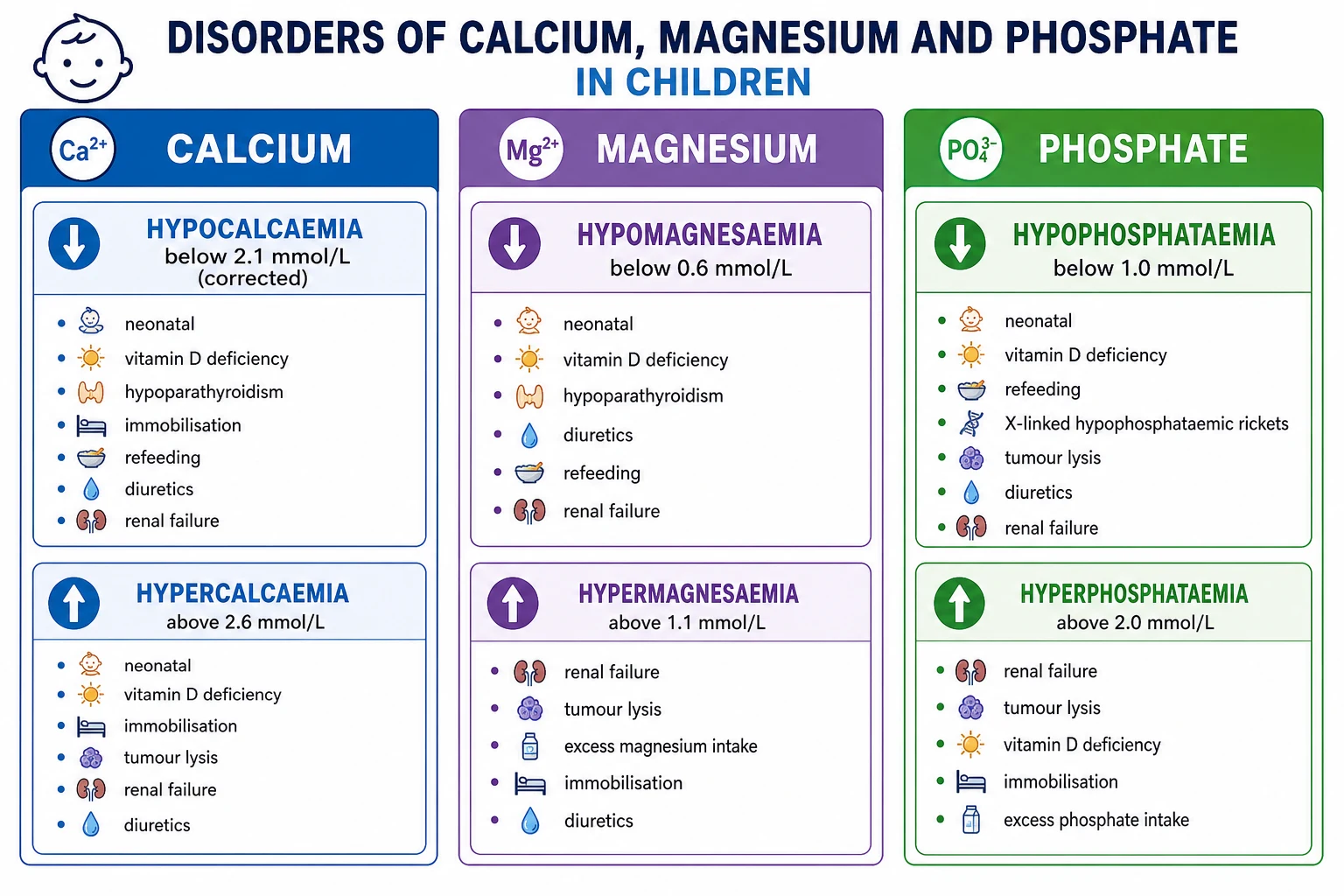
Calcium, magnesium and phosphate travel together. About 99 percent of body calcium and 60 percent of body magnesium sit in bone, and phosphate is their structural partner in hydroxyapatite, so the small serum fraction is a tightly regulated snapshot of a much larger reservoir. [3] [8] Three hormones hold the serum in range: parathyroid hormone raises calcium by resorbing bone and reabsorbing calcium in the distal tubule while it dumps phosphate; activated vitamin D (calcitriol) increases gut absorption of all three; and fibroblast growth factor 23 lowers phosphate by forcing the kidney to excrete it and by switching off calcitriol. [9] When one mineral moves, the others follow, which is why a single abnormal value almost always disturbs its neighbours.
This page treats the three minerals as one system, then splits them by the direction of the disturbance and the mechanism that drove it. Calcium sets the urgency, because a low ionised calcium can stop the heart through a prolonged QT and obstruct the airway through laryngospasm, and a high calcium can dehydrate and destabilise. [1] Magnesium is the hidden cause, because it gates both parathyroid hormone secretion and the renal potassium channel, so a deficit silently breaks calcium and potassium homeostasis at once. [7] Phosphate is the chronic bone problem and the acute refeeding and tumour lysis problem, and its value swings with age, because growing children hold far more phosphate than adults to build skeleton. [9] [12]
Overview & Definition
The first task at the bedside is to read the calcium correctly. Total calcium is reported as a concentration bound to albumin, so hypoalbuminaemia makes a normal calcium look low. The corrected calcium adds back the missing albumin: corrected calcium equals measured calcium plus 0.02 multiplied by 40 minus the albumin in grams per litre, which is the same as adding 0.4 mmol/L for every 10 g/L that albumin sits below 40. [3] [4] The ionised calcium is the active, free fraction and is the value to trust in critical illness, after massive transfusion, in alkalosis and whenever the albumin is unreliable. [2]
Hypocalcaemia is a corrected calcium below 2.1 mmol/L or an ionised calcium below about 1.1 mmol/L; symptoms usually appear below 1.9 mmol/L total and become dangerous below 1.5. Hypercalcaemia is a corrected calcium above 2.6 mmol/L, and it is severe and a potential emergency above 3.0, with a level above 3.5 carrying a real risk of arrhythmia and altered consciousness. [1] [3] Magnesium is normal between 0.7 and 1.0 mmol/L, with hypomagnesaemia below 0.6 and hypermagnesaemia above 1.1. Phosphate is age-dependent and must be read against the reference range: a neonate legitimately runs a phosphate of 1.6 to 2.6 mmol/L, while a teenager sits near 1.0 to 1.5, and hypophosphataemia in a child is generally below 1.0 mmol/L with severe disease below 0.3. [8] [9]
Classification
These disorders are grouped by the mineral that is abnormal and by the mechanism that moved it, because the mechanism dictates the investigation and the fix. The most useful split separates a deficit from an excess of each mineral, then asks whether the problem lies in intake, gut absorption, hormonal regulation, renal handling, or a shift between compartments. [1]
Hypocalcaemia
corrected below 2.1 mmol/L
- Neonatal early (under 72 hours): prematurity, perinatal asphyxia, maternal diabetes, sepsis.
- Neonatal late (5 to 14 days): phosphate load from cow-milk formula, maternal vitamin D deficiency, DiGeorge (22q11) hypoparathyroidism, hypomagnesaemia.
- Hypoparathyroidism: post-surgical, autoimmune (APECED), DiGeorge, activating calcium-sensing receptor mutations.
- Vitamin D deficiency: nutritional rickets, malabsorption, anticonvulsants, chronic kidney disease with low calcitriol.
- Acute shift or chelation: alkalosis, massive citrated transfusion, pancreatitis, sepsis, bisphosphonate.
Hypercalcaemia
corrected above 2.6 mmol/L
- Primary hyperparathyroidism: parathyroid adenoma or MEN syndromes.
- Immobilisation: rapid bone resorption in a bedfast teenager or a child in traction.
- Vitamin D or vitamin A intoxication, granulomatous disease (sarcoid, TB) making excess calcitriol.
- Williams syndrome, subcutaneous fat necrosis of the newborn, familial hypocalciuric hypercalcaemia.
- Malignancy, thiazide diuretics, milk-alkali syndrome.
Hypomagnesaemia
below 0.6 mmol/L
- Renal loss: loop and thiazide diuretics, aminoglycosides, cisplatin, calcineurin inhibitors, proton pump inhibitors.
- Inherited: Gitelman and Bartter syndromes, familial hypomagnesaemia with secondary hypocalcaemia (TRPM6).
- Gut loss: prolonged diarrhoea, malabsorption, short bowel, refeeding.
- The hidden culprit: hypomagnesaemia suppresses parathyroid hormone release and so causes refractory hypocalcaemia, and it disinhibits ROMK to cause refractory hypokalaemia.
Phosphate disorders
age-dependent range
- Hypophosphataemia by shift: refeeding, diabetic ketoacidosis treatment, respiratory alkalosis, recovering sepsis.
- Renal phosphate wasting: hyperparathyroidism, Fanconi syndrome, post-transplant, X-linked hypophosphataemic rickets (FGF23 excess).
- Gut loss: vitamin D deficiency, phosphate-binding antacids, chronic diarrhoea.
- Hyperphosphataemia: renal failure, tumour lysis syndrome, rhabdomyolysis, acidosis, hypoparathyroidism, phosphate enemas.
Epidemiology & Risk Factors
Vitamin D deficiency is the commonest cause of symptomatic hypocalcaemia and rickets worldwide, and the Global Consensus on nutritional rickets places the highest risk in exclusively breast-fed infants whose mothers are themselves deficient, dark-skinned children living at high latitude, and any child with limited sunlight exposure and low dietary intake. [5] The 2024 Endocrine Society vitamin D guideline reinforces that supplementation in infancy prevents rickets and that deficiency remains prevalent across populations that under-fortify food. [6]
Neonates are a distinct high-risk group. Early neonatal hypocalcaemia in the first 72 hours clusters in premature infants, those with perinatal asphyxia, infants of diabetic mothers, and sick neonates with sepsis, because the calcium-supplying mechanisms that should activate at birth are immature or overwhelmed. [2] Late neonatal hypocalcaemia, peaking at the end of the first week, is driven by a phosphate load from cow-milk formula, by maternal vitamin D deficiency, by hypoparathyroidism including DiGeorge syndrome, and by hypomagnesaemia. [2]
In hospitalised children the risk concentrates in three groups: the critically ill child on the intensive care unit where alkalosis, citrate from transfusion and sepsis all depress ionised calcium; the oncology patient at risk of tumour lysis after cytotoxic therapy for a high-burden lymphoma or leukaemia; and the malnourished child or adolescent with anorexia nervosa entering a refeeding programme. [11] [12] Children on loop or thiazide diuretics, calcineurin inhibitors, aminoglycosides, cisplatin and chronic proton pump inhibitors are quietly losing magnesium and may present with refractory hypocalcaemia before the magnesium is even checked. [7]
Pathophysiology
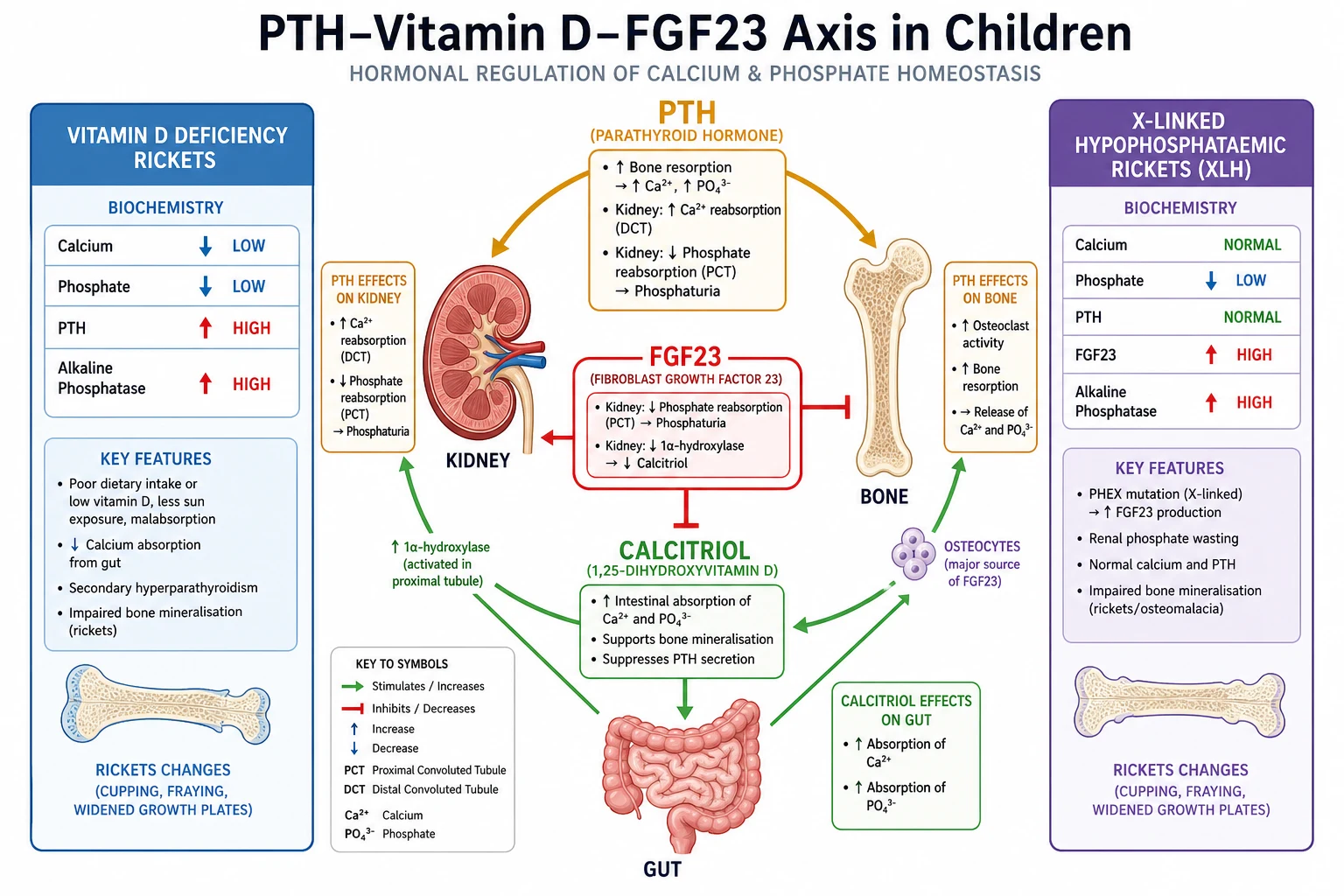
The serum level of each mineral is governed by the same three-way hormonal conversation between parathyroid hormone, vitamin D and fibroblast growth factor 23, played out across bone, gut and kidney. Understanding this loop predicts how a disturbance behaves and how it responds to treatment. [3] [9]
Parathyroid hormone is the emergency responder to a falling calcium. It is released when the calcium-sensing receptor on the parathyroid chief cell detects a low ionised calcium, and it acts within minutes to resorb calcium from bone and to reabsorb calcium in the distal renal tubule while it simultaneously phosphaturic, dumping phosphate to stop a high phosphate from complexing the calcium it just rescued. [3] Parathyroid hormone also drives the kidney to convert 25-hydroxyvitamin D to calcitriol, which then increases gut absorption of calcium and phosphate over hours to days. Vitamin D deficiency therefore presents with low calcium, low or low-normal phosphate, a high parathyroid hormone and a high alkaline phosphatase — the biochemical signature of nutritional rickets. [5]
Fibroblast growth factor 23 is the phosphate governor and the vitamin D brake. Released by osteocytes in response to a high phosphate, it forces the proximal tubule to stop reabsorbing phosphate and to switch off the enzyme that makes calcitriol. [9] In X-linked hypophosphataemic rickets the defect in PHEX drives excess fibroblast growth factor 23, so the kidney wastes phosphate, calcitriol stays inappropriately low, and the calcium and parathyroid hormone remain normal while the alkaline phosphatase climbs — the signature that separates this inherited rickets from the vitamin D deficiency form. [9] [10]
Magnesium holds the gates on this system. It is required for parathyroid hormone release and for adenylate cyclase signalling in the target tissues, so a low magnesium silently suppresses parathyroid hormone and produces a functional hypoparathyroidism that no amount of calcium will fix until the magnesium is restored. [7] Magnesium also governs the ROMK potassium channel in the distal nephron, which is why hypomagnesaemia and refractory hypokalaemia travel together: until magnesium is replaced, the kidney keeps wasting potassium regardless of the dose given. [7] [8]
A compartment shift explains the acute phosphate and calcium swings. Refeeding drives insulin into a starved body, and insulin shifts phosphate into cells to build adenosine triphosphate and phosphorylate glucose, so the serum phosphate falls just as the metabolic demand explodes. [12] Tumour lysis releases a torrent of intracellular phosphate and potassium from lysed malignant cells, and the rising phosphate complexes with calcium to drive the ionised calcium down even though total calcium may look near normal. [11] Alkalosis increases calcium binding to albumin and so lowers the ionised fraction without changing the total, which is why an anxious, hyperventilating adolescent can develop symptomatic tetany with a normal total calcium. [3]
Clinical Presentation
Hypocalcaemia presents with neuromuscular excitability and the landmarks are the bedside signs. Carpopedal spasm is the involuntary contraction of the hand and wrist, and when a blood-pressure cuff inflated above systolic for three minutes reproduces it, that is a positive Trousseau sign. [2] Tapping the facial nerve in front of the ear twitches the ipsilateral facial muscles in a positive Chvostek sign, but it is unreliable — present in some normal children and absent in many with genuine hypocalcaemia. The serious presentations are laryngospasm with stridor, generalised seizures, and a prolonged QT interval that can progress to torsades de pointes and cardiac arrest. [1]
Neonates show the same excitability less clearly. A hypocalcaemic neonate may present with jitteriness, a high-pitched cry, apnoea, poor feeding, or frank seizures, and the threshold to measure ionised calcium in any seizing or irritable neonate is low. [2] Older children with chronic hypocalcaemia, as in hypoparathyroidism or vitamin D deficiency, may show rachitic features: frontal bossing, craniotabes, widened wrists, bowing of the legs, and delayed dentition. [5]
Hypercalcaemia presents with the opposite tone. The child is constipated, anorexic, nauseated and tired, with polyuria and polydipsia as the kidney loses its concentrating ability, which in turn produces dehydration and drives the calcium higher in a vicious circle. [3] Severe hypercalcaemia above 3.5 mmol/L causes confusion, lethargy and a shortened QT interval, and chronic hypercalcaemia in children deposits calcium in the renal medulla as nephrocalcinosis or as renal stones, presenting with haematuria or colic. [1]
Hypomagnesaemia looks exactly like hypocalcaemia, because it produces the same tetany, seizures and prolonged QT, and it usually coexists with hypokalaemia, so muscle weakness, cramps and an ileus may be added. [7] Hypermagnesaemia is rare and almost always iatrogenic, seen in the neonate whose mother received magnesium sulfate for eclampsia or preterm labour, and it presents with hypotonia, hyporeflexia or areflexia, respiratory depression, hypotension and bradycardia. [8]
Hypophosphataemia weakens every cell that depends on adenosine triphosphate. The acute picture in refeeding or severe deficiency is proximal muscle weakness that can progress to respiratory failure as the diaphragm fails, rhabdomyolysis, haemolysis, impaired myocardial contractility and leucocyte dysfunction. [12] Chronic hypophosphataemia, as in X-linked hypophosphataemic rickets, presents with short stature, progressive leg bowing, bone pain, dental abscesses and enthesopathy. [9] Hyperphosphataemia is usually silent itself and announces its presence through the hypocalcaemia and ectopic calcification it causes. [11]
Differential Diagnosis
The differential narrows fast once the biochemistry returns, and the pattern of calcium with phosphate, parathyroid hormone, alkaline phosphatase and vitamin D usually points to a single mechanism. [3] [5]
Low calcium with high phosphate
points to hypoparathyroid
- Hypoparathyroidism of any cause: post-surgical, autoimmune (APECED), DiGeorge (22q11) syndrome.
- Pseudohypoparathyroidism: high parathyroid hormone but end-organ resistance.
- Chronic kidney disease: phosphate retained and calcitriol low, so calcium falls.
- Tumour lysis and rhabdomyolysis: phosphate released, calcium complexed.
Low calcium with low phosphate
points to vitamin D
- Nutritional vitamin D deficiency rickets: low calcium and phosphate, high parathyroid hormone and alkaline phosphatase, low 25-hydroxyvitamin D.
- Malabsorption: coeliac disease, cystic fibrosis, short bowel.
- Liver failure or anticonvulsant therapy impairing vitamin D activation.
- Acute shift: alkalosis, massive transfusion citrate, pancreatitis.
Low phosphate with normal calcium
points to renal wasting
- X-linked hypophosphataemic rickets: normal calcium and parathyroid hormone, high alkaline phosphatase and fibroblast growth factor 23.
- Fanconi syndrome and other proximal tubulopathies: glycosuria, aminoaciduria, acidosis.
- Refeeding and recovery from diabetic ketoacidosis: insulin-driven shift.
- Hyperparathyroidism: phosphate wasted with calcium high.
The hidden cause of refractory hypocalcaemia
check the magnesium
- Hypomagnesaemia suppresses parathyroid hormone release and produces functional hypoparathyroidism.
- Diuretic, proton pump inhibitor, calcineurin inhibitor and cisplatin exposure.
- Gitelman syndrome: hypomagnesaemia with hypokalaemic alkalosis and hypocalciuria.
- Inherited TRPM6 defect: familial hypomagnesaemia with secondary hypocalcaemia and seizures.
The single most reliable discriminator in the rickets phenotypes is the combination of calcium and parathyroid hormone. Vitamin D deficiency lowers calcium, which raises parathyroid hormone, which in turn lowers phosphate; X-linked hypophosphataemic rickets keeps calcium and parathyroid hormone normal because the excess fibroblast growth factor 23 independently drives the phosphate down. [5] [9] The alkaline phosphatase is high in both, because both produce rickets, so it cannot separate them — but the calcium, phosphate and parathyroid hormone together can. [10]
Clinical & Bedside Assessment
Begin with airway, breathing and circulation and attach cardiac monitoring, because a dangerously low ionised calcium can prolong the QT and arrest the heart, and a dangerously high magnesium or calcium can do the same. [1] An electrocardiogram is the single most useful bedside test: a prolonged QT interval with a flattened T wave and a prominent U wave points to hypocalcaemia or hypokalaemia, while a shortened QT interval suggests hypercalcaemia. [2]
Elicit the excitability signs deliberately. Inflate a blood-pressure cuff above systolic for three minutes and watch for carpopedal spasm (Trousseau sign), the more reliable of the two, and tap the facial nerve to look for facial muscle twitching (Chvostek sign), interpreting it cautiously. [2] Assess volume status from capillary refill, pulse, blood pressure and the mucous membranes, because the dehydrated hypercalcaemic child needs saline before anything else, and the volume-depleted hypocalcaemic child with sepsis needs resuscitation in parallel with the electrolyte correction. [1]
Take a history aimed at the mechanism. Ask about diet and sunlight exposure, maternal vitamin D intake, the type of infant formula, recent surgery especially thyroid or parathyroid, chemotherapy and the day it started, diuretic or antacid use, chronic proton pump inhibitor therapy, a family history of hypocalcaemia or rickets, and any features of DiGeorge syndrome such as cardiac murmur, cleft palate or recurrent infections. [2] [5]
Bedside and laboratory anchors
Investigations
The core panel is a set of interlocking values that must be read together: corrected and ionised calcium, phosphate, magnesium, albumin, renal function, alkaline phosphatase, a venous gas, and a 12-lead electrocardiogram. [1] These alone answer most of the questions, because the pattern of the minerals with the renal function and the acid-base status points to the mechanism.
The second tier adds the hormones. Measure parathyroid hormone with a simultaneous calcium: an inappropriately low parathyroid hormone in the face of hypocalcaemia points to hypoparathyroidism, a high parathyroid hormone to secondary hyperparathyroidism of vitamin D deficiency or chronic kidney disease, and a high parathyroid hormone with a high calcium to primary hyperparathyroidism. [3] Measure 25-hydroxyvitamin D to confirm deficiency, and consider fibroblast growth factor 23 where X-linked hypophosphataemic rickets is suspected. [9]
Hypocalcaemia work-up — CALCIUM
CALCIUM
correct the calcium for albumin, and check the ionised fraction
albumin for the correction, a blood gas for pH and bicarbonate
hypomagnesaemia is the hidden cause — check it early
renal function, phosphate, alkaline phosphatase
the active fraction — trust it in critical illness
urinary calcium and phosphate, and the tubular reabsorption of phosphate
QT interval — a prolonged QT signals danger
Targeted tests follow the screen. A 25-hydroxyvitamin D below 50 nmol/L confirms insufficiency and below 25 nmol/L deficiency; parathyroid hormone is high in vitamin D deficiency and chronic kidney disease and low in hypoparathyroidism; fibroblast growth factor 23 is elevated in X-linked hypophosphataemic rickets and tumour-induced osteomalacia. [5] [9] Genetic testing confirms inherited tubulopathies and hypomagnesaemias, and a renal ultrasound looks for nephrocalcinosis in hypercalcaemia and for nephrocalcinosis risk in Bartter, the calcium direction that separates it from Gitelman. [7]
Management — Resuscitation
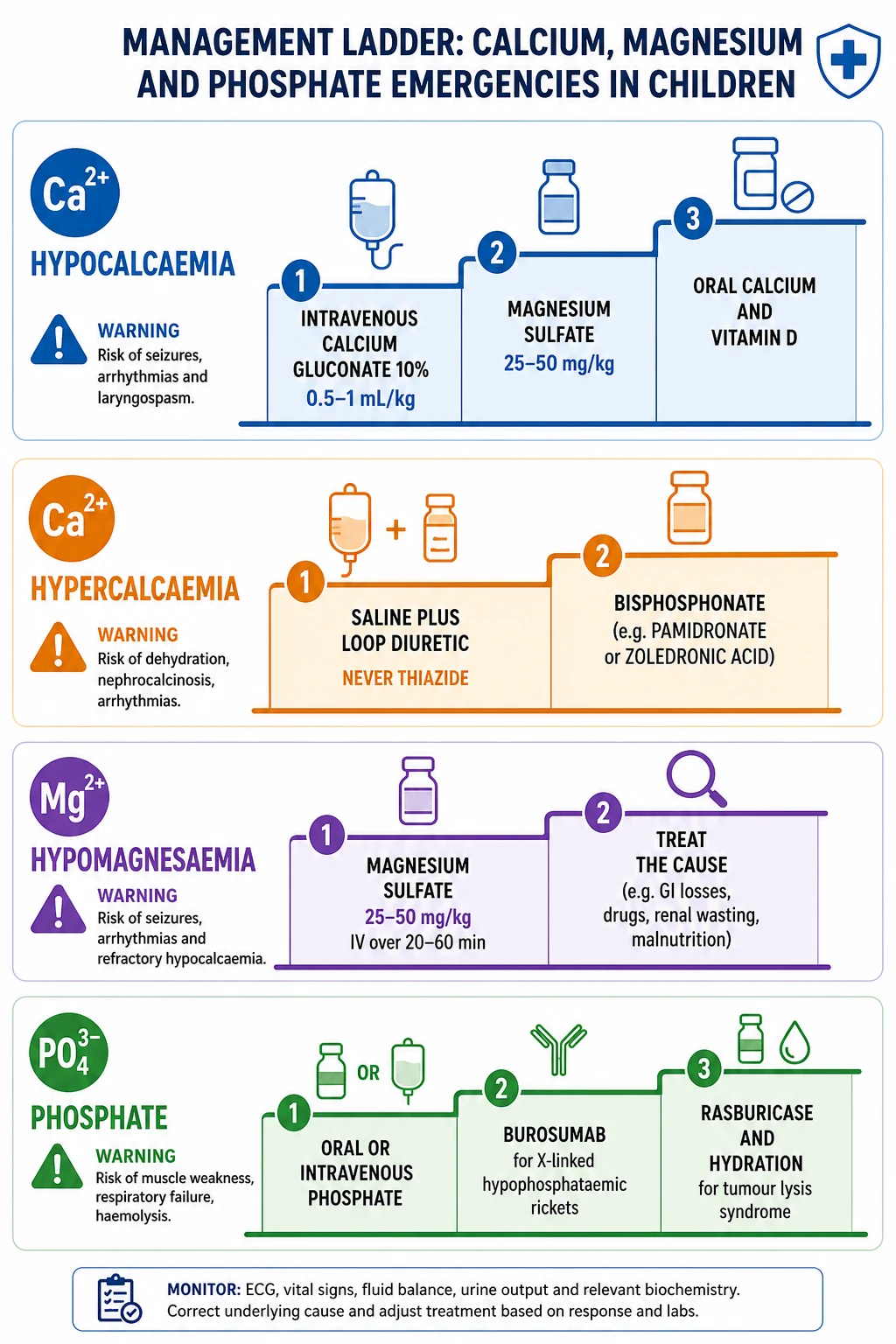
Symptomatic hypocalcaemia with seizures, stridor, carpopedal spasm or a prolonged QT is an emergency. Secure intravenous access and give 10 percent calcium gluconate at 0.5 to 1 mL per kilogram, to a maximum of about 20 mL, slowly over 5 to 10 minutes with continuous cardiac monitoring. [1] [2] Calcium gluconate rather than calcium chloride is preferred peripherally because it is less vesicant if it extravasates; a central line is required for calcium chloride. Stop the infusion if the heart rate drops, because rapid calcium itself causes bradycardia and arrhythmia. [3]
Before or alongside the calcium, check and replace magnesium. Give magnesium sulfate at 25 to 50 mg per kilogram, to a maximum of 2 g, intravenously over 2 to 4 hours with monitoring, because hypocalcaemia will not resolve until magnesium is restored. [7] This single step — magnesium first when the calcium will not correct — is the most reliable intervention in refractory hypocalcaemia and the one most often missed. [8]
Severe hypercalcaemia above 3.0 mmol/L, and any level with dehydration or altered consciousness, is resuscitated with isotonic saline at 10 to 20 mL per kilogram, then a loop diuretic such as furosemide 1 mg per kilogram once rehydrated, to force a sodium-linked calcium diuresis. [1] [3] Never give a thiazide, which reabsorbs calcium in the distal tubule and worsens hypercalcaemia. A bisphosphonate — pamidronate or zoledronic acid — is added for hypercalcaemia of malignancy or immobilisation that does not settle with saline, and calcitonin gives a faster but short-lived calcium-lowering effect while the bisphosphonate takes hold over days. [3]
Hypermagnesaemia in the neonate is managed by stopping the source, supporting respiration, and giving intravenous calcium gluconate as the direct antagonist to the magnesium at the neuromuscular junction. [8] Severe cases need hydration with or without a loop diuretic and, rarely, dialysis. Acute severe hypophosphataemia with respiratory failure or rhabdomyolysis needs intravenous phosphate, given as sodium or potassium phosphate at 0.08 to 0.24 mmol per kilogram over 6 hours, watching the calcium because over-rapid replacement can trigger hypocalcaemia. [12]
Management — Definitive & Stepwise
Once the acute disturbance is controlled, definitive management treats the mechanism and restores the steady state. The framework is to identify and remove the cause, replace the deficit by the appropriate route and rate, and prevent recurrence with the right maintenance. [1]
Hypocalcaemia: from emergency to maintenance
Stabilise the myocardium with intravenous 10 percent calcium gluconate 0.5 to 1 mL/kg over 5 to 10 minutes with cardiac monitoring. <Cite id="1" />
Check and replace magnesium with magnesium sulfate 25 to 50 mg/kg before expecting the calcium to correct. <Cite id="7" />
Confirm the cause: parathyroid hormone, 25-hydroxyvitamin D, renal function, phosphate, magnesium. <Cite id="3" /><Cite id="5" />
Begin oral calcium and the relevant vitamin D: cholecalciferol for deficiency, calcitriol (1,25-dihydroxyvitamin D) for hypoparathyroidism or chronic kidney disease. <Cite id="4" /><Cite id="6" />
Arrange surveillance and transition: serial minerals, renal ultrasound for nephrocalcinosis, and genetic or endocrine follow-up for inherited causes. <Cite id="9" />
For vitamin D deficiency rickets, give a loading dose of cholecalciferol (vitamin D3) and then maintenance supplementation, together with oral calcium, because remineralisation of bone will draw down serum calcium and can provoke hunger fractures if calcium is not supplied. [5] The 2024 Endocrine Society guideline and the global consensus recommend that all infants receive vitamin D supplementation from birth and that deficiency is treated until the biochemistry and radiographs normalise. [6]
For permanent hypoparathyroidism the management is different, because parathyroid hormone cannot be replaced easily, so the standard is calcitriol (active 1,25-dihydroxyvitamin D) with oral calcium, titrated to keep calcium in the low-normal range to avoid hypercalciuria and nephrocalcinosis, with a target 24-hour urinary calcium monitored. [3] [4] Thiazide diuretics reduce urinary calcium and help, the opposite of their danger in hypercalcaemia. Recombinant parathyroid hormone is an emerging therapy for difficult cases. [4]
For X-linked hypophosphataemic rickets the modern standard is burosumab, a monoclonal antibody that binds and neutralises fibroblast growth factor 23, which restores phosphate reabsorption and calcitriol, heals the rickets, and improves linear growth and bowing compared with conventional oral phosphate and calcitriol. [9] [10] Conventional therapy with oral phosphate in divided doses plus calcitriol remains an option where burosumab is unavailable, but it is harder to titrate and carries a risk of secondary hyperparathyroidism and nephrocalcinosis. [10]
Tumour lysis syndrome is prevented and treated as a bundle: aggressive isotonic hydration at 3 litres per square metre per day or 125 mL per square metre per hour started before chemotherapy where possible, allopurinol for low and medium risk, and rasburicase for high-risk disease with a high tumour burden, because rasburicase breaks down uric acid directly and is far faster than allopurinol. [11] The calcium needs watching, because the released phosphate complexes it and the ionised calcium falls; avoid calcium supplementation unless the patient is symptomatic, because adding calcium to a high phosphate risks metastatic calcification. [11]
[1]Specific Subtypes & Scenarios
Neonatal hypocalcaemia
Early neonatal hypocalcaemia in the first 72 hours reflects an immature parathyroid response and a surge of phosphate from tissue breakdown in prematurity, asphyxia and sepsis, and in the infant of a diabetic mother. [2] Late neonatal hypocalcaemia, at the end of the first week into the second, is driven by phosphate loading from cow-milk formula, by maternal vitamin D deficiency that left the infant depleted, by hypomagnesaemia, and by hypoparathyroidism including DiGeorge syndrome. [2] Any neonate with seizures, jitteriness or apnoea should have an ionised calcium measured, and DiGeorge should be considered whenever hypocalcaemia accompanies a congenital cardiac lesion, a cleft palate or characteristic facies. [2]
DiGeorge syndrome (22q11 deletion)
The 22q11.2 deletion disrupts development of the third and fourth pharyngeal pouches, producing hypoparathyroidism with hypocalcaemia alongside conotruncal cardiac defects (tetralogy of Fallot, interrupted aortic arch), cleft palate, immune deficiency from thymic hypoplasia, and characteristic facies. [2] [3] Hypocalcaemia may be the presenting feature, and any infant with hypocalcaemia and a heart murmur warrants a fluorescence in-situ hybridisation or chromosomal microarray for the deletion. [2]
Vitamin D deficiency rickets
The classic nutritional rickets of infancy and childhood presents with bowing of the legs, widened wrists, frontal bossing, craniotabes, delayed dentition, and a waddling gait, on a background of exclusive breast-feeding without supplementation, dark skin, limited sunlight, or malabsorption. [5] The biochemistry is low calcium, low or low-normal phosphate, high parathyroid hormone and high alkaline phosphatase, with a low 25-hydroxyvitamin D, and wrist radiographs show widened, frayed, cupped metaphyses. [5] [6] Treatment is cholecalciferol loading plus oral calcium, then maintenance supplementation. [5]
X-linked hypophosphataemic rickets
The commonest inherited rickets, caused by mutations in PHEX that drive excess fibroblast growth factor 23. It presents with short stature, severe progressive leg bowing, bone pain, dental abscesses and enthesopathy, and the biochemistry separates it from vitamin D deficiency: normal calcium, normal or slightly high parathyroid hormone, low phosphate, high alkaline phosphatase, and elevated fibroblast growth factor 23. [9] [10] Burosumab is now the international guideline-recommended standard for children, replacing conventional oral phosphate and calcitriol. [10]
Tumour lysis syndrome
A metabolic emergency within 12 to 72 hours of starting cytotoxic therapy for a high-burden haematological malignancy such as Burkitt lymphoma or T-cell acute lymphoblastic leukaemia. The lysed cells release potassium and phosphate, the rising phosphate complexes calcium to produce hypocalcaemia, and nucleic-acid breakdown generates uric acid. [11] The hyperphosphataemia, hyperkalaemia, hypocalcaemia and hyperuricaemia together produce acute kidney injury, and the bundle of aggressive hydration with rasburicase for high risk prevents and treats the syndrome. [11]
Refeeding hypophosphataemia
The malnourished child or the adolescent with anorexia nervosa who is refed too rapidly suffers an insulin surge that drives phosphate, potassium and magnesium into cells, just as the metabolic demand for adenosine triphosphate explodes. [12] The result is hypophosphataemia with respiratory failure, cardiac collapse, rhabdomyolysis and haemolysis, and the systematic review evidence in adolescents with anorexia confirms that starting calories low and escalating slowly with phosphate, potassium and magnesium replacement prevents the syndrome. [12]
Hypercalcaemia of immobilisation and Williams syndrome
A bedfast teenager or a child in traction resorbs bone faster than it is laid down, and the calcium rises; the same mechanism explains the hypercalcaemia that occasionally follows fracture healing and the hypercalciuria of immobilisation that causes renal stones. [1] Williams syndrome, caused by a 7q11.23 microdeletion, produces a characteristic elfin facies, supravalvular aortic stenosis, a friendly loquacious personality, and hypercalcaemia in infancy, managed with a low-calcium diet and, if severe, bisphosphonate. [3]
Complications & Pitfalls
Cardiac
the fastest killer
- Hypocalcaemic prolonged QT degenerates to torsades de pointes and ventricular fibrillation.
- Hypercalcaemia shortens the QT and at very high levels causes arrhythmia and arrest.
- Rapid intravenous calcium causes bradycardia and arrhythmia — give it slowly with monitoring.
- Hypermagnesaemia produces heart block and cardiac arrest — antagonise with calcium gluconate.
Neurological and airway
easily missed
- Laryngospasm can obstruct the airway in severe hypocalcaemia.
- Seizures in a neonate with a normal total calcium may still reflect a low ionised fraction.
- Hypomagnesaemia and hypocalcaemia both lower the seizure threshold.
- Over-rapid phosphate replacement causes symptomatic hypocalcaemia.
Renal and bone
chronic
- Hypercalcaemia and hypercalciuria cause nephrocalcinosis and renal stones.
- Treated hypoparathyroidism risks hypercalciuria — target low-normal calcium and monitor urine calcium.
- Conventional oral phosphate for X-linked hypophosphataemic rickets causes secondary hyperparathyroidism and nephrocalcinosis.
- Untreated vitamin D deficiency leaves permanent skeletal deformity and short stature.
Treatment pitfalls
avoid
- Treating hypocalcaemia without checking magnesium — it will not correct.
- Giving a thiazide for hypercalcaemia — it worsens it; thiazides are reserved for the hypercalciuria of hypoparathyroidism.
- Adding calcium during tumour lysis — it risks metastatic calcification; reserve it for symptomatic hypocalcaemia.
- Refeeding a malnourished child at full calories — it triggers hypophosphataemia.
Prognosis & Disposition
The prognosis depends almost entirely on whether the cause is reversible. Acute hypocalcaemia from a correctable insult — sepsis, alkalosis, transient neonatal immaturity, a single refeeding event — resolves fully once the trigger is treated and the mineral replaced, with no long-term sequelae if caught before a seizure or arrhythmia. [2] Nutritional vitamin D deficiency rickets heals completely with calcium and vitamin D, though established skeletal deformity may take years to remodel. [5]
Permanent hypoparathyroidism and the inherited hypophosphataemias require lifelong management but carry an excellent prognosis when the biochemistry is held in range, with normal growth, neurodevelopment and life expectancy. [3] [9] The hazards shift to over-treatment: hypercalciuria and nephrocalcinosis in hypoparathyroidism, and secondary hyperparathyroidism with conventional phosphate in X-linked hypophosphataemic rickets, both of which are minimised by guideline-directed therapy and surveillance. [4] [10]
Disposition follows the severity. Symptomatic hypocalcaemia, severe hypercalcaemia, tumour lysis and refeeding hypophosphataemia need admission to a ward or intensive care for cardiac monitoring, intravenous therapy and serial biochemistry. Asymptomatic mild disturbances, controlled hypoparathyroidism and treated rickets are managed as outpatients with a clear safety-net for tetany, seizures, palpitations, weakness or collapse. [1]
Special Populations
Neonates
The neonate is the highest-risk group, with immature renal and parathyroid function and a high phosphate requirement for growth. Early and late neonatal hypocalcaemia each have distinct mechanisms and need different investigations, and the threshold to measure ionised calcium in any unwell or seizing neonate is low. [2] Maternal magnesium sulfate for eclampsia or preterm labour causes neonatal hypermagnesaemia with hypotonia and respiratory depression that resolves as the magnesium clears. [8]
Adolescents with anorexia nervosa
Adolescents with anorexia nervosa are the classic refeeding group, and the systematic review evidence confirms that the rate of refeeding determines the risk of hypophosphataemia. [12] Start calories low, escalate over days, and replace phosphate, potassium and magnesium prophylactically, monitoring the serum at least daily in the first week. [12]
Children with chronic kidney disease
In chronic kidney disease the retained phosphate drives secondary hyperparathyroidism, the failing kidney cannot make calcitriol, and the result is the combined mineral and bone disorder of chronic kidney disease: hypocalcaemia, hyperphosphataemia, high parathyroid hormone and vascular calcification. [1] Management is phosphate restriction with phosphate binders, calcitriol or its analogues, and control of the parathyroid hormone. [4]
Oncology patients at risk of tumour lysis
Children with high-burden Burkitt lymphoma, T-cell acute lymphoblastic leukaemia and other rapidly proliferating tumours need tumour lysis prophylaxis before and during the first days of chemotherapy, stratified by risk: hydration and allopurinol for low and medium risk, and rasburicase for high risk, with close monitoring of potassium, phosphate, calcium, uric acid and renal function. [11]
Children on diuretics and nephrotoxic drugs
Loop and thiazide diuretics, calcineurin inhibitors, aminoglycosides, cisplatin and chronic proton pump inhibitors all promote renal magnesium loss, and the child on long-term therapy may present with refractory hypocalcaemia or hypokalaemia before the magnesium is checked. [7]
Evidence, Guidelines & Regional Differences
Landmark evidence and guidelines
The Global Consensus Recommendations on Prevention and Management of Nutritional Rickets (Munns et al, 2016) set the international standard for vitamin D and calcium intake in infancy and childhood, defining deficiency and its prevention. [5] The 2024 Endocrine Society guideline on vitamin D for the prevention of disease updates the supplementation and screening recommendations across the lifespan. [6] The international hypoparathyroidism guideline (Khan et al, 2022) defines the targets and monitoring for permanent hypoparathyroidism, and the X-linked hypophosphataemia working-group guidelines establish burosumab as the standard of care in children. [4] [10]
Guidelines
The Coiffier et al evidence-based tumour lysis syndrome guideline (2008) remains the framework for risk stratification and prophylaxis, defining the high-risk groups that warrant rasburicase. [11] The O'Connor and Nicholls systematic review of refeeding hypophosphataemia in adolescents with anorexia nervosa (2013) underpins the refeeding-slowly doctrine. [12]
Regional and practice deltas
- ANZ: RACP and RCPCH-aligned practice favours ionised calcium in acute settings, rasburicase for high-risk tumour lysis, and burosumab as the funded standard for X-linked hypophosphataemic rickets in children. [10]
- North America: the Endocrine Society vitamin D guideline drives supplementation and screening, with burosumab widely available. [6]
- South Asia and high-prevalence regions: nutritional vitamin D deficiency rickets remains common, and the emphasis is on maternal and infant supplementation and sunlight. [5]
Controversies
The exact rate of refeeding in anorexia nervosa remains debated, with a historical trend toward lower starting calories balanced by evidence that cautious but adequate feeding shortens admission without excess refeeding risk. [12] The choice between conventional oral phosphate and burosumab for X-linked hypophosphataemic rickets is narrowing as access and funding for burosumab expand, and the target calcium range in hypoparathyroidism is debated in the context of hypercalciuria risk. [10] [4]
Exam Pearls
- Correct the calcium for albumin, and trust the ionised calcium in critical illness. [3]
- Symptomatic hypocalcaemia gets intravenous 10 percent calcium gluconate at 0.5 to 1 mL/kg slowly with cardiac monitoring. [1]
- Refractory hypocalcaemia is hypomagnesaemia until proven otherwise — replace magnesium first. [7]
- Vitamin D deficiency rickets: low calcium, low phosphate, high parathyroid hormone, high alkaline phosphatase, low 25-hydroxyvitamin D. [5]
- X-linked hypophosphataemic rickets: normal calcium, normal parathyroid hormone, low phosphate, high alkaline phosphatase, high fibroblast growth factor 23. [9]
- Hypercalcaemia gets saline and a loop diuretic, never a thiazide; add a bisphosphonate if severe or persistent. [3]
- Tumour lysis gives hyperphosphataemia, hypocalcaemia, hyperkalaemia and hyperuricaemia — hydrate and give rasburicase for high risk. [11]
- Refeeding drops phosphate as insulin drives it into cells — start low and replace. [12]
- DiGeorge (22q11) presents with neonatal hypocalcaemia, conotruncal heart disease, cleft palate and thymic hypoplasia. [2]
- Burosumab, an anti-fibroblast-growth-factor-23 monoclonal antibody, is now standard for X-linked hypophosphataemic rickets. [10]
- Calcium gluconate is preferred over calcium chloride peripherally because it is less vesicant. [2]
- Hypomagnesaemia causes refractory hypokalaemia as well as refractory hypocalcaemia, through ROMK disinhibition and parathyroid hormone suppression. [7]
Exam application bank (RACP / MRCPCH)
One-line answer
Calcium, magnesium and phosphate disorders share the parathyroid hormone, vitamin D and fibroblast growth factor 23 axis across bone, gut and kidney. Correct the calcium for albumin and trust the ionised fraction. Symptomatic hypocalcaemia gets intravenous 10 percent calcium gluconate 0.5 to 1 mL/kg slowly, and refractory hypocalcaemia is hypomagnesaemia until proven otherwise. Hypercalcaemia gets saline and a loop diuretic, never a thiazide. Vitamin D deficiency rickets has low calcium, low phosphate, high parathyroid hormone and high alkaline phosphatase, while X-linked hypophosphataemic rickets has normal calcium and parathyroid hormone with low phosphate and high fibroblast growth factor 23. Tumour lysis gives hyperphosphataemia with hypocalcaemia and needs rasburicase; refeeding drops phosphate and needs slow calories. [1]
Worked stems (answer without another resource)
Stem 1 — Classic presentation. [1] Map symptoms to mechanism; name the first investigation and first treatment step with dose and route.
Stem 2 — Unstable or complicated. [1] List red flags that force immediate resuscitation, intensive care or theatre, and what you do in the first 15 minutes.
Stem 3 — Atypical group. [1] Neonate, adolescent with anorexia, oncology patient, chronic kidney disease: how presentation and thresholds change.
Stem 4 — Differential trap. [1] Name the three closest rickets phenotypes and the biochemical discriminator for each.
Stem 5 — Disposition. [1] Who goes home with safety-netting, who is admitted, who needs intensive care, and what follow-up is mandatory.
Rapid viva checklist
- Corrected calcium formula and when to use the ionised value. [3]
- Acute management of symptomatic hypocalcaemia with the calcium gluconate dose and rate. [1]
- Why refractory hypocalcaemia points to magnesium and the magnesium sulfate dose. [7]
- The biochemistry that separates vitamin D deficiency rickets from X-linked hypophosphataemic rickets. [5][9]
- Hypercalcaemia resuscitation and the absolute contraindication of a thiazide. [3]
- Tumour lysis tetrad and the role of rasburicase versus allopurinol. [11]
- Refeeding hypophosphataemia and the feeding-rate doctrine. [12]
- DiGeorge syndrome and neonatal hypocalcaemia. [2]
- The mechanism by which fibroblast growth factor 23 controls phosphate and calcitriol. [9]
- Burosumab as the standard for X-linked hypophosphataemic rickets. [10]
Coverage self-check
If you cannot answer any stem above from this page alone [1], re-read the matching section — the page is intended to be self-sufficient for fellowship and RACP and MRCPCH questions on disorders of calcium, magnesium and phosphate in children.
References
- [1]Zieg J; Ghose S; Raina R Electrolyte disorders related emergencies in children. BMC Nephrol, 2024.PMID 39215244
- [2]Kim GK; Siller AF; Craven M; Bansal N Neonatal Endocrine Emergencies. Adv Pediatr, 2025.PMID 40582748
- [3]Mannstadt M; Bilezikian JP; Thakker RV; Hannan FM Hypoparathyroidism. Nat Rev Dis Primers, 2017.PMID 28857066
- [4]Khan AA; Guyatt G; Ali DS; Bilezikian JP Management of Hypoparathyroidism. J Bone Miner Res, 2022.PMID 36161671
- [5]Munns CF; Shaw N; Kiely M; Specker BL Global Consensus Recommendations on Prevention and Management of Nutritional Rickets. J Clin Endocrinol Metab, 2016.PMID 26745253
- [6]Demay MB; Pittas AG; Bikle DD; Diab DL Vitamin D for the Prevention of Disease: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab, 2024.PMID 38828931
- [7]Tseng MH; Konrad M; Ding JJ; Lin SH Clinical and genetic approach to renal hypomagnesemia. Biomed J, 2022.PMID 34767995
- [8]Kröse JL; de Baaij JHF Magnesium biology. Nephrol Dial Transplant, 2024.PMID 38871680
- [9]Haffner D; Emma F; Eastwood DM; Biosse Duplan M Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol, 2019.PMID 31068690
- [10]Ali DS; Carpenter TO; Imel EA; Ward LM X-Linked Hypophosphatemia Management in Children: An International Working Group Clinical Practice Guideline. J Clin Endocrinol Metab, 2025.PMID 39960858
- [11]Coiffier B; Altman A; Pui CH; Younes A Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J Clin Oncol, 2008.PMID 18509186
- [12]O'Connor G; Nicholls D Refeeding hypophosphatemia in adolescents with anorexia nervosa: a systematic review. Nutr Clin Pract, 2013.PMID 23459608